

Abstract
The active form of vitamin D, 1α,25-dihydroxyvitamin D [1α,25(OH)2D], is synthesized from its precursor 25 hydroxyvitamin D [25(OH)D] via the catalytic action of the 25(OH)D-1α-hydroxylase [1α(OH)ase] enzyme. Many roles in cell growth and differentiation have been attributed to 1,25(OH)2D, including a central role in calcium homeostasis and skeletal metabolism. To investigate the in vivo functions of 1,25(OH)2D and the molecular basis of its actions, we developed a mouse model deficient in 1α(OH)ase by targeted ablation of the hormone-binding and heme-binding domains of the 1α(OH)ase gene. After weaning, mice developed hypocalcemia, secondary hyperparathyroidism, retarded growth, and the skeletal abnormalities characteristic of rickets. These abnormalities are similar to those described in humans with the genetic disorder vitamin D dependent rickets type I [VDDR-I; also known as pseudovitamin D-deficiency rickets (PDDR)]. Altered non-collagenous matrix protein expression and reduced numbers of osteoclasts were also observed in bone. Female mutant mice were infertile and exhibited uterine hypoplasia and absent corpora lutea. Furthermore, histologically enlarged lymph nodes in the vicinity of the thyroid gland and a reduction in CD4- and CD8-positive peripheral T lymphocytes were observed. Alopecia, reported in vitamin D receptor (VDR)-deficient mice and in humans with VDDR-II, was not seen. The findings establish a critical role for the 1α(OH)ase enzyme in mineral and skeletal homeostasis as well as in female reproduction and also point to an important role in regulating immune function.
Vitamin D is a major regulator of mineral ion homeostasis and can also have a significant influence on the growth and differentiation of a variety of tissues (1–4). 1α,25-dihydroxyvitamin D [1α,25(OH)2D] is the most potent metabolite of vitamin D (5–7) and is believed to exert most of its actions via the 1α,25(OH)2D receptor (VDR) (8, 9), a member of the nuclear hormone receptor superfamily. The synthesis of 1α,25(OH)2D from its precursor 25-hydroxyvitamin D [25(OH)D] is catalyzed by the mitochondrial cytochrome P450 enzyme 25-hydroxyvitamin D-1α-hydroxylase [1α(OH)ase] or CYP27B1. Although a number of tissues can synthesize 1α,25(OH)2D, the kidney is the principal site of the circulating hormone. Recently cDNAs encoding the mouse (10), rat (11, 12), and human (13, 14) enzyme have been cloned, and the structures of the human (11, 14, 15) and mouse (16, 17) gene have been reported.
Vitamin D-dependent rickets type I (VDDR-I), also known as pseudovitamin D deficiency rickets (PDDR), is an autosomal recessive disorder characterized by low or undetectable levels of 1α,25(OH)2D, secondary hyperparathyroidism, hypocalcemia, hypophosphatemia, and severe rachitic lesions (18–21). VDDR-I is assumed to result from impaired synthesis of 1α,25(OH)2D, and, indeed, a number of 1α(OH)ase gene mutations have been reported in this disorder that result in diminished or absent 1α(OH)ase activity (13, 22–26).
To further investigate the functional role of the 1α(OH)ase enzyme, we generated mice deficient in 1α(OH)ase by gene targeting.
Materials and Methods
Methods including construction of the 1α(OH)ase targeting vector; transfection of embryonic stem (ES) cells and generation of 1α(OH)ase-deficient mice; Southern blot and PCR analysis of ES cell and mouse tail DNA; Northern blot analysis; biochemical and hormonal analyses; histological analysis; computer-assisted image analysis; immunohistochemistry; and fluorescence-activated cell sorter (FACS) lymphocyte phenotyping are presented in the supplemental data (which is published on the PNAS web site, www.pnas.org).
Results
The targeting vector shown in Fig. 1A was used to inactivate one allele of the 1α(OH)ase gene in ES cells. The inactivated allele lacked both the hormone-binding domain and the heme-binding domain of the enzyme. Two independent ES cell clones were used to generate two lines of mice heterozygous for the mutation, which were then interbred to generate 1α(OH)ase null (−/−) mice (Fig. 1B). Litter sizes were no different from normal, and the mutated allele was transmitted to the progeny with the expected Mendelian frequency. Thus, haploinsufficiency of the 1α(OH)ase did not affect embryonic survival. By reverse transcription (RT)-PCR, renal expression of the kidney 1α(OH)ase mRNA in (+/−) mice was reduced relative to that in (+/+) mice, and, in (−/−) mice, it was undetectable (Fig. 1C).
Figure 1.

Gene targeting of mouse 1α(OH)ase. (A) Schematic representation of the genomic region encoding the 1α(OH)ase and the creation of a mutant allele by homologous recombination. The neomycin resistance cassette (1.1 kb), which is introduced in the antisense orientation, is flanked by ≈1.4 and 2.8 kb of 5′ and 3′ 1α(OH)ase gene sequences and replaces exons VI and VII encoding the hormone-binding domain, exon VIII encoding the heme-binding domain, and part of exon IX. Genomic organization of the mouse 1α(OH)ase region was determined by cloning the gene from a 129sv/J library as described (17). (B) Analysis of genomic DNA isolated from pups born to two heterozygotes. For Southern blot, purified DNA was digested with BamHI. The mutated allele (3.4 kb) is distinguished from the wild-type allele (5.4 kb) using the 5′ probe indicated in A. For multiplex PCR, a 500-bp neomycin gene product and a 376-bp 1α(OH)ase exons VI and VII gene product were amplified. The positions of the primers are shown in A. (C) Reverse-transcriptase (RT)-PCR of total kidney RNA (20 μg) isolated from wild-type (+/+), heterozygous (+/−), or homozygous (−/−) 1α(OH)ase-deleted littermates with primers generating a 176-bp 1α(OH)ase product and a 508-bp glyceraldehyde-3-phosphate dehydrogenase (GAPDH) product. Densitometric analysis values of 1α(OH)ase/GAPDH for (+/+) = 100 ± 4%; (+/−) = 60 ± 5%; (−/−) = 0, mean ± SEM, n = 3. (D) Analysis of target gene expression in wild-type (+/+), heterozygous (+/−), and (−/−) 1α(OH)ase mice. Northern blot analyses of 24(OH)ase, calbindin D9K, and calbindin D28K were performed on intestinal and/or kidney RNA of mice of each genotype at 7 weeks. Densitometric analysis values for specific mRNA/18S of (+/+) vs. (+/−) vs. (−/−): intestine Calb9k, 100 ± 5% vs. 85 ± 5% vs. 2 ± 2%; kidney 24(OH)ase, 100 ± 4% vs. 103 ± 5% vs. 4 ± 2%; kidney Calb9k, 100 ± 5% vs. 91 ± 5% vs. 36 ± 4%; kidney Calb28k, 100 ± 5% vs. 96 ± 4% vs. 61 ± 3%, mean ± SEM, n = 3.
Circulating concentrations of 1,25(OH)2D were undetectable in the homozygous null mice and were somewhat lower (although not significantly so) in the heterozygotes relative to normals at 7 weeks of age (Table 1). Serum 25(OH)D concentrations were elevated in (−/−) mice relative to the heterozygotes and normals. Both serum calcium and phosphate concentrations were reduced in (−/−) mice relative to the (+/−) mice that were normal, and urinary phosphate was increased in the homozygous null mice. Serum parathyroid hormone concentrations were markedly elevated, the alkaline phosphatase concentrations were twice normal, and the body weight was substantially reduced in the homozygous null mice at this time (Table 1). The null mutant mice appeared grossly normal from birth until weaning; however, after weaning at 3 weeks, they displayed marked growth retardation (see Fig. 5, which is published as supplemental data).
Table 1.
Serum and urine profiles and body weight of 1α(OH)ase+/+, 1α(OH)ase+/−, and 1α(OH)ase−/− mice
| 1α(OH)ase+/+ | 1α(OH)ase+/− | 1α(OH)ase−/− | |
|---|---|---|---|
| Serum Ca2+, mM | 2.42 ± 0.23 | 2.29 ± 0.03 | 1.35 ± 0.01* |
Serum PO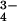 , mM , mM |
3.03 ± 0.13 | 3.44 ± 0.18 | 2.57 ± 0.19* |
| Serum 1,25(OH)2D, pg/ml | 86.33 ± 13.9 | 68 ± 20.1 | Undetectable† |
| Serum 25(OH)D, ng/ml | 18 ± 1.2 | 16.5 ± 3.5 | 32 ± 2** |
| Serum PTH, pg/ml | 29.83 ± 4.88 | 29.8 ± 3.02 | 440 ± 129.8** |
| Serum alkaline phosphatase, units/liter | 255.7 ± 60 | 247.7 ± 31.27 | 506.1 ± 31.7** |
Urinary PO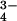 , mmol/mmol creatinine , mmol/mmol creatinine |
9.8 ± 0.7 | 10.8 ± 0.8 | 18.1 ± 0.9** |
| Body weight, g | 20.7 ± 0.7 | 23.3 ± 1.4 | 13.7 ± 1.0** |
Results are means ± SE of determinations in three mice of the same genotype.
, P < 0.05 compared with 1α(OH)ase+/+ mice;
, P < 0.005 compared to 1α(OH)ase+/+ mice. All tests were done on 7-week-old mice.
Less than 4 pg/ml.
The expression of the target genes for 1,25(OH)2D, 24(OH)ase, calbindin D9k, and calbindin D28k were assessed at 7 weeks by Northern blot analysis. There were decreased levels of intestinal calbindin D9k mRNA in the heterozygous mice, and it was absent in the null mutant mice (Fig. 1D). In the kidney, the expression of the 24(OH)ase mRNA was almost completely ablated in the null mutant mice, and the expression of renal calbindin D9k and calbindin D28k was reduced.
Typical features of advanced rickets were observed histologically in bone (Fig. 2 A and B). These features included widening of the epiphyseal growth plates predominantly because of an increase in the width of the hypertrophic zone, which was also disorganized; inadequate mineralization of cartilage, of the primary spongiosa, and of cortical bone; as well as an increase in osteoid in both trabecular and cortical bone (Fig. 2 A and B, and see Fig. 6A, which is published as supplemental data). Osteoblasts lining bone surfaces were increased (Fig. 2 A and B), and the trabecular bone area in the primary spongiosa was greater in null mutant than in wild-type mice (see Fig. 6B, which is published as supplemental data). Osteocalcin was decreased, however, in cortical bone (see Fig. 7 Upper, which is published as supplemental data) whereas type I collagen levels were normal (see Fig. 7 Lower, which is published as supplemental data). Osteoclast numbers and size appeared to be reduced in bone from the null mutant mice when compared with that of normal mice (Fig. 2C), and this finding was confirmed by the image analysis (see Fig. 6C which is published as supplemental data). Parathyroid glands, which stained positively for the calcium-sensing receptor (CaSR), were clearly enlarged in the homozygous mutant mice compared with wild-type mice (Fig. 3).
Figure 2.

Histology of bone from 1α(OH)ase null mutant mice (−/−) and wild-type littermates (+/+). (A) Proximal tibial epiphysis, (GP) growth plate, and metaphysis at 4 weeks, hematoxylin and eosin. (Upper) Bar = 500 μm; (Lower) Bar = 250 μm. (B) Von Kossa stain of undemineralized sections at 4 weeks. Counterstaining with toluidine blue. (DF) distal femur, bar = 500 μm; (GP) growth plate, bar = 100 μm; (TB) trabecular and (CB) cortical bone, bar = 50 μm, *, osteoid. (C) Tartrate-resistant acid phosphatase (TRAP) staining of osteoclasts in the primary spongiosa. Counterstaining with methylene blue. (Upper) Bar = 250 μm; (Lower) Bar = 50 μm
Figure 3.

Histology of parathyroid glands and adjacent thyroid tissue of 1α(OH)ase null mutant mice (−/−) and wild-type littermates (+/+). (Upper), Hematoxylin and eosin; (Lower), Immunostaining for calcium-sensing receptor (CaSR). Bar = 250 μm. Parathyroid glands are denoted by arrows and lymph nodes by arrowheads.
Large ectopic lymph nodes, which stained positive for CD4, were observed in the neck of the null mutant mice and were not seen in the wild-type (Fig. 3, and see Fig. 8, which is published as supplemental data). A reduction in the CD4- and CD8-positive peripheral T lymphocyte populations was also observed in null mutant mice compared with wild-type mice (Table 2, and see Fig. 9, which is published as supplemental data).
Table 2.
Analysis of peripheral T cells in littermate 1α(OH)ase+/+ and 1α(OH)ase−/− mice
| 1α(OH)ase+/+ | 1α(OH)ase−/− | |
|---|---|---|
| CD4+ | 42.9 ± 3.2 | 23.3 ± 3.4* |
| CD8+ | 15.19 ± 0.92 | 10.11 ± 0.95** |
| CD4+CD8+ | 1.32 ± 0.53 | 0.47 ± 0.01 |
| CD4−CD8− | 39.27 ± 3.1 | 66.14 ± 4.3** |
Results are means ± SE of determinations in three 7-wk-old mice of the same genotype.
, P < 0.01;
, P < 0.02;
, P < 0.005, compared with 1α(OH)ase+/+ mice.
Wild-type female mice were demonstrated to be cycling normally by examining vaginal smears, and the mice were killed at the estrus phase of the cycle. In contrast, the null mutant mice were acyclic and did not ovulate. The mutant mice were infertile; attempts to mate them were unsuccessful.
By gross anatomical analysis, the ovary and uterus of female 1α(OH)ase null mutant mice were smaller (Fig. 4) [ratio of ovary plus uterine wet weight (mg) to body weight (g): 1α(OH)ase+/+, 7.82 ± 1.12 (mean ± SE); 1α(OH)ase−/−, 2.04 ± 0.58; P < 0.01]. Histological analysis of female 1α(OH)ase null mutant mice showed uterine hypoplasia at 7 weeks, with a poorly developed endometrium (Fig. 4). Ovaries of these mice were smaller than in wild-type, ovarian follicles were immature, interstitial tissue was increased, and there were no corpora lutea (Fig. 4). Male reproductive organs were grossly normal in 1α(OH)ase null mutant mice (data not shown). Alopecia and loss of whiskers were not observed (see Fig. 5, which is published as supplemental data).
Figure 4.

Comparison of uterus and ovary of wild-type mice (+/+) (Left) and 1α(OH)ase mutant littermates (−/−) (Right). Histology, hematoxylin and eosin staining. Uterus and ovary (Upper), bar = 250 μm; ovary (Lower, Insets), bar = 50 μm. In the uterus, the arrow indicates the normal endometrium. In the ovary Upper panels, the Insets (enlarged in the Lower panels) show the normal corpora lutea in (+/+) mice and that they are absent in the (−/−) mice, which have hypertrophied interstitial cells.
Discussion
Mice lacking the capacity to synthesize 1,25(OH)2D demonstrated hypocalcemia, secondary hyperparathyroidism, hypophosphatemia, and phosphaturia. These abnormalities undoubtedly resulted from the diminished capacity of vitamin D-deficient animals to optimally absorb calcium and the subsequent stimulus for parathyroid gland enlargement, overproduction of parathyroid hormone and consequent effects on renal phosphate handling. In the 1α(OH)ase mutant mice, circulating 1,25(OH)2D3 levels were undetectable by RIA, emphasizing the nonredundant role played by the renal 1α(OH)ase in producing the hormonally active metabolite of vitamin D. It has been suggested that other enzymes such as the vitamin D-25-hydroxylase (CYP27) can catalyze 1α-hydroxylation of 25(OH)D (27), and this might occur in the kidney (28). However, this phenomenon clearly does not occur to any major extent in the homozygous mice although the ability of other enzyme systems to carry out local 1α-hydroxylation of vitamin D metabolites can now be studied further in the 1α(OH)ase knock-out model.
The well-established ligand dependency of the VDR was demonstrated by the marked reduction in mRNA levels encoding intestinal calbindin D9k, renal 24(OH)ase, renal calbindin D9k, and renal calbindin D28k mRNA levels, which was manifested by the 1α(OH)ase null mutant mice relative to wild-type littermates. Further studies can now be done to assess the relative contribution of 1,25(OH)2D relative to serum calcium in expression of these genes. The reduction in 24(OH)ase levels was especially marked. It had been speculated that 24,25(OH)2D might serve physiological roles, and, indeed, intramembranous bone formation is impaired in 24(OH)ase null mutant mice. However, this phenotype was rescued by mating these animals with VDR null mutant mice (29), pointing to the abnormally elevated 1,25(OH)2D levels as the cause of the defect. This observation provided evidence that 24,25(OH)2D is dispensable for bone development and that 24-hydroxylation of vitamin D metabolites represents the essential first step in their catabolism. This view is reinforced by the present study, in which, despite minimal 24(OH)ase activity, bone development was apparently normal up until weaning at sites of intramembranous bone formation such as calvarium and mandible (data not shown).
Typical rachitic lesions were observed in bone of the 1α(OH)ase-deficient mice after weaning, which resulted in the diminished growth of these animals. Inactivation of 1α(OH)ase in the null mice therefore produced a phenotype that was similar to that of the human genetic disease, VDDR-I, in which several mutations of the human 1α(OH)ase gene have now been described, and confirms that deficient 1α(OH)ase activity underlies this disorder. A rachitic phenotype has also been observed in mice homozygous for deletion of the VDR gene and in the human disorder VDDR-II, in which VDR gene mutations have been detected. The skeletal lesions in the VDR null mutant mice were largely reversed by normalizing ambient calcium and phosphate (30, 31). Further studies will be required to assess the role of calcium per se in the genesis of skeletal dysfunction in the 1α(OH)ase-deficient mice.
Elevated serum alkaline phosphatase levels in the 1α(OH)ase null mutant mice likely reflected increased osteoblast activity, and this increased activity was corroborated by the increased trabecular area in the null mutant bone, which was, however, poorly mineralized. The alterations in the levels of osteocalcin may reflect direct effects of 1,25(OH)2D deficiency on osteoblastic gene transcription, indirect effects such as those of hypocalcemia on the production or processing of matrix proteins, or the altered capacity of demineralized matrix to retain such proteins.
Osteoclasts were reduced in the 1α(OH)ase null mutant bone, and this may be a manifestation of the important role that 1,25(OH)2D plays in osteoclastogenesis (32). Nevertheless such a decrease was not observed in the VDR null mutant mouse bones (32, 33). The reason for this discrepancy is unclear but could suggest that 1,25(OH)2D functions through an alternate pathway in mediating its effects on osteoclast development.
Alopecia was noted in VDR knockout mice, and this is often, but not invariably, found in patients of some VDDR-II kindreds (34). It is not seen in patients with dietary vitamin D deficiency or VDDR-I, and, consistent with this, alopecia was not observed in the 1α(OH)ase null mutant mice. Hypocalcemia and/or hypophosphatemia per se are not causal of this lesion inasmuch as normalization of serum calcium and phosphate in VDR null mutant mice did not prevent the alopecia (30). Temporally controlled ablation of RXRα in keratinocytes generated a transgenic mouse model with alopecia similar to that observed in VDR knockout mice, suggesting a role of RXR/VDR heterodimers in directing normal hair cycling (35). Although the VDR has yet to be shown to have ligand-independent transcriptional activity, the present studies may indicate that it does have such effects in the hair follicle or that an alternate ligand may direct VDR action in integumentary tissues.
Two independent strains of VDR-deficient mice have been generated by different targeting strategies, and, although both similarly manifest a skeletal and integumentary phenotype, differences have been observed. In one study, the homozygous mutant mice were viable and fertile, and had no additional developmental abnormalities (36), in the other, the mice had a decreased lifespan after weaning, and female mice had uterine hypoplasia because of impaired ovarian folliculogenesis (33). In our 1α(OH)ase null mutant mice, similar to one of the VDR-ablated models, uterine hypoplasia and decreased ovarian size were found, folliculogenesis was clearly compromised, and the mice were not ovulating and were infertile. The fertility and reproductive capacity of the vitamin D-deficient female rat are markedly diminished (37), and this is not corrected by normalizing the hypocalcemia, but requires 1,25(OH)2D (38). The mechanism of action of 1,25(OH)2D in this process remains to be determined.
The reproductive organs of male 1α(OH)ase null mutant mice appeared grossly normal. Vitamin D deficiency does reduce the reproductive effectiveness of the male rat (39); however, diminished male fertility is largely corrected by restoring the plasma calcium level to normal.
Additional important findings in our study were the presence of ectopic lymph nodes in the neck of 1α(OH)ase null mutant mice, as well as in the heterozygous animals, and evidence for a redistribution in T cell populations in peripheral blood lymphocytes. No such findings have been reported in VDR null mutant mice although 1α,25(OH)2D3 has previously been reported to have profound effects on the immune system (40–42). Whether this represents further evidence that 1α,25(OH)2D may not exert all its effects via the VDR or that the 1α(OH)ase modulates immune function via metabolites other than 1α,25(OH)2D remains to be determined.
In summary, our studies have provided a mouse model of 1α(OH)ase deficiency that phenotypically is similar to the human disease, VDDR-I. The alterations in reproductive and immune function noted in the mice may be masked in the human by early introduction of vitamin D replacement therapy, or may be species specific. Our model should prove useful, however, in determining the precise role played by 1α,25(OH)2D in the physiology of several cells and organs, whether this role can be substituted by the calcium ion, whether the effects of 1α,25(OH)2D occur solely via the VDR, and whether other vitamin D metabolites or other products of the 1α(OH)ase exist and function.
Supplementary Material
Acknowledgments
We thank M. Gratton and K. McDonald for technical assistance, Dr. M. D. McKee for facilitating the image analysis and Dr. T. Owens for his helpful insights into the phenotype of the mice. This work was supported by Grants MT-5775 and MT-9315 from the Canadian Institutes of Health Research (CIHR; D.G. and G.N.H., respectively), Grant 00731 from the National Cancer Institute of Canada (D.G.), and from the Kidney Foundation of Canada (G.N.H.). D.M. is the recipient of a CIHR postdoctoral fellowship.
Abbreviations
- 1α(OH)ase
25(OH)D-1α-hydroxylase
- VDDR-I
vitamin D dependent rickets type I
- VDR
vitamin D receptor
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Bouillon R, Okamura W H, Norman A W. Endocr Rev. 1995;16:200–257. doi: 10.1210/edrv-16-2-200. [DOI] [PubMed] [Google Scholar]
- 2.Studzinski G P, McLane J A, Uskokovic M R. CRC Crit Rev Eukaryotic Gene Exp. 1993;3:279–312. [PubMed] [Google Scholar]
- 3.Walters M R. Endocr Rev. 1992;13:719–764. doi: 10.1210/edrv-13-4-719. [DOI] [PubMed] [Google Scholar]
- 4.Jones G, Strugnell S A, DeLuca H F. Physiol Rev. 1998;78:1193–1231. doi: 10.1152/physrev.1998.78.4.1193. [DOI] [PubMed] [Google Scholar]
- 5.Holick M F, Schnoes H K, DeLuca H F, Suda T, Cousins R J. Biochemistry. 1971;10:2799–2804. doi: 10.1021/bi00790a023. [DOI] [PubMed] [Google Scholar]
- 6.Lawson D E M, Fraser D R, Kodicek E, Morris H R, Williams D H. Nature (London) 1971;230:228–230. doi: 10.1038/230228a0. [DOI] [PubMed] [Google Scholar]
- 7.Norman A W, Myrtle J F, Midgett R J, Nowicki H G, Williams V, Popjak G. Science. 1971;173:51–54. doi: 10.1126/science.173.3991.51. [DOI] [PubMed] [Google Scholar]
- 8.Haussler M R, McCain T A. N Engl J Med. 1977;297:974–983. doi: 10.1056/NEJM197711032971804. [DOI] [PubMed] [Google Scholar]
- 9.Liao J, Ozone K, Sone T, McDonnell D P, Pike J W. Proc Natl Acad Sci USA. 1990;87:9751–9755. doi: 10.1073/pnas.87.24.9751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takeyama K-I, Kitanaka S, Sato T, Kobori M, Yanagisawa J, Kato S. Science. 1997;277:1827–1830. doi: 10.1126/science.277.5333.1827. [DOI] [PubMed] [Google Scholar]
- 11.St-Arnaud R, Messerlian S, Moir J M, Omdahl J L, Glorieux F H. J Bone Miner Res. 1997;12:1552–1559. doi: 10.1359/jbmr.1997.12.10.1552. [DOI] [PubMed] [Google Scholar]
- 12.Shinki T, Shimada H, Wakino S, Anazawa H, Hayashi M, Saruta T, DeLuca H F, Suda T. Proc Natl Acad Sci USA. 1997;94:12920–12925. doi: 10.1073/pnas.94.24.12920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu G K, Lin D, Zhang M Y H, Bikle D D, Shackleton C H L, Miller W L, Portale A A. Mol Endocrinol. 1997;11:1961–1970. doi: 10.1210/mend.11.13.0035. [DOI] [PubMed] [Google Scholar]
- 14.Monkawa T, Yoshida T, Wakino S, Shinki T, Anazawa H, DeLuca H F, Suda T, Hayashi M, Saruta T. Biochem Biophys Res Commun. 1997;239:527–533. doi: 10.1006/bbrc.1997.7508. [DOI] [PubMed] [Google Scholar]
- 15.Fu G K, Portale A A, Miller W L. Cell Biol. 1997;16:1499–1507. doi: 10.1089/dna.1997.16.1499. [DOI] [PubMed] [Google Scholar]
- 16.Kimmel-Jehan C, DeLuca H F. Biochim Biophys Acta. 2000;1475:109–113. doi: 10.1016/s0304-4165(00)00065-9. [DOI] [PubMed] [Google Scholar]
- 17.Panda D K, Al Kawas S, Seldin M F, Hendy G N, Goltzman D. J Bone Miner Res. 2001;16:46–56. doi: 10.1359/jbmr.2001.16.1.46. [DOI] [PubMed] [Google Scholar]
- 18.Prader A, Illig R, Heierli E. Helv Paediatr Acta. 1961;16:452–468. [PubMed] [Google Scholar]
- 19.Scriver C R, Fraser D, Kooh S W. In: Calcium Disorders. Heath D, Marx S J, editors. London: Butterworth; 1982. pp. 1–46. [Google Scholar]
- 20.Glorieux F H, St-Arnaud R. In: Vitamin D. Feldman D, Glorieux F H, Pike J W, editors. San Diego: Academic; 1997. pp. 755–764. [Google Scholar]
- 21.Liberman U A, Marx S J. In: Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. 4th Ed. Favus M J, editor. Philadelphia: Lippincott Williams & Wilkins; 1999. pp. 323–328. [Google Scholar]
- 22.Kitanaka S, Takeyama K, Murayama A, Sato T, Okumura K, Nogami M, Hasegawa Y, Niimi H, Yanagisawa J, Tanaka T, Kato S. N Engl J Med. 1998;338:653–661. doi: 10.1056/NEJM199803053381004. [DOI] [PubMed] [Google Scholar]
- 23.Wang J T, Lin C J, Burridge S M, Fu G K, Labuda M, Portale A A, Miller W L. Am J Hum Genet. 1998;63:1694–1702. doi: 10.1086/302156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoshida T, Monkawa T, Tenenhouse H S, Goodyer P, Shinki T, Suda T, Wakino S, Hoyashi M, Saruta T. Kidney Int. 1998;54:1437–1443. doi: 10.1046/j.1523-1755.1998.00133.x. [DOI] [PubMed] [Google Scholar]
- 25.Kitanaka S, Murayama A, Sakaki T, Inouye K, Seino Y, Fukumoto S, Shima M, Yukizane S, Takayanagi M, Niimi H, et al. J Clin Endocrinol Metab. 1999;84:4111–4117. doi: 10.1210/jcem.84.11.6131. [DOI] [PubMed] [Google Scholar]
- 26.Smith S J, Rucka A K, Berry J L, Davies M, Mylchreest S, Paterson C R, Heath D A, Tassabehji M, Read A P, Mee A P, Mawer E B. J Bone Miner Res. 1999;14:730–739. doi: 10.1359/jbmr.1999.14.5.730. [DOI] [PubMed] [Google Scholar]
- 27.Axen E, Postlind H, Sjoberg H, Wikvall K. Proc Natl Acad Sci USA. 1994;91:10014–10018. doi: 10.1073/pnas.91.21.10014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Araya Z, Norlin M, Postlind H. FEBS Lett. 1996;390:10–14. doi: 10.1016/0014-5793(96)00617-5. [DOI] [PubMed] [Google Scholar]
- 29.St-Arnaud R, Arabian A, Travers R, Barletta F, Ravel-Pandya M, Chapin K, Depovere J, Mathieu C, Christakos S, Demay M B, Glorieux F H. Endocrinology. 2000;141:2658–2666. doi: 10.1210/endo.141.7.7579. [DOI] [PubMed] [Google Scholar]
- 30.Li Y C, Amling M, Pirro A E, Priemel M, Meuse J, Baron R, Delling G, Demay M B. Endocrinology. 1998;139:4391–4396. doi: 10.1210/endo.139.10.6262. [DOI] [PubMed] [Google Scholar]
- 31.Amling M, Priemel M, Holzmann T, Chapin K, Rueger J M, Baron R, Demay M B. Endocrinology. 1999;140:4982–4987. doi: 10.1210/endo.140.11.7110. [DOI] [PubMed] [Google Scholar]
- 32.Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, et al. Proc Natl Acad Sci USA. 1998;95:3597–3602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshizawa T, Handa Y, Uematsu Y, Takeda S, Sekine K, Yoshihara Y, Kawakami T, Arioka K, Sato H, Uchiyama Y, et al. Nat Genet. 1997;16:391–396. doi: 10.1038/ng0897-391. [DOI] [PubMed] [Google Scholar]
- 34.Fraher L J, Karmali R, Hinde F R J, Hendy G N, Jani H, Nicholson L, Grant D, O'Riordan J L H. Eur J Pediatr. 1986;145:389–395. doi: 10.1007/BF00439245. [DOI] [PubMed] [Google Scholar]
- 35.Li M, India A K, Warot X, Brocard J, Messaddeq N, Kato S, Metzger D, Chambon P. Nature (London) 2000;407:633–636. doi: 10.1038/35036595. [DOI] [PubMed] [Google Scholar]
- 36.Li Y C, Pirro A E, Amling M, Delling G, Baron R, Bronson R, Demay M B. Proc Natl Acad Sci. USA. 1997;94:9831–9835. doi: 10.1073/pnas.94.18.9831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Halloran B P, Deluca H F. J Nutr. 1980;110:1573–1580. doi: 10.1093/jn/110.8.1573. [DOI] [PubMed] [Google Scholar]
- 38.Kwiecinski G G, Petrie G I, Deluca H F. Am J Physiol. 1989;256:483–487. doi: 10.1152/ajpendo.1989.256.4.E483. [DOI] [PubMed] [Google Scholar]
- 39.Kwiecinski G G, Petrie G I, Deluca H F. J Nutr. 1989;119:741–744. doi: 10.1093/jn/119.5.741. [DOI] [PubMed] [Google Scholar]
- 40.Abe E, Miyaura C, Sakagami H, Takeda M, Konno K, Yamazaki T, Yoshika S, Suda T. Proc Natl Acad Sci USA. 1981;78:4990–4994. doi: 10.1073/pnas.78.8.4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nunn J D, Katz D R, Barker S, Fraher L J, Hewison M, Hendy G N, O'Riordan J L H. Immunology. 1986;59:479–484. [PMC free article] [PubMed] [Google Scholar]
- 42.Provvedini D M, Rulot C M, Sobol R E, Tsoukas C D, Manolagas S C. J Bone Miner Res. 1987;2:239–241. doi: 10.1002/jbmr.5650020311. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.