

Abstract
A new series of phosphodiesterase-9 (PDE9) inhibitors that contain a scaffold of 6-amino-pyrazolopyrimidinone have been discovered by a combination of structure-based design and computational docking. This procedure significantly saved load of chemical synthesis and is an effective method for the discovery of inhibitors. The best compound 28 has an IC50 of 21 nM and 3.3 µM respectively for PDE9 and PDE5, and about three orders of magnitude of selectivity against other PDE families. The crystal structure of the PDE9 catalytic domain in complex with 28 has been determined and shows a hydrogen bond between 28 and Tyr424. This hydrogen bond may account for the 860-fold selectivity of 28 against PDE1B, in comparison with about 30-fold selectivity of BAY73-6691. Thus, our studies suggest that Tyr424, a unique residue of PDE8 and PDE9, is a potential target for improvement of selectivity of PDE9 inhibitors.
The second messengers adenosine and guanosine 3’,5’-cyclic monophosphate (cAMP and cGMP) modulate many physiological processes, such as cardiac and smooth muscle contraction, steroid hormone function, platelet aggregation, apoptosis, leukocyte migration, adrenal hyperplasia, inflammation, axon guidance and regeneration, memory, and circadian regulation.1–6 Cyclic nucleotide phosphodiesterases (PDEs) hydrolyze intracellular cAMP and cGMP and thus play key roles in the cellular processes.7–10 Family-selective PDE inhibitors have been widely studied as therapeutics for treatment of human diseases.11–17 The most successful example of this drug class is the PDE5 inhibitor sildenafil that has been marketed as drugs (Viagra & Revatio) for the treatments of male erectile dysfunction and pulmonary hypertension.11,12
PDE9 is a family of cGMP-specific enzymes and has the lowest KM among cGMP-specific PDE families.7–10 Selective PDE9 inhibitors have been studied as potential therapeutics for treatment of diabetes and CNS diseases such as Alzheimer′s disease.18–30 The crystal structures of the PDE9A catalytic domain in complex with the inhibitors20,31–33 and the substrate cGMP34 have not only provided insight into the catalytic mechanism, but also served as templates to lead to discovery of PDE9 inhibitors with high affinity.18,20–22,30 However, some of these PDE9 inhibitors showed only moderate selectivity against PDE1,18,20 which might cause side effects for the treatment of CNS diseases since PDE1 is abundant in brain.30 To explore improvement of the selectivity, we designed a novel series of PDE9 inhibitors (scheme 1) that contain a scaffold of 6-amino-pyrazolopyrimidinone and are capable of directly forming a hydrogen bond with Tyr424 that is unique to PDE9 and PDE8. The best compound in this series of the PDE9 inhibitors showed about three orders of magnitude of selectivity against PDE1. We also reported a combined procedure of structure-based inhibitor design and computational docking, which significantly saved load of chemical synthesis and is thus an effective method for discovery of PDE9 inhibitors.
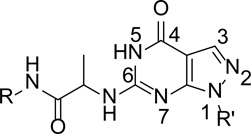
Scheme 1.

Chemical structure of our compounds
Results
Design of a new series of PDE9 inhibitors
Since most current PDE9 inhibitors contain a pharmacophore of pyrazolopyrimidinone,18, 20–23 we chose 6-amino-pyrazolopyrimidinone as the scaffold to design a new series of PDE9 inhibitors by using the template structure of PDE9 in complex with (R)-BAY73-669132 (10r, 1-(2-chlorophenyl)-6-(3,3,3-trifluoro-2-methylpropyl)-1H-pyrazolo[3,4-d]pyrimidine-4(5H)-one). As revealed by the crystal structure, 10r forms two hydrogen bonds with invariant Gln453 and stacks against Phe456 (Fig. 1A), thus accounting for its IC50 of 22 nM against PDE9.32 The structure also shows that Tyr424 of PDE9A2 does not form a hydrogen bond with 10r, but is located at another site to the small pocket where the fluoromethyl group of 10r occupies (Fig. 1A). To target interaction with Tyr424, we manually constituted a linker chain with various lengths and different substitution groups in a graphic terminal. The linker contains an alanine, in hope of that the methyl side chain or the Cβ atom would interact with Gly452 and Phe441 to prevent the substitution from entering the neighboring small subpocket (top right in Fig. 1B) so as to steer the chain toward Tyr424. The amide group of the alanine linker would form a hydrogen bond with Tyr424 for improvement of selectivity against other PDE families. Finally, a phenyl group at the end of the substitution is designed to occupy the partially open hydrophobic pocket that is composed of Met365, Phe441, and Val460 of PDE9A2 for further enhancement of the inhibitor affinity (Fig. 1B). This idea was originated on the basis of BAYER’s compound BR4872 (Drs. Andreas Knorr and Frank Wunder, personal communication) and was then verified by docking various fragments into the binding pocket of PDE9A2.
Figure 1.

Design of a new type of PDE9 inhibitors. (A) Surface presentation of 10r binding to the pocket of PDE9A2. Color codes are white for carbon, blue for nitrogen, red for oxygen of PDE9A2, and orange for chlorine. Inhibitor 10r is shown in both surface and stick models (light green). (B) Design of a scaffold that may potentially form a hydrogen bond with Tyr424 and fit tightly to the PDE9 pocket.
With the Surflex-Dock35,36 program, the X-ray crystal structure of PDE9A in complex with the inhibitor (S)-BAY73-6691 (10s, PDB code: 3K3H)32was chosen to validate our docking. In the docking with the bound inhibitor, the top 30 poses of the docked conformations have the RMSDs in a range of 0.4 to 1.9 Å from the true position in the crystal structure and eight of them were less than 1.5 Å, thus indicating that the Surflex-Dock program could produce reliable docking results. Under identical docking conditions, the manually designed fragments were docked into the binding pocket of PDE9A. This manual design and computational docking led to identification of the basic fragment shown in scheme 1 and significantly saved load of synthesis.
Chemical synthesis
A series of compounds with various substituents of R and R’ (Scheme 1) were synthesized by following the similar protocols in literature.37,38 Thus, the commercially available arylhydrazines (1) were converted to the corresponding pyrazoles (3) by reacting with ethoxymethyl-enemalononitrile (2). Subsequent oxidation yielded carboxamide (4) that then underwent intermolecular cyclization by urea to deliver 1-aryl (akyl)-4,6-dihydroxypyrazolo[3,4-d]pyrimidine (5). Chlorination of 5 by phosphorus oxychloride in the presence of phosphorus pentachloride yielded 1-aryl(akyl)-4,6-dichloropyrazolo[3,4-d] pyrimidine (6). Hydrolysis of 6 by KOH produced 1-aryl(alkyl)-6-chloro-4-hydroxypyrazolo[3,4-d] pyrimidines (7–8) (Scheme 2).
Scheme 2.

Syntheses of intermediates 7–8. (a) EtOH, 60°C; (b) 30% H2O2/25% NH3 .H2O =1:3 (v/v), rt; (c) Urea, 200°C; (d) PCl5, POCl3, reflux; (e) 2 N (aq) KOH, 60°C.
The reaction of 2-((tert-butoxycarbonyl)amino)propanoic acid (11) with aromatic amines (12) yielded compounds 13. Subsequent deprotection of 13 in HCl (g)-methanol solution afforded the corresponding compounds 14–21 (Scheme 3).
Scheme 3.

Syntheses of intermediates 14–21. (a) DMF, 1-methyllimidazole, then DECP, rt. (b) MeOH/HCl, 0°C.
The desired compounds 26–35 were prepared according to well-documented methods,37,38 as outlined in scheme 4.
Scheme 4.

Syntheses of targeted compounds
Enzymatic properties of PDE9A inhibitors
Among compounds 26–35, 28 shows the highest potency with an IC50 of 21 nM against the PDE9A catalytic domain (Table 1). The chlorine atom of the 2-chlorophenyl group on the R' substitution is important for the inhibitor binding, as shown by the over 300-fold affinity loss when the 2-chlorophenyl group is replaced with the phenyl group (see comparison between compounds of 28 and 34, Table 1). However, the same Cl substitution at R’ has less impact when methoxylphenyl at R is changed to an ethoxylphenyl group, as shown by only 11-fold difference in the potency between 29 and 35 (Table 1). Replacement of the methoxylphenyl group at the R position with an ethoxylphenyl group reduced the inhibitory potency of the compound containing the o-Cl-phenyl at R’ by ~35-fold (comparison between 28 and 29). The explanation to these changes is not clear, but might be due to that the 2-Cl group plays a role in maintaining intra-molecular relationship between the Cl-phenyl group at R’ and oxy-phenyl group at R to pick up hydrophobic interactions within the PDE9 pocket, as revealed by the structure described in the next section. The R substitutions at the m-, o-, and p-positions of the phenyl ring are also important for binding of the inhibitors. A comparison of 28 with 31 and 32 shows that substitution of the methoxyl at the p-position (28) rather than at the o- or m-position increases inhibitory potency by 10- to 15-fold (Table 1). This result may be reasonably explained by the crystal structure, in which the m- and o-substitutions might disturb the van der Waals’ interaction with Met365. Among the p-substitutions tested here, the methoxyl group is most potent (compare 26–30 and 33). This is perhaps due to the hydrophilic nature of the nearby binding pocket so that too much hydrophobicity would not favor the inhibitor binding.
Table 1.
Chemical structures and affinity of compounds with PDE9A
 | |||
|---|---|---|---|
| Compounds | R | R′ | IC50 (µM) |
| 26 | 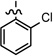 |
0.721 ± 0.096 | |
| 27 | 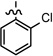 |
0.052 ± 0.008 | |
| 28 | 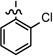 |
0.021 ± 0.005 | |
| 29 | 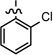 |
0.696 ± 0.059 | |
| 30 | 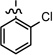 |
0.595 ± 0.071 | |
| 31 | 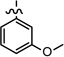 |
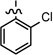 |
0.237 ± 0.055 |
| 32 | 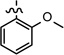 |
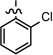 |
0.308 ± 0.057 |
| 33 | 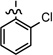 |
0.204 ± 0.022 | |
| 34 | 7.51 ± 0.88 | ||
| 35 | 7.47 ± 0.93 | ||
In term of inhibitory selectivity, compound 28 shows at least 150-fold selectivity against other PDE families (Table 2). Especially worth mentioning is the 860-fold selectivity against PDE1B, which is significantly better than those of other PDE9 inhibitors such as 10r (about 30 folds)18 and PF-04447943 (9, about 170 folds)30 that has been the best PDE9 inhibitor having great pharmacological potency for the treatment of CNS diseases.22,28,30 Compound 28 barely inhibits PDE8 (IC50 of ~ 100 µM, Table 2), although PDE8 also contains a tyrosine at the corresponding position of Tyr424 of PDE9A2, implying that the inhibitory selectivity would be jointly determined by multiple residues at the active sites of the PDE families.
Table 2.
Affinity of inhibitor 28 with PDE families.
| Proteins | IC50 (µM) |
|---|---|
| PDE9A2 (181–506) | 0.021 ± 0.005 |
| PDE1B(1–516) | 18.0 ± 1.6 |
| PDE2A(580–941) | >50 |
| PDE3A(679–1087) | >50 |
| PDE4D2 (86–413) | 15.7 ± 2.4 |
| PDE5A1 (535–860) | 3.3 ± 0.5 |
| PDE7A1(130–482) | >100 |
| PDE8A1 (480–820) | ~100 |
| PDE10A2(448–789) | >50 |
Binding of 28 to the PDE9 catalytic domain
As expected by the structure-based design and computational docking, compound 28 binds to the active site of PDE9 in a similar pattern as other PDE9 inhibitors.20,32 While the overall orientation of 28 is comparable with that of 10r, the pyrazolopyrimidinone ring shows about 0.7 Å positional translation (Fig. 2). This movement should not be surprising because 10r does not fully occupy the binding pocket of PDE9 (Fig. 1A) and thus implies a feasibility to design inhibitors with various substitution that would lock the orientation of pyrazolopyrimidinone. Our compound 28 is a good example for design of PDE9 inhibitors, in which 28 not only retains the affinity with PDE9 but also shows the significantly improved selectivity against other PDE families (Table 2).
Figure 2.

Binding of 28 to PDE9A2. (A) Surface presentation on binding of inhibitor 28 (green sticks) to the active site of PDE9. The chlorine atom of 28 is in orange color. Dotted lines represent the hydrogen bonds between 28 and PDE9 residues Tyr424 and Gln453. (B) Superposition of PDE9-28 (green and cyan) over PDE9-10r (yellow and salmon). Thr451 and Ala452 show significant positional differences between PDE9-28 and PDE9-10r. The pyrazolopyrimidinone ring has about 0.7 Å difference between two structures. (C) Electron density for 28 bound to chain A of the PDE9 dimer. The mesh (Fo - Fc) map was drawn from the PDE9 structure with omission of 28 and contoured at 2.5σ. (D) Density for two molecules of 28 bound to chain B. The lower 28 binds to the active site of PDE9.
The electron density maps of (2Fo-Fc) and (Fo-Fc) clearly revealed that the (S)-enantiomer of 28 binds to the PDE9 pocket (Fig. 2C). This is consistent with the use of (L)-2-aminopropanoic acid as the source material to synthesize 11. The amide oxygen of 28 forms a hydrogen bond with tyrosyl OH of Tyr424 as originally designed. The O4 and N5 atoms of pyrimidine of 28 are involved in two hydrogen bonds with the nitrogen and oxygen of side chain amide of the invariant Gln453 (2.8 Å and 2.9 Å), which are comparable with 2.7 and 2.9 Å for the same bonds in the structure of PDE9A-10r.32 The pyrazolopyrimidine ring of 28 forms aromatic π-stacking against Phe456 of PDE9A (Fig. 2). In addition, compound 28 contacts via van der Waals’ interaction with residues Phe251, His252, Met365, Ile403, Asn405, Leu420, and Phe441.
An unusual observation of the structural study is that a third molecule of 28 binds to molecule B in the dimer of the PDE9A catalytic domain (Fig. 2D). This molecule has weaker electron density than the two molecules that bind to the active site of PDE9 (Fig. 2D), indicating its partial occupancy. Its B-factor of 81.6 Å2 is significantly higher than the average of 50.2 Å2 for the two molecules of 28 bound at the active site (Table 3), consistently indicating its partial occupancy. The third molecule of 28 forms van der Waals’ interactions with Pro440, Phe441, Thr451, Ala452, Ile454, and Gly455 of molecule B in the PDE9 dimer, and Ala499 and Glu502 of symmetry-related molecule A. It also contacts 28 that binds to the active site of molecule B in the PDE9 dimer (Fig. 2D). A structural superposition by using comparable residues 190–493 showed no substantial conformational differences between molecules A and B of the PDE9 dimer (RMSD of 0.24 Å), but the intermolecular interactions in the crystal lattice. Thus, the partial occupancy of the third 28 in molecule B is probably an artifact resulted from the crystal lattice packing.
Table 3.
Statistics on diffraction data and structure refinement
| Data collection | PDE9-28 |
| Space group | P41212 |
| Unit cell (a, c, Å) | 104.3, 270.1 |
| Resolution (Å) | 2.7 |
| Total measurements | 216,409 |
| Unique reflections | 41,120 |
| Completeness (%) | 99.7 (98.2)* |
| Average I/σ | 17.4 (4.4)* |
| Rmerge | 0.093 (0.37)* |
| Structure Refinement | |
| R-factor | 0.212 |
| R-free | 0.245 (10.0%)‡ |
| Resolution (Å) | 15–2.7 |
| Reflections | 38,971 |
| RMS deviation for | |
| Bond length | 0.007 Å |
| Bond angle | 1.3° |
| Average B-factor (Å2) | |
| Protein | 52.8 (5390)§ |
| Inhibitor | 50.2 (62) & 81.6 (31)# |
| Zn | 61.0 (2) |
| Mg | 39.9 (2) |
| Water | 38.9 (18) |
The numbers in parentheses are for the highest resolution shell.
The percentage of reflections omitted for calculation of R-free.
The number of atoms in the crystallographic asymmetric unit.
The average B-factor of 50.2 Å2 is for the two molecules of 28 that bind at the active site, while 81.6 Å2 is for the third 28.
Discussion
Implication on inhibitor selectivity
While several PDE9 inhibitors have been shown to have high affinity with PDE9,18, 20–22 a concern for these inhibitors is the selectivity against PDE1. Since PDE9 inhibitors have been extensively studied as the therapeutics for treatment of the CNS diseases19–23,25,27,28,30 and PDE1 is abundant in brain,7–9 improvement on the selectivity against PDE1 appears to be very important for lowering risk of side effects.30 The moderate selectivity of several PDE9 inhibitors against PDE118,20 might be explained by the comparability between the active sites of PDE9 and PDE1, as revealed by the structural superposition (Fig. 3A). The sequence alignment shows that the residues involved in binding of inhibitor 28 are well conserved, such as Tyr424 of PDE9A2 versus Phe392 of PDE1B, Phe251 ~ Tyr222, Leu421 ~ Met389, and Phe441 ~ Leu409, in addition to the identical residues of Met365, Ile403, Leu420, Gln453, and Phe456. Since Tyr424 is unique to PDE9 and PDE8, it may be speculated that an inhibitor that is capable of forming a hydrogen bond with the tyrosyl OH of Tyr424 of PDE9 would increase its selectivity against PDE1 as well as other PDE families.20,21,30 This idea was illustrated by the design of 9 that forms a hydrogen bond indirectly with Tyr424 via a bridging water and shows improved selectivity (170-fold) against PDE1C.30 In comparison, our inhibitor 28 that directly interacts with Tyr424 shows about 860-fold selectivity against PDE1B.
Figure 3.

Structural comparison. (A) Superposition of PDE9A (green sticks) over PDE1B (cyan), and PDE8A (orange). Inhibitor 28 of PDE9A is shown as purple sticks. IBMX of PDE8A is shown as yellow sticks. (B) A surface presentation for comparison on binding of 28 (green sticks) and cGMP (cyan) to the PDE9A active site. Residues were obtained from the superposition of the PDE9 complex structures. (C) Comparison on binding of 28 and IBMX (salmon). (D) Comparison on 28 and Pfizer inhibitor 7 (PF7, yellow). Residues Thr451 and Ala452 showed significant movement in the PDE9-PF7 structure.
On the other hand, since PDE8 also contains a tyrosine at the corresponding position of Tyr424, the selectivity against PDE8 would be a concern. The enzymatic measurement shows that inhibitor 28 barely inhibits PDE8A (IC50 = ~100 µM versus 21 nM for PDE9, Table 2). This might be explained by a distant relationship between the active sites of PDE8 and PDE9. The structure comparison shows that the selectivity of PDE9 inhibitors against PDE8 may be determined by several factors: (1) the poorly comparable positions of several active site residues such as Phe441 of PDE9A2 versus Val768/Phe769 of PDE8A, (2) the different conformations of the M-loops, and (3) the mutation of amino acids such as Phe785 of PDE8 to Val460 of PDE9A2.
Hints on further improvement of PDE9 inhibitors
While our inhibitor 28 shows high affinity to PDE9 and good selectivity against other PDE families, it does not seem to fully occupy the active site of PDE9 (Fig. 3B), implying a room for further improvement. The surface presentation of the PDE9 structure reveals empty space near the metal binding pocket (left of Fig. 3B). Although this space could in principle accommodate modifications of the current PDE9 inhibitors to improve their affinity and selectivity, occupation of this space would require a huge molecule. Since larger inhibitors have poorer capacity of penetration through the blood barrier, inhibitors that can fully occupy the active site pocket would be expected to have poor bioavailability and to be unrealistic. Nevertheless, there is a small space near the Cβ atom of alanine of 28 (top right of Fig. 3B) and an extension from the chiral carbon of alanine, such as replacement of the methyl group with an ethyl or propyl group would be expected to fill the space and thus improve the affinity and selectivity.
In addition, there is another small pocket (top of Fig. 3B) neighboring the main pocket of the active site, which is composed of Leu421 and the M-loop residues Val447 and Lys449 and gated by Phe441 and Ala452. The structural superposition shows that significant positional changes on the residues in the M-loop and N-terminal to the invariant Gln453 were induced by some but not all PDE9 inhibitors. Inhibitors with large substitutions of 10r (PDE code 3E3K), 10s (3K3H) and Pfizer inhibitor 7 (3JSW)20 pushed away the Cα atoms of Thr451 and Ala452 by over 1 Å (3–4 times the RMSD, Figs. 2B & 3D). In comparison, IBMX (2HD1), cGMP (3DYQ), and inhibitor 28 have no direct or weak interaction with Ala452 and Phe441, and thus show no substantial positional movement (Figs. 3B & 3C). The positional differences of these residues appear to be the consequence of the inhibitor binding because the M-loop is not involved in crystallographic lattice packing. Thus, the above comparison implies that the gatekeepers of Phe441 and Ala452 could be pushed slightly apart and a suitable substitution on the side chain of alanine of 28 might be able to enter the small neighboring pocket of Leu421, Val447 and Lys449, thus leading to a new type of PDE9 inhibitors.
Experimental Section
Protein expression and purification
The cDNA of the wild type PDE9A2 for expression of the catalytic domain (residues 181–506) was subcloned and purified following the similar protocols previously reported.31 In brief, the recombinant plasmid was transferred into E. coli. strain BL21 (Codonplus, Stratagene). The E. coli. cells carrying the pET-PDE9A2 plasmids were grown in LB medium at 37°C to absorption A600 = 0.6 - 0.8, and then 0.1 mM isopropyl β-D-thiogalactopyranoside was added to induce the expression. The cells after induction were grown at 15°C overnight. Recombinant PDE9A2 proteins were purified by the column chromatography of Ni-NTA affinity (Qiagen), Q-sepharose ionic exchange, and gel filtration. A typical batch of purification yielded 20–60 mg PDE9A2 from a 1-liter cell culture. The PDE9A2 proteins had purity greater than 90% as shown by SDS-PAGE.
The catalytic domains of PDE4D2 (86–413), PDE5A1 (535–860), PDE7A1 (130–482), PDE8A2 (480–820), and PDE10A2 (448–789) were purified by using similar protocols previously published.40–43 PDE1B (1–516) was partially purified at UNC, and PDE2A and PDE3A were prepared by refolding (unpublished data).
Enzymatic assay
The enzymatic activities of the catalytic domains of PDE9A and other PDEs were measured by using cGMP or cAMP as substrates. The assay buffer contains 20–50 mM Tris-HCl (pH 7.5 – 8.0), 10 mM MgCl2 or 4 mM MnCl2, 1 mM DTT, 10–20 nM 3H-cGMP or 3H-cAMP (20,000–30,000 cpm/assay, GE Healthcare). The reaction was carried out at room temperature for 15 min and then terminated by addition of 0.2 M ZnSO4. The reaction product 3H-GMP was precipitated out by 0.25 M BaSO4, whereas unreacted 3H-cGMP remained in the supernatant. Radioactivity in the supernatant was measured in 2.5 ml Ultima Gold liquid scintillation cocktails (PerkinElmer) by a PerkinElmer 2910 liquid scintillation counter. For the measurement of IC50 of inhibitors, at least eight concentrations of inhibitors were used in the presence of 3H-cAMP or 3H-cGMP. The enzyme concentration in a range of 10–100 ng/ml, which hydrolyzed up to 70% of the substrate, was chosen for each inhibitory assay. Each measurement was repeated at least three times. The IC50 values were calculated by nonlinear regression.
Molecular docking
Fragments output from the manual design were docked into the active site of PDE9A by using the Surflex-Dock protocol of program Tripos SYBYL.35,36 For Surflex-Dock in SYBYL 7.3.5, energy-based protocol approach was used to locate the binding site. The threshold and bloat values were set to 0.5 and 0, respectively. The search grid extends 6 Å beyond the protein dimensions. Ring flexibility and soft grid treatment were turned on. The obtained poses were evaluated by the consensus scores of ChemScore, G_Score, D_score, PMF_Score. Thirty configurations of the ligands were output from the docking program for analysis.
Crystallization and structure determination
The crystals of PDE9A2 were grown by using hanging drop vapor diffusion method. The PDE9A2 (181–506) (10–15 mg/mL) in a buffer of 50 mM NaCl, 20 mM Tris.HCl pH 7.5, 1 mM β-mercaptoethanol and 1 mM EDTA was mixed with 2 mM IBMX for 2h before setting-up of crystallization against the well buffer of 2.0 M Na formate, 0.1 M HEPES pH 7.5, 5% xylitol at 4°C. The PDE9A-IBMX complex crystals were formed after 1d and reached the maximum size in 2 weeks. Crystals of the PDE9A-28 complex were prepared by soaking PDE9-IBMX co-crystals in the crystallization buffer plus 5 mM 28 at 25°C for 3 days. The crystals were flash-frozen in liquid nitrogen by using the well buffer plus saturated xylitol as the cryosolvent. X-ray diffraction data were collected at 100 K on Beamline BL17U of Shanghai Synchrotron Radiation Facility, China (Table 3) and processed by HKL2000.44 The structure of the PDE9A2-28 complex was solved by the molecular replacement, using the PDE9A2 catalytic domain as the initial model. The resulted model was rebuilt by program O45 and refined by CNS.46
Analysis of synthesized compounds
1H NMR and 13C NMR spectra were recorded at room temperature on a Mercury-Plus 300 instrument with TMS as an internal reference. LC/MS was run on a LCMS-2010A. GC/MS was run on a Finnigan Voyager with an electron impact (70 eV) mass selective detector and an innowax 30 m × 0.25 mm × 0.25 um capillary apolar column. GC-MS method: initial temperature, 50°C; initial time, 3 min; ramp, 20°C /min; final temperature, 250°C; final time, 2 min. EI mass spectra were recorded on the Thermo DSQ mass spectrometer. Thin-layer chromatography was performed on precoated silica gel F-254 plates (0.25 mm, E. Merck) and was visualized with UV light. Element analyses were carried out on an element vario EL series element analyzer and have errors within ± 0.3% for CHN elements. Melting points were determined on a WRS-1B digital melting point apparatus and were not corrected. All starting materials and reagents were purchased from commercial suppliers and used directly without further purification.
Synthesis of compounds 26–35
To a 50 mL round-bottom flask were added iso-PrOH (25 mL), compound 7 or 8 (0.4 mmol), 2-amino-N-arylpropan-amide (compounds 14–21) (0.48 mmol) and TEA (0.4 mmol, 0.04 g). The mixture was refluxed for 6 h, then allowing the mixture to cool to room temperature. After removal of the solvent, the residue was purified by flash column chromatography on silica gel (eluting with DCM: MeOH = 25:1) to provide the pure target compounds. The following are the parameters for compounds 26–35.
2-((1-(2-chlorophenyl)-4-hydroxy-1H-pyrazolo[3,4-d]pyrimidin-6-yl)amino)-N-phenylpropanam ide (26)
yield: 41%. Mp: 138–139 °C. 1H NMR (300 MHz, CDCl3) δ: 10.80 (s, 1H), 8.64 (br s, 1H), 8.13 (s, 1H), 7.51-7.46 (m, 2H), 7.35-7.24 (m, 6H), 7.06 (d, J = 6.3 Hz, 2H), 4.56-4.49 (m,1H), 1.52 (d, J = 7.3 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ: 170.9, 159.4, 155.2, 152.8, 137.1, 136.7, 131.5, 130.3, 130.1, 129.4, 128.7, 127.2, 124.5, 119.9, 100.5, 51.5, 17.5. ESI-MS: m/z = 407[M-H]−. IR (KBr, cm−1): 3310, 2980, 1690, 1610, 1548, 1253. Anal. Calcd for C20H17ClN6O2: C: 58.75; H: 4.19; N: 20.56; Found: C: 58.56; H: 4.31; N: 20.59.
2-((1-(2-chlorophenyl)-4-hydroxy-1H-pyrazolo[3,4-d]pyrimidin-6-yl)amino)-N-(p-tolyl)propanamide (27)
yield: 45%. Mp: 141–143 °C. 1H NMR (300 MHz, CDCl3) δ: 10.70 (s, 1H), 8.33 (br s, 1H), 8.11 (s, 1H), 7.50-7.26 (m, 4H), 7.08-6.96 (m, 5H), 4.54-4.45 (m, 1H), 2.29 (s, 3H), 1.49 (d, J = 6.9 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ: 170.6, 159.4, 155.2, 152.9, 136.8, 135.3, 134.5, 134.3, 131.6, 130.4, 130.1, 129.4, 129.3, 127.3, 120.1, 113.9, 100.6, 51.4, 20.9, 17.4. ESI-MS: m/z = 421[M-H]−. IR (KBr, cm−1): 3310, 2980, 1690, 1610, 1548, 1246. Anal. Calcd for C21H19ClN6O2: C: 59.65; H: 4.53; N: 19.87; Found: C: 59.86; H: 4.35; N: 19.69.
2-((1-(2-chlorophenyl)-4-hydroxy-1H-pyrazolo[3,4-d]pyrimidin-6-yl)amino)-N-(4-methoxypheny l)propanamide (28)
yield: 45%. Mp: 140–141°C. 1H NMR (300 MHz, CDCl3) δ: 10.57 (s, 1H), 8.14 (s, 1H), 8.13 (br s, 1H), 7.52-7.29 (m, 4H), 7.11-6.77 (m, 5H), 4.55-4.46 (m, 1H), 3.78 (s, 3H), 1.52 (d, J = 6.9 Hz, 3H) 13C NMR (75 MHz, CDCl3) δ: 170.6, 158.4, 156.6, 155.2, 152.9, 136.7, 135.3, 131.6, 130.4, 130.1, 130.0, 129.5, 127.3, 121.9, 113.9, 100.6, 55.5, 51.3, 17.5. ESI-MS: m/z = 437[M-H]−. IR (KBr, cm−1): 3310, 2980, 1680, 1548, 1240. Anal. Calcd for C21H19ClN6O3: C: 57.47; H: 4.36; N: 19.15; Found: C: 57.56; H: 4.21; N: 18.95.
2-((1-(2-chlorophenyl)-4-hydroxy-1H-pyrazolo[3,4-d]pyrimidin-6-yl)amino)-N-(4-ethoxyphenyl) propanamide (29)
yield: 50%. Mp: 169–171°C. 1H NMR (300 MHz, CDCl3) δ: 8.37 (br s, 1H), 8.07 (s, 1H), 7.47-7.43 (m, 2H), 7.36-7.06 (m, 4H), 6.95 (d, J = 6.6 Hz, 1H), 6.76 (d, J = 9.0 Hz, 2H), 4.49-4.46 (m,1H), 3.99-3.93 (q, J = 6.9 Hz, 2H), 1.43 (d, J = 7.5 Hz, 3H), 1.39 (d, J =7.2 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ:170.7, 159.3, 155.9, 152.2, 136.7, 135.3, 131.6, 130.3, 130.1, 129.9, 127.3, 121.8, 114.5, 100.6, 63.7, 51.3, 17.4, 14.8. ESI-MS: m/z = 451 [M-H]−. IR (KBr, cm−1): 3310, 2980, 1690, 1610, 1552, 1240, 1050. Anal. Calcd for C22H21ClN6O3: C, 58.34; H, 4.67; N, 18.56. Found: C: 58.23; H: 4.70; N: 18.63.
2-((1-(2-chlorophenyl)-4-hydroxy-1H-pyrazolo[3,4-d]pyrimidin-6-yl)amino)-N-(4-isopropoxyph enyl)propanamide (30)
yield: 47%. Mp: 120–122 °C. 1H NMR (300 MHz, CDCl3) δ: 8.39(br s, 1H), 8.08 (s, 1H), 7.47-7.23 (m, 4H), 7.09-6.97 (m, 3H), 6.76-6.73 (d, J = 8.7 Hz, 2H), 4.50-4.42(m,2H), 1.46 (d, J = 6.9 Hz, 3H), 1.31 (d, J = 6.0 Hz, 6H).13C NMR (75 MHz, CDCl3) δ: 170.6, 159.4, 155.2, 154.7, 152.9, 136.7, 135.2, 131.5, 130.3, 130.1, 129.9, 121.8, 116.1, 100.5, 51.3, 22.0, 17.5. ESI-MS: m/z = 465 [M-H]−. IR (KBr, cm−1): 3300, 2980, 1690, 1610, 1548, 1230. Anal. Calcd for C23H23ClN6O3: C, 59.16; H, 4.97; N, 18.00 Found: C: 58.89; H: 4.93; N: 17.83.
2-((1-(2-chlorophenyl)-4-hydroxy-1H-pyrazolo[3,4-d]pyrimidin-6-yl)amino)-N-(3-methoxypheny l)propanamide (31)
yield: 42%. Mp: 230–232 °C (lit. Mp: °C). 1H NMR (300 MHz, CDCl3) δ: 8.40(br s, 1H), 8.10 (s, 1H), 7.48-7.45 (m, 2H), 7.36-7.07 (m, 4H), 7.01 (d, J = 6.3 Hz, 1H), 6.62 (d, J = 8.1 Hz, 2H), 4.53-4.45 (m, 1H), 3.74 (s, 3H), 1.50 (d, J = 6.9 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ:170.7, 159.9, 159.5, 155.2, 152.8, 138.3, 136.7, 135.2, 131.6, 130.4, 130.1, 129.4, 127.3, 112.1, 109.9, 106.1, 100.5, 55.3, 51.6, 51.4, 17.5. ESI-MS: m/z = 437 [M-H]−. IR (KBr, cm−1) : 3310, 2968, 1683, 1610, 1547, 1290, 1060. Anal. Calcd for C21H19ClN6O3: C: 57.47; H: 4.36; N: 19.15; Found: C: 57.39; H: 4.37; N: 19.25.
2-((1-(2-chlorophenyl)-4-hydroxy-1H-pyrazolo[3,4-d]pyrimidin-6-yl)amino)-N-(2-methoxypheny l)propanamide (32)
yield: 36%. P: 115–117 °C. 1H NMR (300 MHz, CDCl3) δ: 8.18 (s, 1H), 8.14 (br s, 1H), 7.42-7.27 (m, 3H), 7.16-7.04 (m, 3H), 6.98-6.84 (m, 3H), 4.61-4.52 (m, 1H), 3.76 (s, 3H), 1.57 (d, J = 6.9 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ:170.8, 159.6, 155.5, 152.6, 148.1, 136.7, 135.4, 131.7, 130.2, 129.7, 129.3, 127.0, 126.8, 124.3, 121.0, 120.2, 109.9, 100.6, 55.7, 51.7, 18.34. ESI-MS: m/z = 437 [M-H]−. IR (KBr, cm−1) : 3310, 2990, 1690, 1610, 1539, 1288, 1020. Anal. Calcd for C21H19ClN6O3: C: 57.47; H: 4.36; N: 19.15; Found: C: 57.61; H: 4.31; N: 19.01.
2-((1-(2-chlorophenyl)-4-hydroxy-1H-pyrazolo[3,4-d]pyrimidin-6-yl)amino)-N-(4-(methylthio)p henyl)propanamide (33)
yield: 44%. Mp: 161–163 °C. 1H NMR (300 MHz, CDCl3) δ: 10.70 (s, 1H), 8.33 (br s, 1H), 8.13 (s, 1H), 7.52-7.47 (m, 2H), 7.39-7.32 (m, 3H), 7.31-6.97 (m, 4H), 4.56-4.46 (m, 1H), 2.45 (s, 3H), 1.53 (d, J = 6.9 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ: 170.4, 159.4, 155.1, 152.8, 136.8, 135.3, 134.6, 134.1, 131.6, 130.4, 130.2, 129.4, 127.6, 127.4, 120.5, 100.6, 51.5, 17.4, 16.6. ESI-MS: m/z = 453 [M-H]−. IR (KBr, cm−1) : 3300, 2984, 1684, 1610, 1542, 1256, 1040. Anal. Calcd for C21H19ClN6O2S: C, 55.44; H, 4.21; N, 18.47. Found: C: 55.59; H: 4.05; N: 18.41.
2-((4-hydroxy-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-6-yl)amino)-N-(4-methoxyphenyl)propana mide (34)
yield: 41%. Mp: 283–285 °C. 1H NMR (300 MHz, DMSO-d6) δ: 10.67(s, 1H), 10.17(br s, 1H), 8.06 (d, J = 6.3 Hz, 3H), 7.54 (d, J = 9.0 Hz, 2H), 7.36-7.23 (m, 3H), 7.11 (d, J = 6.3 Hz, 1H), 6.89 (d, J = 9.0 Hz, 2H), 4.55-4.46 (m, 1H), 3.71(s, 1H). 13C NMR (75 MHz, DMSO-d6) δ: 170.2, 157.0, 155.0, 153.6, 152.8, 138.7, 135.9, 131.7, 128.7, 125.6, 120.5, 120.3, 113.7, 101.2, 55.0, 50.7, 18.4. ESI-MS: m/z = 403[M-H]−. IR (KBr, cm−1) : 3280, 2960, 1690, 1504, 1407, 1268, 945. Anal. Calcd for C21H20N6O3: C: 62.37; H: 4.98; N: 20.78; Found: C: 62.56; H: 4.81; N: 20.59.
N-(4-ethoxyphenyl)-2-((4-hydroxy-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-6-yl)amino)propanam ide (35)
yield: 37%. Mp: 277–279 °C. 1H NMR (300 MHz, DMSO-d6) δ: 10.67(s, 1H), 10.16 (br s, 1H), 8.06 (d, J = 6.3 Hz, 3H), 7.53 (d, J = 7.8 Hz, 2H), 7.35-7.31 (m, 3H), 7.25 (d, J = 6.9 Hz, 1H), 6.87 (d, J = 7.8 Hz, 2H), 4.53-4.48 (m, 1H), 3.97 (d, J = 6.9 Hz, 2H), 1.48 (d, J = 5.7 Hz, 3H), 1.48 (d, J = 6.3 Hz, 3H). 13C NMR (75 MHz, DMSO-d6) δ: 170.1, 157.0, 154.3, 153.6, 152.8, 138.7, 135.8, 131.6, 128.7, 125.6, 120.4, 120.3, 114.2, 101.2, 62.9, 50.6, 18.4, 14.6. ESI-MS: m/z = 417[M-H]−. IR (KBr, cm−1) : 3416, 2984, 1690, 1500, 1399, 1240. Anal. Calcd for C22H22N6O3: C: 63.15; H: 5.30; N: 20.08; Found: C: 63.06; H: 5.36; N: 19.78.
Supplementary Material
Acknowledgments
This work is partially supported by US NIH GM59791 (H. Ke), National High Technology Research and Development Program of China (863 Program, No. 2006AA09Z446), Natural Science Foundation of China (20872182, 20802095, 21103234), Natural Science Foundation of Guangdong Province (S2011030003190), Natural Science Foundation of Department of Education in Guangdong Province (CXZD1006), Natural Science Foundation of Guangzhou City (2010Y1-C531), and Fundamental Research Funds for the Central Universities (11ykzd05). The diffraction data were collected on beamline BL17U of Shanghai Synchrotron Radiation Facility.
Abbreviations Used
- PDE
Phosphodiesterase
- cAMP
adenosine 3’,5’-cyclic monophosphate
- cGMP
guanosine 3’,5’-cyclic monophosphate
Footnotes
Supporting Information Available: Sequence alignment of the active site residues of 21 PDE genes. This material is available free of charge via the Internet at http://pubs.acs.org.
PDB ID Code: The atomic coordinates and structure factors have been deposited into the RCSB Protein Data Bank with accession number 4GH6.
References
- 1.Antoni F. Molecular diversity of cyclic AMP signaling. Front. Neuroendocrin. 2000;21:103–132. doi: 10.1006/frne.1999.0193. [DOI] [PubMed] [Google Scholar]
- 2.De Felice FG, Wasilewska-Sampaio AP, Barbosa AC, Gomes FC, Klein WL, Ferreira ST. Cyclic AMP enhancers and Abeta oligomerization blockers as potential therapeutic agents in Alzheimer's disease. Curr. Alzheimer. Res. 2007;4:263–271. doi: 10.2174/156720507781077287. [DOI] [PubMed] [Google Scholar]
- 3.Jarnaess E, Taskén K. Spatiotemporal control of cAMP signalling processes by anchored signalling complexes. Biochem. Soc. Trans. 2007;35:931–937. doi: 10.1042/BST0350931. [DOI] [PubMed] [Google Scholar]
- 4.O'Neill JS, Maywood ES, Chesham JE, Takahashi JS, Hastings MH. cAMP-dependent signaling as a core component of the mammalian circadian pacemaker. Science. 2008;320:949–953. doi: 10.1126/science.1152506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piper M, van Horck F, Holt C. The role of cyclic nucleotides in axon guidance. Adv. Exp. Med. Biol. 2007;621:134–143. doi: 10.1007/978-0-387-76715-4_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zaccolo M, Movsesian MA. cAMP and cGMP signaling cross-talk: role of phosphodiesterases and implications for cardiac pathophysiology. Circ. Res. 2007;100:1569–1578. doi: 10.1161/CIRCRESAHA.106.144501. [DOI] [PubMed] [Google Scholar]
- 7.Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol. Rev. 2006;58:488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- 8.Omori K, Kotera J. Overview of PDEs and their regulation. Circ. Res. 2007;100:309–327. doi: 10.1161/01.RES.0000256354.95791.f1. [DOI] [PubMed] [Google Scholar]
- 9.Conti M, Beavo JA. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: Essential components in cyclic nucleotide signaling. Ann. Rev. Biochem. 2007;76:481–511. doi: 10.1146/annurev.biochem.76.060305.150444. [DOI] [PubMed] [Google Scholar]
- 10.Ke H, Wang H. Crystal structures of phosphodiesterases and implications on substrate specificity and inhibitor selectivity. Curr. Top. Med. Chem. 2007;7:391–403. doi: 10.2174/156802607779941242. [DOI] [PubMed] [Google Scholar]
- 11.Rotella DP. Phosphodiesterase 5 inhibitors: current status and potential applications. Nat. Rev. Drug Discov. 2002;1:674–682. doi: 10.1038/nrd893. [DOI] [PubMed] [Google Scholar]
- 12.Galie N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N. Eng. J. Med. 2005;353:2148–2157. doi: 10.1056/NEJMoa050010. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt CJ. Phosphodiesterase inhibitors as potential cognition enhancing agents. Curr. Top. Med. Chem. 2010;10:222–230. doi: 10.2174/156802610790411009. [DOI] [PubMed] [Google Scholar]
- 14.Lipworth BJ. Phosphodiesterase-4 inhibitors for asthma and chronic obstructive pulmonary disease. Lancet. 2005;365:167–175. doi: 10.1016/S0140-6736(05)17708-3. [DOI] [PubMed] [Google Scholar]
- 15.Blokland A, Schreiber R, Prickaerts J. Improving memory: a role for phosphodiesterases. Curr. Pharm. Des. 2006;12:2511–2523. doi: 10.2174/138161206777698855. [DOI] [PubMed] [Google Scholar]
- 16.Menniti FS, Faraci WS, Schmidt CJ. Phosphodiesterases in the CNS: targets for drug development. Nat. Rev. Drug Discov. 2006;5:660–670. doi: 10.1038/nrd2058. [DOI] [PubMed] [Google Scholar]
- 17.Houslay MD, Schafer P, Zhang KY. Keynote review: phosphodiesterase-4 as a therapeutic target. Drug Discov. Today. 2005;10:1503–1519. doi: 10.1016/S1359-6446(05)03622-6. [DOI] [PubMed] [Google Scholar]
- 18.Wunder F, Tersteegen A, Rebmann A, Erb C, Fahrig T, Hendrix M. Characterization of the first potent and selective PDE9 inhibitor using a cGMP reporter cell line. Mol. Pharmacol. 2005;68:1775–1781. doi: 10.1124/mol.105.017608. [DOI] [PubMed] [Google Scholar]
- 19.van der Staay FJ, Rutten K, Bärfacker L, Devry J, Erb C, Heckroth H, Karthaus D, Tersteegen A, van Kampen M, Blokland A, Prickaerts J, Reymann KG, Schröder UH, Hendrix M. The novel selective PDE9 inhibitor BAY 73-6691 improves learning and memory in rodents. Neuropharmacology. 2008;55:908–918. doi: 10.1016/j.neuropharm.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 20.Verhoest PR, Proulx-Lafrance C, Corman M, Chenard L, Helal CJ, Hou X, Kleiman R, Liu S, Marr E, Menniti FS, Schmidt CJ, Vanase-Frawley M, Schmidt AW, Williams RD, Nelson FR, Fonseca KR, Liras S. Identification of a brain penetrant PDE9A inhibitor utilizing prospective design and chemical enablement as a rapid lead optimization strategy. J. Med. Chem. 2009;52:7946–7949. doi: 10.1021/jm9015334. [DOI] [PubMed] [Google Scholar]
- 21.Deninno MP, Andrews M, Bell AS, Chen Y, Eller-Zarbo C, Eshelby N, Etienne JB, Moore DE, Palmer MJ, Visser MS, Yu LJ, Zavadoski WJ, Michael GE. The discovery of potent, selective, and orally bioavailable PDE9 inhibitors as potential hypoglycemic agents. Bioorg. Med. Chem. Lett. 2009;19:2537–2541. doi: 10.1016/j.bmcl.2009.03.024. [DOI] [PubMed] [Google Scholar]
- 22.Hutson PH, Finger EN, Magliaro BC, Smith SM, Converso A, Sanderson PE, Mullins D, Hyde LA, Eschle BK, Turnbull Z, Sloan H, Guzzi M, Zhang X, Wang A, Rindgen D, Mazzola R, Vivian JA, Eddins D, Uslaner JM, Bednar R, Gambone C, Le-Mair W, Marino MJ, Sachs N, Xu G, Parmentier-Batteur S. The selective phosphodiesterase 9 (PDE9) inhibitor PF-04447943(6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-1-(tetrahydro-2H-pyran-4-yl)-1,5-dihydro-4H-pyrazolo[3,4-d]pyri midin-4-one) enhances synaptic plasticity and cognitive function in rodents. Neuropharmacology. 2011;61:665–676. doi: 10.1016/j.neuropharm.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 23.Vardigan JD, Converso A, Hutson PH, Uslaner JM. The selective phosphodiesterase 9 (PDE9) inhibitor PF-04447943 attenuates a scopolamine-induced deficit in a novel rodent attention task. J. Neurogenet. 2011;25:120–126. doi: 10.3109/01677063.2011.630494. [DOI] [PubMed] [Google Scholar]
- 24.Nagasaki S, Nakano Y, Masuda M, Ono K, Miki Y, Shibahara Y, Sasano H. Phosphodiesterase type 9 (PDE9) in the human lower urinary tract: an immunohistochemical study. BJU Int. 2012;109:934–940. doi: 10.1111/j.1464-410X.2011.10429.x. [DOI] [PubMed] [Google Scholar]
- 25.Liddie S, Anderson K, Paz A, Itzhak Y. The effect of phosphodiesterase inhibitors on extinction of cocaine-induced conditioned place preference in mice. J. Psychopharmacol. 2012 May 16; doi: 10.1177/0269881112447991. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 26.Saravani R, Karami-Tehrani F, Hashemi M, Aghaei M, Edalat R. Inhibition of phosphodiestrase 9 induces cGMP accumulation and apoptosis in human breast cancer cell lines, MCF-7 and MDA-MB-468. Cell Prolif. 2012;45:199–206. doi: 10.1111/j.1365-2184.2012.00819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kroker KS, Rast G, Giovannini R, Marti A, Dorner-Ciossek C, Rosenbrock H. Inhibition of acetylcholinesterase and phosphodiesterase-9A has differential effects on hippocampal early and late LTP. Neuropharmacology. 2012;62:1964–1974. doi: 10.1016/j.neuropharm.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 28.Kleiman RJ, Chapin DS, Christoffersen C, Freeman J, Fonseca KR, Geoghegan KF, Grimwood S, Guanowsky V, Hajós M, Harms JF, Helal CJ, Hoffmann WE, Kocan GP, Majchrzak MJ, McGinnis D, McLean S, Menniti FS, Nelson F, Roof R, Schmidt AW, Seymour PA, Stephenson DT, Tingley FD, Vanase-Frawley M, Verhoest PR, Schmidt CJ. Phosphodiesterase 9A regulates central cGMP and modulates responses to cholinergic and monoaminergic perturbation in vivo. J. Pharmacol. Exp. Ther. 2012;341:396–409. doi: 10.1124/jpet.111.191353. [DOI] [PubMed] [Google Scholar]
- 29.Hanna CB, Yao S, Wu X, Jensen JT. Identification of phosphodiesterase 9A as a cyclic guanosine monophosphate-specific phosphodiesterase in germinal vesicle oocytes: a proposed role in the resumption of meiosis. Fertil. Steril. 2012;98:487–495. doi: 10.1016/j.fertnstert.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verhoest PR, Fonseca KR, Hou X, Proulx-Lafrance C, Corman M, Helal CJ, Claffey MM, Tuttle JB, Coffman KJ, Liu S, Nelson FR, Kleiman RJ, Menniti FS, Schmidt CJ, Vanasse-Frawley MA, Liras S. Design and discovery of 6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-1-(tetrahydro-2H-pyran4-yl)-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one (PF-04447943) a selective brain penetrant PDE9A inhibitor for the treatment of cognitive disorders. J. Med. Chem. 2012 Jul 25; doi: 10.1021/jm3007799. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 31.Huai Q, Wang H, Zhang W, Colman R, Robinson H, Ke H. Crystal structure of phosphodiesterase 9 shows orientation variation of inhibitor 3-isobutyl-1-methylxanthine binding. Proc. Natl. Acad. Sci. USA. 2004;101:9624–9629. doi: 10.1073/pnas.0401120101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang H, Luo X, Ye M, Hou J, Robinson H, Ke H. Insight into binding of phosphodiesterase-9a selective inhibitors by crystal structures and mutagenesis. J. Med. Chem. 2010;53:1726–1731. doi: 10.1021/jm901519f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hou J, Xu J, Liu M, Zhao R, Luo H-B, Ke H. Structural asymmetry of phosphodiesterase-9, potential protonation of a glutamic acid, and role of the invariant glutamine. PLoS ONE. 2011;6:e18092. doi: 10.1371/journal.pone.0018092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu S, Mansour MN, Dillman KS, Perez JR, Danley DE, Aeed PA, Simons SP, Lemotte PK, Menniti FS. Structural basis for the catalytic mechanism of human phosphodiesterase 9. Proc. Natl. Acad. Sci. USA. 2008;105:13309–13314. doi: 10.1073/pnas.0708850105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jain AN. Surflex-Dock 2.1: Robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. J. Comput. Aided. Mol. Des. 2007;21:281–306. doi: 10.1007/s10822-007-9114-2. [DOI] [PubMed] [Google Scholar]
- 36.Fang JS, Huang D, Zhao W, Ge H, Luo H-B, Xu J. A new protocol for predicting novel GSK-3β ATP competitive inhibitors. J. Chem. Inf. Model. 2011;51:1431–1438. doi: 10.1021/ci2001154. [DOI] [PubMed] [Google Scholar]
- 37.Das J, Moquin RV, Pitt S, Zhang R, Shen DR, McIntyre KW, Gillooly K, Doweyko AM, Sack JS, Zhang H. Pyrazolo-pyrimidines: A novel heterocyclic scaffold for potent and selective p38α inhibitors. Bioorg. Med. Chem. Lett. 2008;18:2652–2657. doi: 10.1016/j.bmcl.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 38.Sykes BM, Atwell GJ, Hogg A, Wilson WR, O’Connor CJ, Denny WA. N-Substituted 2-(2,6-Dinitrophenylamino)propanamides: Novel prodrugs that release a primary amine via nitroreduction and intramolecular cyclization. J. Med. Chem. 1999;42:346–355. doi: 10.1021/jm960783s. [DOI] [PubMed] [Google Scholar]
- 39.Wang H, Liu Y, Huai Q, Cai J, Zoraghi R, Francis SH, Corbin JD, Robinson H, Xin Z, Lin G, Ke H. Multiple conformations of phosphodiesterase-5: implications for enzyme function and drug development. J. Biol. Chem. 2006;281:21469–21479. doi: 10.1074/jbc.M512527200. [DOI] [PubMed] [Google Scholar]
- 40.Wang H, Liu Y, Chen Y, Robinson H, Ke H. Multiple elements jointly determine inhibitor selectivity of cyclic nucleotide phosphodiesterases 4 and 7. J. Biol. Chem. 2007;280:30949–30955. doi: 10.1074/jbc.M504398200. [DOI] [PubMed] [Google Scholar]
- 41.Wang H, Yan Z, Yang S, Cai J, Robinson H, Ke H. Kinetic and structural studies of phosphodiesterase-8A and implication on the inhibitor selectivity. Biochemistry. 2008;47:12760–12768. doi: 10.1021/bi801487x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang H, Liu Y, Hou J, Zheng M, Robinson H, Ke H. Structural insight into substrate specificity of phospodiesterase 10. Proc. Natl. Acad. Sci., USA. 2007;104:5782–5787. doi: 10.1073/pnas.0700279104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huai Q, Wang H, Sun Y, Kim HY, Liu Y, Ke H. Three dimensional structures of PDE4D in complex with roliprams and implication on inhibitor selectivity. Structure. 2003;11:865–873. doi: 10.1016/s0969-2126(03)00123-0. [DOI] [PubMed] [Google Scholar]
- 44.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 45.Jones TA, Zou J-Y, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 46.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR System: A New Software Suite for Macromolecular Structure Determination. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.