

Abstract
Cyclooxygenase-2 (COX-2) is upregulated in pancreatic ductal adenocarcinomas (PDAC). However, how COX-2 promotes PDAC development is unclear. While previous studies have evaluated the efficacy of COX-2 inhibition via the use of non steroidal anti-inflammatory drugs (NSAIDs) or the COX-2 inhibitor celecoxib in PDAC models, none have addressed the cell intrinsic vs. microenvironment roles of COX-2 in modulating PDAC initiation and progression. We tested the cell intrinsic role of COX-2 in PDAC progression, using both loss-of-function and gain-of-function approaches. Cox-2 deletion in Pdx1+ pancreatic progenitor cells significantly delays the development of PDAC in mice with K-ras activation and Pten haploinsufficiency. Conversely, COX-2 over-expression promotes early onset and progression of PDAC in the K-ras mouse model. Loss of PTEN function is a critical factor in determining lethal PDAC onset and overall survival. Mechanistically, COX-2 over-expression increases P-AKT levels in the precursor lesions of Pdx1+;K-rasG12D/+;Ptenlox/+ mice in the absence of Pten LOH. In contrast, Cox-2 deletion in the same setting diminishes P-AKT levels and delays cancer progression. These data suggest an important cell intrinsic role for COX-2 in tumor initiation and progression through activation of the PI3K/AKT pathway. PDAC that is independent of intrinsic COX-2 expression eventually develops with decreased FKBP5 and increased GRP78 expression, two alternate pathways leading to AKT activation. Together, these results support a cell intrinsic role for COX-2 in PDAC development and suggest that, while anti-COX-2 therapy may delay the development and progression of PDAC, mechanisms known to increase chemoresistance through AKT activation must also be overcome.
Keywords: PTEN, pancreatic cancer, COX-2, GRP78, FKBP5, chemoresistance
Introduction
Pancreatic ductal adenocarcinoma (PDAC), the 4th leading cause of cancer-related deaths in the United States, has a 5-year survival rate of 4% (1). The poor outcome of PDAC has been attributed to late detection, the aggressive nature of the disease, and poor response to local and systemic therapies. A better understanding of the underlying mechanisms that lead to PDAC initiation, progression, and chemoresistance would help the development of more effective treatment regimens.
Cyclooxygenases (COXs), also known as prostaglandin (PG) endoperoxide synthases, are enzymes essential in the conversion of arachidonic acid to prostaglandins. COXs exist as two isoforms, COX-1 and COX-2 (2). COX-1 is constitutively expressed in most mammalian tissues and is responsible for mediating various normal physiological processes. COX-2, on the other hand, is low or undetectable in most normal tissues, but is induced in response to a wide range of stimuli in many cell types including epithelial cells, endothelial cells, and macrophages (3).
COX-2 levels are often elevated in lung, breast, esophageal, bladder, prostate, and pancreatic cancers (4). Substantial data suggest that COX-2 expression is not simply a by-product, but is a causal factor of tumor development. COX-2 over-expression in transgenic mice led to breast cancer formation, a phenotype greatly reduced by the use of the COX-2 inhibitor celecoxib (5) (Figure 1). Likewise, tumor growth in pancreatic cancers initiated by either COX-2 over-expression (6, 7) or mutant K-ras activation (8), and prostate cancers initiated by the SV40 large T antigen (9), was significantly reduced by celecoxib treatment.
Figure 1. Celecoxib Structure.
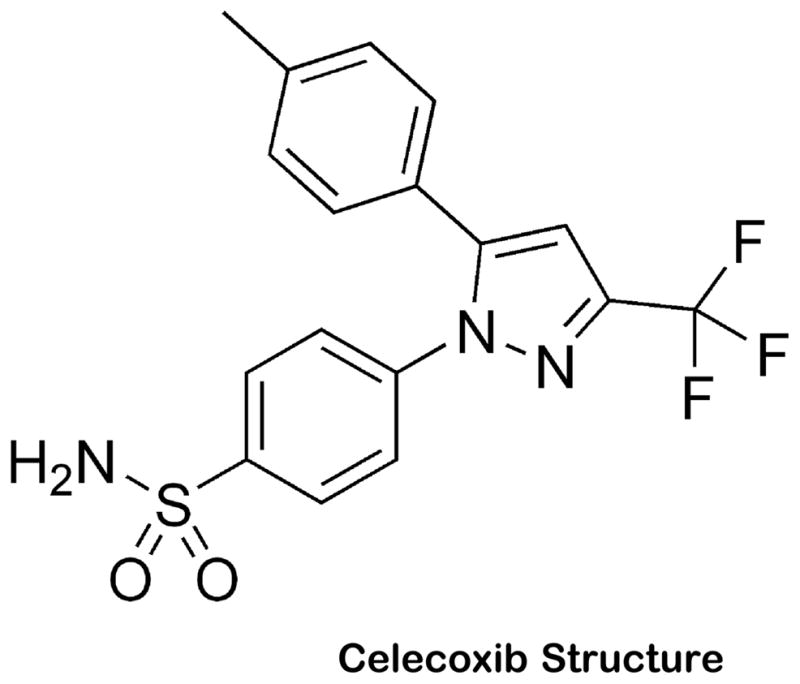
Chemical structure of celecoxib.
Importantly, celecoxib significantly reduced premalignant colon polyp formation in patients at risk for colon cancer (10). However, the addition of celecoxib to gemcitabine therapy, the current standard of care for treatment of PDAC, did not show significant improvement in the survival of patients with metastatic disease (11), suggesting that the efficacy of celecoxib may be cancer- or cancer stage-dependent.
We previously showed that K-ras activation and Pten haploinsufficiency cooperate to activate P-AKT in pre-cancerous lesions and accelerate tumor development in our mouse model of PDAC (12), suggesting that PTEN loss of function is a critical factor for determining the onset of lethal tumor development. Although PTEN mutation is not commonly found in human PDAC, loss of at least one copy of PTEN or gain/amplification of AKT2 has been reported in 32.8% of PDAC xenografts derived from primary patient samples (13). Moreover, elevated P-AKT was observed in 68.5% of PDAC tissue microarray samples (13), suggesting that mechanisms other than PTEN deletion or AKT2 gain/amplification contribute to the elevated P-AKT levels seen in PDAC. In addition, our recent integrative, survival-based, molecular profiling of human PDAC provided multiple lines of correlative data showing that dysregulation of the PI3K/AKT pathway is linked to clinical disease progression (14). Taken together, these data provide strong rationale for further studies designed to identify mechanisms through which the PI3K/AKT pathway is regulated in PDAC development in the absence of Pten LOH or mutation. We hypothesized that COX-2 expression, which suppresses PTEN activity in cell culture (15), could play a significant role in tumor development through activation of the PI3K/AKT pathway. To investigate this hypothesis we conditionally either over-expressed or deleted Cox-2 in the K-ras/Pten mouse model, to explore the role of COX-2 in regulating PI3K/AKT pathway activation during tumorigenesis.
While it is clear that COX-2 plays an enhancing and/or causal role in the progression of many types of cancer, the role(s) of COX-2 expression in the initiated epithelial cell versus in cells within the tumor microenvironment are unclear. We recently demonstrated that Cox-2 deletion in myeloid and endothelial cells, but not in epithelial cells, exacerbates murine colitis (16). These studies point towards the importance of elucidating the cell-intrinsic versus cell-extrinsic role of COX-2 in tumorigenesis. To address this issue in pancreatic tumorigenesis, we conditionally deleted or over-expressed Cox-2 in the Pdx1+ pancreatic progenitor cells.
Materials and Methods
Mouse strains
The generation of conditional Cox-2 over-expression (Cox-2 COE) and conditional knockout (Cox-2 KO) mice has been reported previously (17, 18). To conditionally over-express COX-2 in the pancreas, we crossed the Pdx1-Cre+ line to the floxed Cox-2 COE transgenic line (17). Primers designed to detect the Cox-2 transgenic cassette were utilized for genotyping (17). The Pdx1-Cre+;Cox-2COE line was further crossed to Pdx1-Cre+;K-rasG12D;Ptenlox/+ mice (12) to produce Pdx1-Cre+;K-rasG12D;Ptenlox/+;Cox-2COE animals and littermate controls on a mixed C57/BL/6,129/BALB/c background. Likewise, Cox-2 conditional knockout mice (18) were utilized to produce Pdx1-Cre+;K-rasG12D;Ptenlox/+;Cox-2KO and Pdx1-Cre+;K-rasG12D;Ptenlox/lox;Cox-2KO animals. All studies were performed under the regulation of the Division of Laboratory Animal Medicine at the University of California at Los Angeles.
Histology and immunohistochemistry
Immunohistochemical analysis was performed on formalin-fixed, paraffin-embedded tissue. Antigen retrieval was performed by heating the slides at 95°C in citrate buffer (pH 6.0) or Tris-EDTA buffer (pH8.0) for 15 minutes prior to staining. The following primary antibodies were used: phospho-AKT(Ser473) (Cell Signaling; 1:50), Cytokeratin 19 (ab15463) (Abcam; 1:100), COX2 (SP21) (Thermo Scientific, ready-to-use), GRP78 (11587-1AP) (ProteinTech Group; 1:50), and FKBP5 (14155-1-AP) (ProteinTech Group, 1:50). Blocking of GRP78 expression with an excess of synthetic GRP78 peptide (ag2188, ProteinTech Group) was done by adding twice the volume of peptide as volume of antibody used for primary incubation. Only unidentifiable human tumor samples, collected by Tissue Procurement Core Laboratory at UCLA under the approval of the UCLA Institutional Review Board, were used in this study. All pathology was graded according to the consensus criteria established at the 2004 Penn Workshop (19).
Laser-Capture Micro-dissection and LOH Analysis
Laser-capture microdissection (LCM) of H&E-stained sections was performed using a Leica LMD7000. 100,000μm2 of tissue from ADMs or PDACs in Pdx1-Cre+;K-rasG12D;Ptenlox/+;Cox-2COE and Pdx1-Cre+;K-rasG12D;Ptenlox/+;Cox-2KO mice were collected and extraction done following protocols from QiaAMP DNA Micro Kit (Qiagen). Four microliter aliquots of DNA were used for PCR analysis. PCR was performed for detection of wild-type, floxed, and recombined alleles of Pten using previously described primers (12).
Statistical Analysis
Array analysis was done by the UCLA Clinical Microarray Core on the Affymetrix Mouse 430 2.0 array. Microarray data are available at the National Center for Biotechnology Information Gene Expression Omnibus (GSE38988). Data normalization, filtering, and hierarchical clustering were done using dChip software. For each gene, its expression in each genotype group was represented by the geometric average of the biological replicated samples (n=4). The log ratio between a pair of two genotypes was then calculated. The error bar represents +/− SEM. Animal survival was determined by the Kaplan–Meier survival method. p < 0.001, log-rank test, for each pair-wise combination.
Results
Targeted Cox-2 deletion delays tumor onset in the K-ras/Pten PDAC model
Increased COX-2 expression is observed in chronic pancreatitis, pancreatic intraepithelial neoplasias (PanINs), and PDACs (20). To address the effect of cell-intrinsic loss of COX-2 activity on tumor formation we genetically eliminated Cox-2 by crossing the Pdx1-Cre+;K-rasG12D/+;Ptenlox/+ PDAC model (12) to mice carrying a floxed, conditional Cox-2 deletion allele (Cox-2lox/lox) (18). Pdx1+ pancreatic progenitor cells can give rise to endocrine, exocrine, and ductal tissue, but not to cells in the microenvironment, such as macrophages, or cells associated with blood vessels (21). Since conditional K-ras activation, Pten deletion, and Cox-2 deletions are all dependent on Cre activity driven by the Pdx1 promoter in these mice, Cox-2 will be deleted in the same cells in which mutant K-rasG12D activation and Pten deletion take place.
We first examined how Cox-2 deletion affected the survival of Pdx1-Cre+;K-rasG12D/+;Ptenlox/lox mice, which succumb to tumor burden by three weeks of age (12) (Figure 2A; left, green line). Removal of endogenous COX-2 expression in Pdx1-Cre+;K-rasG12D/+;Ptenlox/lox;Cox-2lox/lox mice extended the median survival time to 35 days (Figure 2A; left, brown line).
Figure 2. Targeted Cox-2 deletion causes delayed tumor onset and increased life expectancy in the Pdx1-Cre;K-ras;Pten mouse model.

A: Kaplan-Meier analysis comparison of Pdx1-Cre+;K-rasG12D/+;Ptenlox/lox and Pdx1-Cre+;K-rasG12D/+;Ptenlox/lox;Cox-2lox/lox mice. B: Histology of Pdx1-Cre+;K-rasG12D/+;Ptenlox/lox and Pdx1-Cre+;K-rasG12D/+;Ptenlox/lox;Cox-2lox/lox mice. C: Histology and COX-2 IHC or double IF [Cytokeratin-19(CK-19), COX-2], as indicated, on tumors of Pdx1-Cre+;K-rasG12D/+;Ptenlox/lox (enlarged version of Figure 2B panel) and Pdx1-Cre+;K- rasG12D/+;Ptenlox/lox;Cox-2lox/lox mice. H&E, hematoxylin and eosin. Scale bars, 50μm.
At 2 weeks of age, Pdx1-Cre+;K-rasG12D/+;Ptenlox/lox mice developed PDAC (Figure 2B, top panel). In contrast, removal of intrinsic COX-2 activity in Pdx1-Cre+;K-rasG12D/+;Ptenlox/lox;Cox-2lox/lox mice resulted in a predominantly normal pancreas parenchyma (Figure 2B, bottom panel). Although lesions in the Pdx1-Cre+;K-rasG12D/+;Ptenlox/lox;Cox-2lox/lox mice appeared much later, once tumors developed the phenotypes resembled those of Pdx1-Cre+;K-rasG12D/+;Ptenlox/lox mice (Figure 2C, left panels).
In order to confirm that Cox-2 is indeed deleted in tumors from Pdx1-Cre+;K-rasG12D/+;Ptenlox/lox;Cox-2lox/lox mice, we evaluated COX-2 expression by IHC and double immunofluorescence analyses. Compared to the robust COX-2 staining observed in cytokeratin-19 positive (CK19+) neoplastic cells of Pdx1-Cre+;K-rasG12D/+;Ptenlox/lox mice (Figure 2C, red arrow, upper panel), no COX-2 expression can be detected in tumors from Pdx1-Cre+;K-rasG12D/+;Ptenlox/lox;Cox-2lox/lox mice (Figure 2C, red arrow, lower panel) except CK19- cells in the microenvironment (Figure 2C, green arrow, lower panel). These observations suggest that, although genetic deletion of Cox-2 in the Pdx1+ pancreatic progenitor cells is not sufficient to fully abrogate tumorigenesis caused by alterations of the RAS/RAF/MAPK and PTEN/PI3K pathways, there is an important cell intrinsic role for COX-2 in pancreatic tumorigenesis; without intrinsic COX-2 expression, PDAC development is significantly delayed.
COX-2 over-expression alone is insufficient to promote PDAC development
Having established that loss of cell intrinsic COX-2 expression delays PDAC development, we next sought to determine whether cell intrinsic COX-2 over-expression alone had an effect on PDAC development. To examine this question we crossed mice carrying a conditional Cox-2 over expression allele (Cox-2COE) (17) with the Pdx1-Cre+ line (22) to generate Pdx1-Cre+;Cox-2COE mice.
Examination of hematoxylin and eosin (H&E) stained sections of pancreata from Pdx1-Cre+;Cox-2COE mice revealed structures that have the appearance of acinar-to-ductal metaplasia (ADM), a pre-neoplastic ductal structure that emerges from acinar cells (Figure 3A, red arrow) and low grade mPanIN lesions (data not shown) in 58% and 42% of mice analyzed, respectively beginning at nine months (Figure 3B). However, despite the presence of these early lesions, the life span of Pdx1-Cre+;Cox-2COE mice (red line) was not altered when compared to that of wild-type (WT) littermate controls(black line) (Figure 3C) and no further progression could be detected in the Pdx1-Cre+;Cox-2COE mice up to 16 months of age, establishing that intrinsic COX-2 over-expression alone in Pdx1 expressing cells of the pancreas is not sufficient to induce PDAC development.
Figure 3. Pdx1-Cre+ driven expression of COX-2 alone does not alter survival.

A: Histology of wild-type (WT) and Pdx1-Cre+;Cox-2COE mice. Scale bar, 50μm. B: Quantification of lesion development in Pdx1-Cre+;Cox-2COE and wild-type mice. ND=not detected. C: Kaplan-Meier survival curve of Pdx1-Cre+;Cox-2COE mice compared to littermate controls.
COX-2 over-expression promotes earlier onset of PDAC development and reduces survival of Pdx1-Cre+;K-rasG12D/+;Ptenlox/+ mice
Consistent with our finding that COX-2 over-expression alone is not sufficient to promote PDAC development, Guerra et al., (23) demonstrated that injury-induced pancreatitis, in which Cox-2 over-expression plays an essential role, cannot cause PDAC unless a K-ras activation mutation is present. Given this precedent, we tested whether intrinsic COX-2 over-expression would accelerate tumorigenesis in a mouse model capable of developing PDACs.
Mice expressing K-rasG12D from its endogenous locus generate PDACs only after a prolonged latency (22), unless coupled with other genetic alterations commonly found in human PDAC, including loss of Ink4a/Arf (24), TGF-β receptor type 2 (Tgfbr2) (25), Smad4 (26), mutation in p53 (27), or haploinsufficiency for Pten (12). COX-2 over-expression leads to significant reduction of median survival time in the Pdx1Cre+;K-rasG12D mice, from 525 days to 410 days (Figure 4A, comparing black and green lines).
Figure 4. COX-2 over expression in Pdx1-Cre+;K-rasG12D/+;Ptenlox/+ mice causes decreased survival and earlier lesion onset.

A: Kaplan-Meier survival curves of Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2COE, Pdx1-Cre+;K-rasG12D/+;Ptenlox/+, Pdx1-Cre+; K-rasG12D/+;Cox-2COE, and Pdx1-Cre+;K-rasG12D/+ mice. B: Quantification of mPanIN, ADM, and PDAC development in Pdx1-Cre+;K-rasG12D/+;Ptenlox/+ and Pdx1-Cre+;K-rasG12D/+;Cox-2COE mice by H&E examination. ND=not detected. C: Histology of Pdx1-Cre+;K-rasG12D/+;Ptenlox/+ mice and Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2COE mice at 2 months of age. Scale bar, 50μm.
When one allele of Pten was removed from the Pdx1-Cre+;K-rasG12D/+ mice, we observed a substantial reduction in the overall survival in the Pdx1-Cre+; K-rasG12D/+;Ptenlox/+ mice (Figure 4A, blue line). Of note, the Pdx1-Cre+;K-rasG12D/+;Ptenlox/+ mice and Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2COE mice have similar median survival times (Figure 4A, blue and red line, respectively). This genetic evidence suggests that although COX-2 over-expression can collaborate with RAS/RAF/MAPK pathway activation, the biological effects of COX-2 over-expression may overlap with PI3K/AKT activation in promoting PDAC development.
To further investigate the effects of COX-2 over-expression and RAS/RAF/MAPK and PI3K pathway activations, we examined pancreata of each model between age 1–3 months (n=12 for each cohort), and scored the presence of ADMs, mPanINs, or PDACs. As summarized in Figure 4B, Pdx1-Cre+;K-rasG12D/+;Ptenlox/+ and Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2COE mice developed a similar spectrum of lesions. Although Cox-2 over-expression had no significant impact on the incidence of early ADM and mPanIN1A lesions, it promoted the progression of these early lesions, as evidenced by the larger and higher grade lesions found in Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2COE mice when compared to age- and genetically background-matched Pdx1-Cre+;K-rasG12D/+;Ptenlox/+ mice (Figure 4B and Figure 4C). Only 3/12 Pdx1-Cre+;K-rasG12D/+;Ptenlox/+ mice, compared to 10/12 Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2COE mice developed mPanIN2–3s during this time frame. Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2COE mice also displayed an increased incidence of invasive PDACs as quantified in Figure 4B and demonstrated in the lower panel of Figure 4C. The fact that the early progression observed histopathologically does not seem to have a major impact on the overall survival of the two cohorts suggests that the kinetics of disease progression from the early lesions to lethal disease may not be linear because of the timing and nature of secondary mutations acquired by localized lesions in each cohort. Since metastatic incidences are rare in both cohorts (<less than 5%), it is difficult to quantitatively compare the rates and onset of these lesions.
AKT activation is associated with COX-2-mediated tumor progression
Since Pdx1-Cre+;K-rasG12D/+;Ptenlox/+ mice and Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2COE mice have a similar median survival time, we reasoned that COX-2 over-expression may functionally mimic Pten loss of heterozygosity (LOH), an event critical for PDAC progression in the Pdx1-Cre+;K-rasG12D/+;Ptenlox/+ model (12). To test this hypothesis, we assessed both the Pten genomic and functional status in ADM and PDAC lesions in the Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2COE and Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2lox/lox mice. PTEN functional status was determined by P-AKT IHC analysis and Pten genomic status was investigated by genomic PCR, using laser captured lesion tissues. AKT is significantly activated in ADM lesions of Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2COE mice but not in similar lesions of either Pdx1-Cre+;K-rasG12D/+;Ptenlox/+ (Hill et al 2010; and data not shown) or Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2lox/lox mice (Figure 5A; comparing left and right panels). In contrast, PDAC lesions from COX-2 over expressing and Cox-2 deleted mice have comparable enhanced P-AKT staining, further supporting our hypothesis that AKT activation is a limiting step in ADM to PDAC progression (12).
Figure 5. P-AKT expression is associated with COX-2 mediated tumor acceleration.

A: P-AKT IHC in Pdx1-Cre+; K-rasG12D/+;Ptenlox/+;Cox-2COE and Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2lox/lox mice. Scale bar, 50μm. B: PCR analysis of DNA extracted from laser captured samples was performed on ADMs and PDACs of Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2COE and Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2lox/lox mice, to detect PCR products from the wild type Pten allele (WT allele), the floxed Pten allele (lox allele), and the deleted Pten allele (del band).
We then extracted genomic DNA from laser captured ADM and PDAC lesions from cohorts of Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2COE and Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2lox/lox mice (n=4) and performed PCR analysis. The WT Pten allele (in red, Fig. 5B) is maintained at near equal molar ratio to the Pten deleted allele (in blue, Fig. 5B) in both pre-cancer (ADM) and cancer (PDAC) lesions, indicating that majority of cells in these lesions did not acquire Pten LOH. This result suggests that COX-2 over-expression, together with Pten haploinsufficiency, promotes AKT activation - a necessary step for the progression of precursor lesions to PDAC (12). The presence of elevated COX-2 activity, therefore, removes the selective pressure for Pten LOH that is observed in the Pdx1-Cre+;K-rasG12D/+;Ptenlox/+ model (15).
Upregulated GRP78 expression correlates with P-AKT activation in the precursor lesions
Upregulation of GRP78, a member of the Hsp70 protein family, is associated with poor prognosis of a number of human cancers (28, 29). While GRP78 is known to be expressed in the cytoplasm, the recent discovery of GRP78 on the cell surface and its role in activating AKT (28, 29) prompted us to investigate whether GRP78 expression is involved in initiation and progression in human PDAC. While no GRP78 expression was observed in the ducts of wild-type murine samples (Supplemental Figure 1A, green arrow in left panels), GRP78 expression is significantly upregulated in the ductal structures of human ADM and PDAC lesions (Figure 6A, upper panels, high power magnification; Supplemental Figure 1A, right panels, lower power magnification). Importantly, GRP78 is strongly expressed in both the cytoplasm and the membranes of these human lesions (Figure 6A, upper panels).
Figure 6. Increased membrane-associated GRP78 expression is associated with COX-2 mediated tumor acceleration and correlates with P-AKT expression.

A: Representative IHC for GRP78 on ADMs and PDACs from human pancreatic tumor samples and from Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2COE and Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2lox/lox tumors. B: Co-IF for GRP78 and P-AKT in PDAC of Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2lox/lox mice (low power, left and magnified inset of white outlined area, right). Scale bars, 50μm.
We then analyzed GRP78 expression in our murine models. Similar to the human disease, both ADM and PDAC lesions in the Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2COE mice have strongly upregulated GRP78 expression, especially its membrane-associated form (Figure 6A, middle panels), corresponding to AKT activation (Figure 5A, 1st and 2nd panel from left). The specificity of this staining was confirmed by demonstrating that excess GRP78 peptide can block labeling with this antibody (Supplemental Figure 1B). In contrast, only low cytosolic GRP78 expression (Figure 6A, lower left panel) and nearly undetectable AKT activation (Figure 5A, 3rd panel from left) can be observed in the ADM lesions from mice with the targeted Cox-2 deletion.
These data suggest that COX-2 over-expression may lead to upregulated expression of membrane-associated GRP78 and to subsequent AKT activation. However, even in the absence of epithelial COX-2 expression, PDAC lesions in the Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2lox/lox mice also showed upregulated GRP78 expression (lower right panel of Figure 6A, Supplemental Figure 1B), which co-localizes with enhanced P-AKT in the membrane (Figure 6B), suggesting that GRP78 and AKT can be upregulated and activated in a COX-2-independent manner as the disease progresses.
Decreased FK506 binding protein 5 (FKBP5) negative feedback leads to enhanced AKT activation in PDAC of Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2lox/lox mice
Although Cox-2 conditional deletion led to increased median survival of Pdx1-Cre+;K-rasG12D/+;Ptenlox/+ mice (65 days: data not shown), all Cox-2 deleted animals ultimately succumbed to aggressive PDAC without Pten LOH (Figure 5A), suggesting that resistance to COX-2 directed therapy is likely to occur in patients.
Decreased expression of scaffold protein FKBP5 has been identified as one of the mechanisms that enhances AKT activity and causes PDAC chemoresistance (30). FKBP5, by acting as a scaffolding protein for AKT and PH domain and leucine rich repeat protein phosphatase 1 (PHLPP), promotes PHLPP dephosphorylation of AKT at amino acid S473 (31), thereby suppressing AKT activity (30, 32). We examined the expression level of FKPB5 mRNA and found that FKBP5 is down regulated significantly in PDACs of Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2lox/lox mice (Figure 7A; p=0.01; 2-tail t-test, determined as the Log2 ratio of FKBP5 expression of KO [Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2lox/lox over Pdx1-Cre+;K-rasG12D/+;Ptenlox/+] and OE [Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2COE over Pdx1-Cre+;K-rasG12D/+;Ptenlox/+]).
Figure 7. Decreased FKBP5 negative feedback leads to enhanced AKT activation in Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2lox/lox mice.

A: FKBP5 mRNA expression in PDACs of Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2lox/lox (KO, n=4) and Pdx1-Cre+;K-rasG12D/+;Pten lox/+;Cox-2COE (OE, n=4) mice. B: Representative IHC analyses for FKBP5 in PDACs in the three mutant murine lines. C: IHC for P-AKT and FKBP5 in consecutive sections in a PDAC from a Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2lox/lox mouse. Scale bars, 50μm. D: Model of COX-2 mediated activation of P-AKT in tumors with, and without, COX-2 expression. Left, Pdx1-Cre+;K-rasG12D/+;Pten lox/+;Cox-2COE mice have earlier PDAC onset because early P-AKT activation, possibly mediated by increased GRP78 expression, cooperates with Pten haploinsufficiency to initiate tumorigenesis. Right, P-AKT activation in tumors of Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2lox/lox mice could be the result of loss of FKBP5 regulated AKT suppression, which is reduced in the absence of COX-2 expression.
IHC analysis revealed strong FKBP5 staining in PDACs of Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2COE and Pdx1-Cre+;K-rasG12D/+;Ptenlox/+ mice while FKBP5 expression was reduced to almost undetectable levels in Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2lox/lox mice (Figure 7B). Furthermore, enhanced membrane-associated P-AKT correlates with reduced FKBP5 staining in PDACs of Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2lox/lox mice (Figure 7C, region indicated by green arrow, Supplemental Figure 2). These results suggest that, in the absence of Pten LOH, down-regulation of FKBP5 could cause activation of AKT in PDAC with Cox-2 deletion or – in a clinical context, as a result of celecoxib treatment leading to chemoresistance.
Discussion
Using both loss- and gain-of-function genetic approaches, our study establishes a cell intrinsic role for COX-2 in the onset and progression of PDAC. COX-2 over-expression cooperates with Pten haploinsufficiency to activate AKT, a gate-keeping event in PDAC progression, leading to accelerated tumor development. Mechanistically, AKT activation may occur as a result of increased membrane-associated GRP78 expression, induced as a consequence of enhanced COX-2 signaling (Figure 7D, left). Alternatively, AKT can be activated by decreased FKBP5-PHLPP-AKT negative feedback circuitry associated with Cox-2 abrogation (Figure 7D, right) or – perhaps – in response to NSAID or coxib treatment. Our findings suggest that, while anti-COX-2 therapies may delay PDAC onset and progression, tumors undergoing NSAID or coxib treatment may acquire changes that lead to AKT activation and therapeutic resistance. These alternate mechanisms that lead to AKT-mediated resistance must also be overcome in order to achieve therapeutic efficacy (33–35).
Cell intrinsic role for COX-2 in pancreatic tumor development
Previous studies have evaluated the efficacy of COX-2 inhibition, via the use of NSAIDs or celecoxib treatment, in PDAC models (6–8). However, there are no previous studies that have been able to discriminate between the effect of COX-2 inhibition in both the tumor cells and the microenvironment, and COX-2 inhibition in only the tumor cells. The murine models we have generated provided an opportunity to elucidate the cell intrinsic function of COX-2 in PDAC development.
Unlike previous studies, which showed that cytokeratin 5 driven expression of COX-2 led to the development of PDAC, (6), we observed that COX-2 expression in Pdx1+ progenitor cells alone is not sufficient for the development of PDAC in Pdx1-Cre+;Cox-2COE mice. This result raises the question of what is preventing tumor development in Pdx1-Cre+;Cox-2COE mice or in mice with injury-induced pancreatitis, where COX-2 plays a critical role in subsequent tumor development (23). Recent studies indicate K-ras mutation is essential for the progression of pre-malignant lesions to PDAC in mice with injury-induced pancreatitis (23, 36). Consistent with these observations, our genetic study suggests that the cross-talk between COX-2 and RAS/MAPK signaling pathway is essential in promoting PDAC development. Furthermore, the median survival of Pdx1-Cre+;K-rasG12D/+;Cox-2COE mice was significantly longer than that of Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2COE mice, establishing the importance of PTEN pathway function in determining both PDAC progression and overall survival. The similar survival trends of Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2COE and Pdx1-Cre+;K-rasG12D/+;Ptenlox/+ mice also support the hypothesis that the effects of COX-2 expression overlap with the PI3K/AKT pathway and suggest that COX-2 is a key modulator of PI3K/AKT activation in pancreatic tumorigenesis, without the need for Pten loss of heterozygosity.
The PTEN pathway and PDAC progression
Tumors in the K-ras;Pten mice recapitulate the pathologic features of human PDACs. Moreover, these mice model the loss of PTEN function which is now recognized as a frequent, major contributor in human PDAC progression. While loss of both alleles of PTEN is infrequent in PDAC, elevated P-AKT was frequently observed in PDAC tissue microarray samples (13), suggesting that mechanisms other than PTEN deletion contribute to the elevated P-AKT levels seen in PDAC. Elucidating these mechanisms is of critical importance, as our recent work has shown that dysregulated PI3K/AKT signaling is significantly associated with distinct pancreatic cancer patient subgroups with more aggressive disease progression (14). The identification of the driver(s) for PI3K/AKT pathway expression, and what the downstream effects of this PI3K/AKT expression are, should aid the development of more effective therapies. Our previous work shows that Pten heterozygozity leads to earlier precursor lesion development which is associated with increased cell proliferation (12). Moreover, our findings suggest two AKT activating mechanisms, both related to COX-2 expression, may occur during the development of PDAC.
P-AKT activation in Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2COE mice is associated with an increase in membrane-bound GRP78, which was diminished upon Cox-2 deletion, in precursor lesions. Increased GRP78 expression has recently been identified as a critical factor in protecting cells from cell death in response to cellular insults, including inflammation and pancreatitis (37). It seems likely that that increased COX-2 expression regulates GRP78 activity. The GRP78 promoter contains a cAMP responsive element (38). COX-2 signaling via epithelial cell EP receptors may activate GRP78 via the cAMP/PKA pathway (39).
The high level of GRP78 we observed in tumors in our in vivo models raises the question of whether GRP78 is essential for pancreatic tumor progression. Targeted knockout of Grp78 in the prostate epithelium and hematopoietic stem cells inhibited Pten null prostate tumorigenesis, leukemia development and AKT activation (40, 41), suggesting GRP78 as a potential therapeutic target for cancers that result from PTEN loss or inactivation.
PDACs invariably developed in all Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2lox/lox mice, suggesting that tumors can compensate for COX-2 loss through other mechanisms. AKT activation is associated with decreased expression of the FKBP5 scaffold protein in Pdx1-Cre+;K-rasG12D/+;Ptenlox/+;Cox-2lox/lox pancreatic tumors (Fig. 7C). Decreased FKBP5 expression has been identified as one of the mechanisms that enhances AKT activity and results in PDAC chemoresistance (30, 42). These data suggest that FKBP5 may function as a tumor suppressor in PDAC development. Based on our data showing a correlation between loss of COX-2 expression and decreased FKBP5 expression, we suggest that coxib treatment could result in loss of FKBP5 mediated suppression of AKT activation.
Therapeutic implications
Our results and those of others (8) show that therapeutic regimens designed to target COX-2 expression may be most useful for delaying both precursor lesions and PDAC initiation in patients at high risk for pancreatic cancer. Recent studies have shown that dissemination of pancreatic epithelial tumor cells can occur much earlier than previously thought and that these cells can seed distant metastasis (43). Given the fact that PDAC can grow at an exponential rate with micro-metastasis developing from small primary tumors prior to surgical resection (44), it is possible that therapeutics which delay precursor lesion formation, like COX-2 inhibitors, could be administrated in this previously unappreciated therapeutic window to significantly delay/prevent seeding of metastases in patients at high-risk for PDAC development.
Moreover, our results emphasize the importance of AKT activity in the effectiveness of anti-COX-2 therapies. Recent studies have shown that COX-2 inhibition in lung and colon cancer cells was ineffective at eliciting cell death unless COX-2 inhibitors were used in combination with PPARγ ligand ciglitazone (45) and atorvastatin (46). Induction of cell death, in both studies, was associated with decreased P-AKT activation. We identified increased GRP78 and decreased FKBP5 as two mechanisms through which pancreatic tumors could activate AKT. The lack of increased therapeutic benefit from combination celecoxib+gemcitabine treatment of metastatic PDACs (11) could be the result of increased therapeutic resistance resulting from AKT activation. We suggest, therefore, that pathways that cause AKT activation will likely have to be targeted to enhance therapeutic efficacy. Co-targeting the COX-2, RAS/RAF/MAPK, and PI3K/AKT pathways may be necessary to slow progression of late stage PDAC.
Supplementary Material
Acknowledgments
We thank the UCLA Tissue Procurement Core Laboratory for their technical help; Dr. Bangyan Stiles for sharing protocols; Antreas Hindoyan and Katharina Schlacher for helpful discussions, and Ken-Ichiro Kamei and Tomo-o Ishikawa for providing COX-2 COE and Cox-2flox mice and for assistance in their analyses.
Grant Support
This work is supported, in part, by Damon Runyon Cancer Foundation and USHHS Ruth L. Kirschstein Institutional National Research Service Award T32 CA009056 (to R.Hill, L.Tran and J.H.Calvopina), UCLA Jonsson Comprehensive Cancer Center Seed Grant and NIH grants (UO1 CA84128, RO1 CA107166 to H.Wu; P50 CA086306 to H.Herschman. and H.Wu; and R01 CA08452 to H. Herschman.)
Footnotes
No potential conflicts of interest.
References
- 1.Stathis A, Moore MJ. Advanced pancreatic carcinoma: current treatment and future challenges. Nat Rev Clin Oncol. 2010;7:163–72. doi: 10.1038/nrclinonc.2009.236. [DOI] [PubMed] [Google Scholar]
- 2.Herschman HR. Regulation of prostaglandin synthase-1 and prostaglandin synthase-2. Cancer metastasis reviews. 1994;13:241–56. doi: 10.1007/BF00666095. [DOI] [PubMed] [Google Scholar]
- 3.Masferrer JL, Reddy ST, Zweifel BS, Seibert K, Needleman P, Gilbert RS, et al. In vivo glucocorticoids regulate cyclooxygenase-2 but not cyclooxygenase-1 in peritoneal macrophages. J Pharmacol Exp Ther. 1994;270:1340–4. [PubMed] [Google Scholar]
- 4.Koki A, Khan NK, Woerner BM, Dannenberg AJ, Olson L, Seibert K, et al. Cyclooxygenase-2 in human pathological disease. Adv Exp Med Biol. 2002;507:177–84. doi: 10.1007/978-1-4615-0193-0_28. [DOI] [PubMed] [Google Scholar]
- 5.Narko K, Zweifel B, Trifan O, Ristimaki A, Lane TF, Hla T. COX-2 inhibitors and genetic background reduce mammary tumorigenesis in cyclooxygenase-2 transgenic mice. Prostaglandins Other Lipid Mediat. 2005;76:86–94. doi: 10.1016/j.prostaglandins.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 6.Colby JK, Klein RD, McArthur MJ, Conti CJ, Kiguchi K, Kawamoto T, et al. Progressive metaplastic and dysplastic changes in mouse pancreas induced by cyclooxygenase-2 overexpression. Neoplasia. 2008;10:782–96. doi: 10.1593/neo.08330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muller-Decker K, Furstenberger G, Annan N, Kucher D, Pohl-Arnold A, Steinbauer B, et al. Preinvasive duct-derived neoplasms in pancreas of keratin 5-promoter cyclooxygenase-2 transgenic mice. Gastroenterology. 2006;130:2165–78. doi: 10.1053/j.gastro.2006.03.053. [DOI] [PubMed] [Google Scholar]
- 8.Funahashi H, Satake M, Dawson D, Huynh NA, Reber HA, Hines OJ, et al. Delayed progression of pancreatic intraepithelial neoplasia in a conditional Kras(G12D) mouse model by a selective cyclooxygenase-2 inhibitor. Cancer Res. 2007;67:7068–71. doi: 10.1158/0008-5472.CAN-07-0970. [DOI] [PubMed] [Google Scholar]
- 9.Narayanan BA, Narayanan NK, Pttman B, Reddy BS. Adenocarcina of the mouse prostate growth inhibition by celecoxib: downregulation of transcription factors involved in COX-2 inhibition. Prostate. 2006;66:257–65. doi: 10.1002/pros.20331. [DOI] [PubMed] [Google Scholar]
- 10.Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Solomon SD, Kim K, et al. Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med. 2006;355:873–84. doi: 10.1056/NEJMoa061355. [DOI] [PubMed] [Google Scholar]
- 11.Dragovich T, Burris H, 3rd, Loehrer P, Von Hoff DD, Chow S, Stratton S, et al. Gemcitabine plus celecoxib in patients with advanced or metastatic pancreatic adenocarcinoma: results of a phase II trial. Am J Clin Oncol. 2008;31:157–62. doi: 10.1097/COC.0b013e31815878c9. [DOI] [PubMed] [Google Scholar]
- 12.Hill R, Calvopina JH, Kim C, Wang Y, Dawson DW, Donahue TR, et al. PTEN loss accelerates KrasG12D-induced pancreatic cancer development. Cancer Res. 2010;70:7114–24. doi: 10.1158/0008-5472.CAN-10-1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ying H, Elpek KG, Vinjamoori A, Zimmerman SM, Chu GC, Yan H, et al. Pten is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF-kappaB-cytokine network. Cancer Discov. 2011;1:158–69. doi: 10.1158/2159-8290.CD-11-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donahue TR, Tran LM, Hill R, Li Y, Kovochich A, Calvopina JH, et al. Integrative survival-based molecular profiling of human pancreatic cancer. Clin Cancer Res. 2012;18:1352–63. doi: 10.1158/1078-0432.CCR-11-1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Covey TM, Edes K, Fitzpatrick FA. Akt activation by arachidonic acid metabolism occurs via oxidation and inactivation of PTEN tumor suppressor. Oncogene. 2007;26:5784–92. doi: 10.1038/sj.onc.1210391. [DOI] [PubMed] [Google Scholar]
- 16.Ishikawa TO, Oshima M, Herschman HR. Cox-2 deletion in myeloid and endothelial cells, but not in epithelial cells, exacerbates murine colitis. Carcinogenesis. 2011;32:417–26. doi: 10.1093/carcin/bgq268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamei K, Ishikawa TO, Herschman HR. Transgenic mouse for conditional, tissue-specific Cox-2 overexpression. Genesis. 2006;44:177–82. doi: 10.1002/dvg.20199. [DOI] [PubMed] [Google Scholar]
- 18.Ishikawa TO, Herschman HR. Conditional knockout mouse for tissue-specific disruption of the cyclooxygenase-2 (Cox-2) gene. Genesis. 2006;44:143–9. doi: 10.1002/gene.20192. [DOI] [PubMed] [Google Scholar]
- 19.Hruban RH, Rustgi AK, Brentnall TA, Tempero MA, Wright CV, Tuveson DA. Pancreatic cancer in mice and man: the Penn Workshop 2004. Cancer Res. 2006;66:14–7. doi: 10.1158/0008-5472.CAN-05-3914. [DOI] [PubMed] [Google Scholar]
- 20.Maitra A, Ashfaq R, Gunn CR, Rahman A, Yeo CJ, Sohn TA, et al. Cyclooxygenase 2 expression in pancreatic adenocarcinoma and pancreatic intraepithelial neoplasia: an immunohistochemical analysis with automated cellular imaging. Am J Clin Pathol. 2002;118:194–201. doi: 10.1309/TPG4-CK1C-9V8V-8AWC. [DOI] [PubMed] [Google Scholar]
- 21.Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129:2447–57. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- 22.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–50. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 23.Guerra C, Collado M, Navas C, Schuhmacher AJ, Hernandez-Porras I, Canamero M, et al. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell. 2011;19:728–39. doi: 10.1016/j.ccr.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112–26. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ijichi H, Chytil A, Gorska AE, Aakre ME, Fujitani Y, Fujitani S, et al. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev. 2006;20:3147–60. doi: 10.1101/gad.1475506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kojima K, Vickers SM, Adsay NV, Jhala NC, Kim HG, Schoeb TR, et al. Inactivation of Smad4 accelerates Kras(G12D)-mediated pancreatic neoplasia. Cancer Res. 2007;67:8121–30. doi: 10.1158/0008-5472.CAN-06-4167. [DOI] [PubMed] [Google Scholar]
- 27.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–83. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 28.Zhang LH, Yang XL, Zhang X, Cheng JX, Zhang W. Association of elevated GRP78 expression with increased astrocytoma malignancy via Akt and ERK pathways. Brain Res. 2011;1371:23–31. doi: 10.1016/j.brainres.2010.11.063. [DOI] [PubMed] [Google Scholar]
- 29.Lin Y, Wang Z, Liu L, Chen L. Akt is the downstream target of GRP78 in mediating cisplatin resistance in ER stress-tolerant human lung cancer cells. Lung Cancer. 2011;71:291–7. doi: 10.1016/j.lungcan.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 30.Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, et al. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell. 2009;16:259–66. doi: 10.1016/j.ccr.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005;18:13–24. doi: 10.1016/j.molcel.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 32.Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011;19:792–804. doi: 10.1016/j.ccr.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu HG, Ai YW, Yu LL, Zhou XD, Liu J, Li JH, et al. Phosphoinositide 3-kinase/Akt pathway plays an important role in chemoresistance of gastric cancer cells against etoposide and doxorubicin induced cell death. Int J Cancer. 2008;122:433–43. doi: 10.1002/ijc.23049. [DOI] [PubMed] [Google Scholar]
- 34.Sinnberg T, Lasithiotakis K, Niessner H, Schittek B, Flaherty KT, Kulms D, et al. Inhibition of PI3K-AKT-mTOR signaling sensitizes melanoma cells to cisplatin and temozolomide. J Invest Dermatol. 2009;129:1500–15. doi: 10.1038/jid.2008.379. [DOI] [PubMed] [Google Scholar]
- 35.Jiao M, Nan KJ. Activation of PI3 kinase/Akt/HIF-1alpha pathway contributes to hypoxia-induced epithelial-mesenchymal transition and chemoresistance in hepatocellular carcinoma. Int J Oncol. 2012;40:461–8. doi: 10.3892/ijo.2011.1197. [DOI] [PubMed] [Google Scholar]
- 36.Carriere C, Young AL, Gunn JR, Longnecker DS, Korc M. Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem Biophys Res Commun. 2009;382:561–5. doi: 10.1016/j.bbrc.2009.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ye R, Mareninova OA, Barron E, Wang M, Hinton DR, Pandol SJ, et al. Grp78 heterozygosity regulates chaperone balance in exocrine pancreas with differential response to cerulein-induced acute pancreatitis. Am J Pathol. 2010;177:2827–36. doi: 10.2353/ajpath.2010.100368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alexandre S, Nakaki T, Vanhamme L, Lee AS. A binding site for the cyclic adenosine 3′,5′-monophosphate-response element-binding protein as a regulatory element in the grp78 promoter. Mol Endocrinol. 1991;5:1862–72. doi: 10.1210/mend-5-12-1862. [DOI] [PubMed] [Google Scholar]
- 39.Chang SH, Ai Y, Breyer RM, Lane TF, Hla T. The prostaglandin E2 receptor EP2 is required for cyclooxygenase 2-mediated mammary hyperplasia. Cancer Res. 2005;65:4496–9. doi: 10.1158/0008-5472.CAN-05-0129. [DOI] [PubMed] [Google Scholar]
- 40.Fu Y, Wey S, Wang M, Ye R, Liao CP, Roy-Burman P, et al. Pten null prostate tumorigenesis and AKT activation are blocked by targeted knockout of ER chaperone GRP78/BiP in prostate epithelium. Proc Natl Acad Sci U S A. 2008;105:19444–9. doi: 10.1073/pnas.0807691105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wey S, Luo B, Tseng CC, Ni M, Zhou H, Fu Y, et al. Inducible knockout of GRP78/BiP in the hematopoietic system suppresses Pten-null leukemogenesis and AKT oncogenic signaling. Blood. 2011 doi: 10.1182/blood-2011-06-357384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li L, Lou Z, Wang L. The role of FKBP5 in cancer aetiology and chemoresistance. Br J Cancer. 2011;104:19–23. doi: 10.1038/sj.bjc.6606014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–61. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haeno H, Gonen M, Davis MB, Herman JM, Iacobuzio-Donahue CA, Michor F. Computational modeling of pancreatic cancer reveals kinetics of metastasis suggesting optimum treatment strategies. Cell. 2012;148:362–75. doi: 10.1016/j.cell.2011.11.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim BM, Maeng K, Lee KH, Hong SH. Combined treatment with the Cox-2 inhibitor niflumic acid and PPARgamma ligand ciglitazone induces ER stress/caspase-8-mediated apoptosis in human lung cancer cells. Cancer Lett. 2011;300:134–44. doi: 10.1016/j.canlet.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 46.Xiao H, Zhang Q, Lin Y, Reddy BS, Yang CS. Combination of atorvastatin and celecoxib synergistically induces cell cycle arrest and apoptosis in colon cancer cells. Int J Cancer. 2008;122:2115–24. doi: 10.1002/ijc.23315. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.