

Abstract
Accumulating evidence suggests that self-renewal and differentiation capabilities reside only in a subpopulation of tumor cells, termed cancer stem cells (CSCs), whereas the remaining tumor cell population lacks the ability to initiate tumor development or support continued tumor growth. In head and neck squamous cell carcinoma (HNSCC), as with other malignancies, CSCs have been increasingly shown to have an integral role in tumor initiation, disease progression, metastasis, and treatment resistance. In this article, the author summarizes the current knowledge of the role of CSCs in HNSCC and discusses the therapeutic implications and future directions of this field.
Keywords: CSC, field cancerization, HNSCC, metastasis, tumorigenesis
INTRODUCTION
Head and neck squamous cell carcinoma (HNSCC) ranks sixth worldwide for cancer-related mortality, with an estimated 500000 new cases diagnosed yearly. For the past several decades, the mainstay of treatment for HNSCC has been surgery and external beam radiation. More recent clinical trials have demonstrated the benefit of combining chemotherapy and radiation for advanced stage disease, leading to modest improvements in treatment outcomes.[1] Despite this recent improvement, the increase in overall survival has been nominal and cancer recurrence and treatment failures continue to occur in a significant percentage of patients. The biology underpinning why some tumors respond favorably to treatment and others do not is largely unknown. Over the last 15 years, advances in tumor biology have led to the discovery that many cancers, including HNSCC, appear to be supported by cells with stem-like properties. Studies in a wide variety of malignancies have demonstrated that only a distinct subpopulation of tumor cells, termed cancer stem cells (CSCs), contain the ability to undergo self-renewal and differentiation (properties of normal stem cells) and hence have the ability to initiate tumorigenesis and support ongoing tumor growth. Furthermore, it appears that, like their normal stem cell counterparts, CSCs have increased resistance to standard cytotoxic therapies. These findings have coalesced into the CSC theory of tumorigenesis, which has remarkable implications on our understanding of tumor initiation, disease progression, and treatment response [Figure 1]. Here we review the basics of the CSC theory as it applies to HNSCC.
Figure 1.
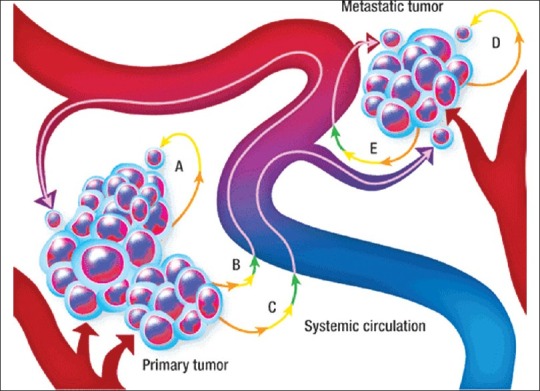
Self-seeding concept of cancer growth and metastasis (a) dislodging and reattachment of a primary tumor cell at the primary site, (b) dislodging, intravasation, circulation, then extravasation back to the primary site, (c) dislodging, intravasation, circulation, then extravasation to a metastatic site, and (d) self-seeding from a metastatic site following path A or path B
EMERGENCE OF THE CANCER STEM CELL THEORY
Stem cells can be divided into main three categories: embryonic, germinal, and somatic. Embryonic stem cells (ESCs) originate from the inner cell mass of the blastocyst. ESCs are omnipotent and have indefinite replicative life span, which is attributable to their telomerase expression. Germinal stem cells are derived from primary germinal layers of embryo. They differentiate into progenitor cells to produce specific organ cells. Somatic/adult stem cells (ASCs) are progenitor cells as they are less totipotent, i.e. less replicative life span than ESCs. They exist in mature tissues such as hematopoietic, neural, gastrointestinal, and mesenchymal tissues.[1,2] The most commonly used ASCs derived from bone marrow are hemopoietic stem cells (HSCs) and other primitive progenitor cells including mesenchymal stem cells (MSCs) and multipotent adult progenitor cells (MAPCs).[3,4] The microRNAs expression has been reported as a requisite to bypass G1/S checkpoint, thus for the self-renewal characteristic of stem cells. Figure 2 shows hierarchy of stem cells with cell determination and differentiation.[3]
Figure 2.
Hierarchy of stem cells with cell determination and differentiation with self-renewal capacity
The cellular and molecular requirements for initiation of tumorigenesis are a series of mutations resulting in the acquisition of replication and growth-factor independence, resistance to growth-inhibitory signals, tissue invasion, and metastasis. The mechanisms underlying these mutations have been extensively interrogated; however, a unifying “model of tumorigenesis” remains to be completely elucidated.[1,4] Tumors have long been recognized to consist of a heterogeneous population of cells differing in proliferative capacity, histologic and immunophenotypic appearance, and tumorigenic potential. Over the last several years however, a new hypothesis has emerged suggesting that tumor heterogeneity is supported by a stem cell hierarchy.
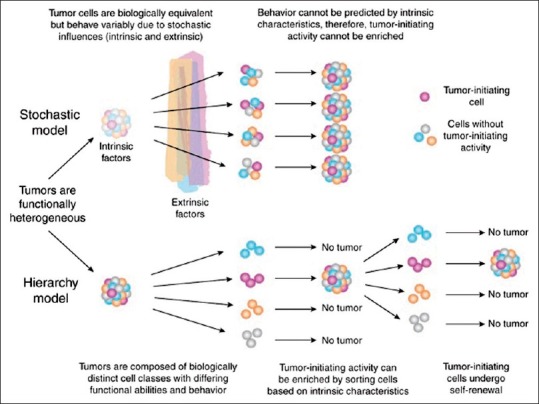
The CSC hypothesis postulates that tumor heterogeneity with regards to initiation, progression, response to therapy, and metastasis is the result of mutations which either render a normal somatic tissue stem cell cancerous, or cause a cancer cell to become stem cell-like. This mutated CSC is then capable of giving rise both to additional CSCs and to a variety of more differentiated and functionally divergent cancer cells, much like a normal somatic tissue stem cell.[5] Unlike in the traditional stochastic tumorigenesis model, the CSC model proposes that tumorigenicity resides in only a small subpopulation of cancer cells and that these cells, rather than the bulk of the tumor, are responsible for tumor initiation and growth [Figure 3].
Figure 3.
CSC model of tumorogenesis
CSCs were first experimentally defined in hematopoietic malignancies by John Dick and colleagues in 1994. Transplantation of a defined subpopulation of human acute myeloid leukemia (AML) cells into immunodeficient mice was not only able to recapitulate AML but it was phenotypically and pathologically similar to the patient's original leukemia. In contrast, the remaining cell populations (CD34low and CD34hi CD38hi) failed to give rise to new leukemia cells.
The identification of the cell population responsible for initiating tumorigenesis has significant implications for the prognosis and treatment of cancer. At present, cytotoxic chemotherapies target the rapidly cycling cells of the tumor and result in impressive reduction in the tumor size, but leave the largely chemotherapy-resistant CSCs untouched. Additionally, both in vitro assays and in vivo monitoring for effectiveness of new experimental cancer therapies are based on reduction in the cell number or in the tumor size. It is therefore theoretically possible that therapies which result in tumor cell death, as currently assayed, will not have any significant effect on the CSC and will therefore not result in long-term disease control or eradication. The ability of the CSC to produce phenotypically diverse tumor cells may also contribute to increased metastatic potential with new mutations selecting for migratory and invasive properties of the tumor [Figures 4 and 5].
Figure 4.
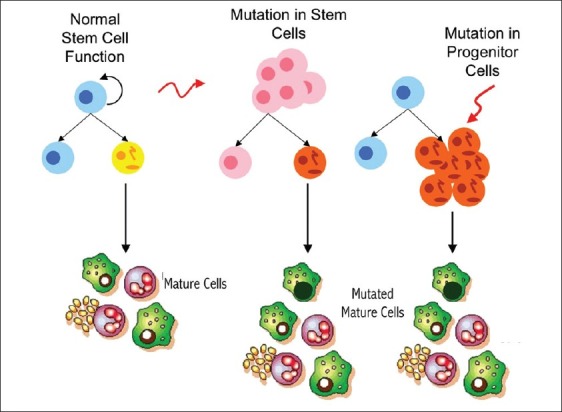
Mutation in stem cell population
Figure 5.
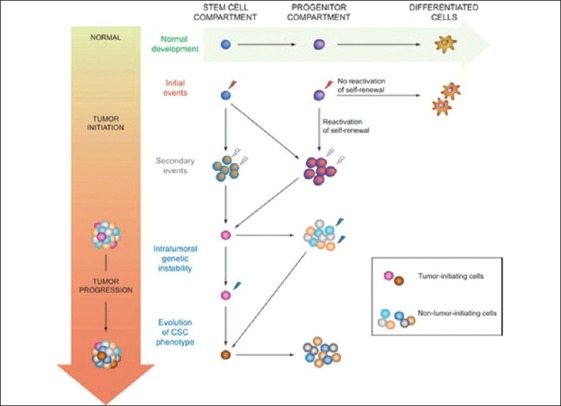
Ability of the CSC to produce phenotypically diverse tumor cells contributing in increased metastatic potential of the tumor
THE CHOICE OF SOURCE OF STEM CELLS FOR CANCER THERAPY
The existence of CSCs has several implications in terms of future cancer treatment and therapies. These include disease identification, selective drug targets, prevention of metastasis, and development of new intervention strategies.
Normal somatic stem cells are naturally resistant to chemotherapeutic agents: they have various pumps (such as MDR) that pump out drugs, DNA repair proteins and they also have a slow rate of cell turnover (chemotherapeutic agents naturally target rapidly replicating cells). CSCs that have mutated from normal stem cells may also express proteins that would increase their resistance toward chemotherapeutic agents. These surviving CSCs then repopulate the tumor, causing relapse. By selectively targeting CSCs, it would be possible to treat patients with aggressive, nonresectable tumors, as well as preventing the tumor from metastasizing. The hypothesis suggests that upon CSC elimination, cancer would regress due to differentiation and/or cell death.[6,7]
Origin of head and neck cancer stem cells
The origin of the cancer-initiating cell has long been presumed to be the normal endogenous tissue stem cell. This is based upon their similar behaviors and the notion that only accumulated mutations within a long-lived cell could ultimately result in tumorigenesis. In colorectal cancer, there is a strong correlation between induced loss of the Wnt signaling molecule APC in a putative stem cell population and the formation of benign intestinal polyps,[8,9] providing evidence that intestinal cancers can arise from a progenitor population. However, it is possible that accumulation of genetic mutations within a differentiated or progenitor cell can allow expression of stem cell behavior, and that this may provide an alternative source of CSCs. With the primary focus on identifying CSC markers in HNSCC, little is known about the identity or the location of the normal endogenous stem cell or the stem cell microenvironment. Several studies have examined the putative HNSCC CSC marker CD44 in normal head and neck epithelia with differing conclusions. In one study, isolated CD44hi normal oral keratinocytes were shown to exhibit a G2-block associated with apoptosis resistance, a potential stem cell feature, suggesting that CD44 is likely expressed in normal head and neck epithelial stem cells. However, a subsequent study demonstrated that 60–95% of the normal epithelia express CD44 (or 60–80% the splice variant CD44 v6), far too many cells to be considered tissue stem cells. While CD44 populations may indeed harbor a subpopulation encompassing stem cells, by itself it does not appear to be an adequate stem cell marker for normal oral mucosa. The head and neck stem cell identity and niche is clearly underexplored; however, key insights from the skin, airway mucosa, and esophagus may guide future investigations into elucidation of this stem cell population.[4,8,10]
Cancer stem cell identification
Methods for the identification of CSCs in solid malignancies mirror those strategies employed to differentiate normal stem cells from their differentiated progeny. These include the efflux of vital dyes by multidrug transporters, the enzymatic activity of aldehyde dehydrogenase, colony and sphere-forming assays utilizing specific culture conditions, and the most widely used method—the expression of specific cell surface antigens known to enrich for stem cells. Once the subpopulation of tumor cells has been identified and isolated, functional characterization through quantitative xenotransplantation assays, the gold-standard for identification of CSCs, are used to assess the tumorigenicity and self-renewing potential of the putative CSC population in vivo [Figure 4].
Surface antigens
By far the most common method of identifying CSCs has relied on the expression of specific cell surface antigens that enrich for cells with CSC properties. Many of these antigens were initially targeted because of their known expression on endogenous stem cells. While a multitude of studies have identified CSC markers across a variety of solid malignancies, relatively few of these markers have been studied in HNSCC.
CD133
A pentaspan transmembrane glycoprotein localized on cell membrane protrusions,[9,11] is a putative CSC marker for a number of epithelial malignancies including brain, prostate, colorectal, and lung.[12,13] In HNSCC cell lines, CD133hi cells display increased clonogenicity, tumor sphere formation, and tumorigenicity in xenograft models when compared to their CD133 low counterparts.[14,15] While CD133 expression has been noted in primary human HNSCC tumors, quantitative xenotransplantation assays utilizing CD133+hi cells from fresh tumors have yet to be performed. Given the artificial environment of cell culture, these findings will need to be substantiated using primary tumor samples before any definitive conclusions can be made about the usefulness of CD133 as a CSC marker in HNSCC.
CD44
One of the most well-recognized CSC markers is a large cell surface glycoprotein that is involved in cell adhesion and migration. It is a known receptor for hyaluronic acid and interacts with other ligands such as matrix metalloproteases. Initially, it was identified as a solid malignancy CSC marker in breast cancer. Many Authors have demonstrated that CD44 expression could also be used to isolate a tumor subpopulation with increased tumorigenicity in HNSCC.
Aldehyde dehydrogenase activity
Aldehyde dehydrogenase (ALDH) is an intracellular enzyme normally present in the liver. Its known functions include the conversion of retinol to retinoic acids and the oxidation of toxic aldehyde metabolites, like those formed during alcohol metabolism and with certain chemotherapeutics such as cyclophosphamide and cisplatin. ALDH activity is known to enrich hematopoetic stem/progenitor cells and more recently has been shown to enrich cells with increased stem-like properties in solid malignancies. Chen et al. showed that ALDH activity correlated with disease staging in HNSCC and that higher enzymatic activity correlated with expression of epithelial-to-mesenchymal transition (EMT) genes as well as enriching cells with CSC properties.
Side population
Hoechst 33342 is a fluorescent DNA-binding dye that preferentially binds to A-T rich regions. It is actively pumped out of cells by members of the ATP-binding cassette (ABC) transporter superfamily. Once stained with Hoechst dye, cells can be sorted by fluorescent-activated cell sorting (FACS) based upon the activity level of these multidrug transporters. Side population (SP) cells from oral squamous cell carcinoma have been shown to have increased clonogenicity and tumorigenicity in xenotransplantation assays.Furthermore, HNSCC SP cells displayed higher expression of known stem cell related genes—Oct4, CK19, BMI-1 and CD44—and lower expression of involucrin and CK13, genes associated with a differentiated status.
Tumor sphere formation
Under serum-free culture conditions, CSCs can be maintained in an undifferentiated state, and when driven toward proliferation by the addition of growth factors, they form clonally derived aggregates of cells termed tumor spheres. In HNSCC, these spheres have been shown to be enriched for stem markers, including CD44hi, Oct-4, Nanog, Nestin, and CD133hi, as well as exhibiting increased tumorigenicity in orthotopic xenografts.
Cancer stem cells and treatment response
Aside from providing a model of disease progression and metastasis, CSCs have important implications regarding cancer treatment. While current chemotherapy and radiation treatment for HNSCC are focused on indiscriminate cytoreduction, the CSC hypothesis suggests that only by eliminating CSCs can cancer be treated effectively [Figure 4]. However, there is substantial evidence that CSCs have inherent drug and radiation resistance, rendering most conventional therapies ineffective and explaining tumor recurrence despite significant reductions in the tumor volume. By definition, stem cells must divide frequently and thus have highly stringent mechanisms to prevent and rapidly correct DNA damage. In the case of CSCs, there is evidence that these enhanced DNA protection and damage repair pathways lead to significant resistance to radiation and chemotherapy.
The presence of a CSC population has thus far been implicated in radioresistance of multiple tumor types. For example, glioblastoma tumors that recur after radiation have been found to be enriched in CD133hi CSCs.[16,17]
Furthermore, this radioresistance of CD133hi CSCs appears to be mediated through enhanced repair of DNA damage via the Chk1 and Chk2 kinases and through enhanced cell longevity via the histone deacetylase SirT1. Similar results have been seen in a murine model of breast cancer, which showed that CSCs have substantially lower amounts of reactive oxygen species, leading to radioresistance. HNSCC CSCs also demonstrated enhanced radioresistance in murine models that could be reduced by knockdown of the transcriptional repressor Bmi-1. However, the radioresistance of CSCs may not only depend on factors intrinsic to the CSC, but may also be driven by the unique CSC microenvironment. Cell cultures of CSCs from glioblastoma, pancreatic, breast, and colorectal carcinoma have been reported to have similar radiosensitivity to cell cultures that are not enriched for CSCs. In addition to radioresistance, CSCs also appear to mediate chemoresistance in multiple tumor types. There is evidence for chemoresistance of CSCs in lung, pancreatic, and breast carcinoma. In HNSCC, CSCs were made more chemosensitive via knockdown of Bmi-1.[18,19]
Moreover, knockdown of CD44 increased the sensitivity of HNSCC cells to cisplatin, indicating that this CSC marker may be involved in meditating the response of these cells to chemotherapy. Other mechanisms of chemoresistance, such as drug efflux pumps, have been postulated but not yet identified in HNSCC. Future studies will be needed to further define resistance mechanisms in HNSCC CSCs to improve therapy and possibly prevent tumor spread or recurrence.
CONCLUSIONS
To date, most studies have focused on the identification of tumor cell populations enriched for cells with stem-like properties. We have little understanding of the significance of the markers used to identify these cells—whether they are simply markers of convenience or whether they have functional significance remains unknown. Examining the role of these molecules in the tumorigenic, metastatic, and treatment resistance properties of CSCs is certainly a logical step in our way to discovering the mechanisms by which CSCs differ from the remaining tumor cell population. The CSC theory offers an intriguing insight into why currently available therapies for head and neck cancer so often fail. While increasing evidence suggests that CSCs display increased resistance to multiple treatment paradigms inclusive of chemotherapy and radiation, the exact mechanisms by which they do this are incompletely understood. Focused research on mechanisms of treatment resistance in CSCs and whether they can be overcome will prove equally important as efforts to improve CSC identification.
Increasing our knowledge of the differences between CSCs, their differentiated progeny and normal endogenous stem cells, will translate into our ability to understand the seemingly complex cellular hierarchy in tumors. Ultimately, an understanding of CSCs has the potential to identify novel targets for therapy and impact patient care.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Sagar J, Chaib B, Sales K, Winslet M, Seifalian A. Role of stem cells in cancer therapy and cancer stem cells: A review. Cancer Cell Int. 2007;7:1–11. doi: 10.1186/1475-2867-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 3.Serakinci N, Guldberg P, Burns JS, Abdallah B, Schrodder H, Jensen T, et al. Adult human mesenchymal stem cell as a target for neoplastic transformation. Oncogene. 2004;23:5095–8. doi: 10.1038/sj.onc.1207651. [DOI] [PubMed] [Google Scholar]
- 4.Sylvester KG, Longaker MT. Stem cells: Review and update. Arch Surg. 2004;139:93–9. doi: 10.1001/archsurg.139.1.93. [DOI] [PubMed] [Google Scholar]
- 5.Kruse AL, Grätz KW. Oral carcinoma after hematopoietic stem cell transplantation – a new classification based on a literature review over 30 years. Head Neck Oncol. 2009;1:29. doi: 10.1186/1758-3284-1-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang SJ, Wong G, De Heer AM, Xia W, Bourguignon LY. CD44 Variant isoforms in head and neck squamous cell carcinoma progression. Laryngoscope. 2009;119:1518–30. doi: 10.1002/lary.20506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xi S, Grandis JR. Gene therapy for the treatment of oral squamous cell carcinoma. J Dent Res. 2003;82:11–6. doi: 10.1177/154405910308200104. [DOI] [PubMed] [Google Scholar]
- 8.Miura M, Gronthos S, Zhao M, Lu B, Fisher LW, Robey PG, et al. SHED: Stem cells from human exfoliated deciduous teeth. Proc Natl Acad Sci U S A. 2003;100:5807–12. doi: 10.1073/pnas.0937635100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qiao B, Johnson NW, Chen X, Li R, Tao Q, Gao J. Disclosure of a stem cell phenotype in an oral squamous cell carcinoma cell line induced by BMP-4 via an epithelial-mesenchymal transition. Oncol Rep. 2011;26:451–61. doi: 10.3892/or.2011.1299. [DOI] [PubMed] [Google Scholar]
- 10.Hatfield SD, Shcherbata HR, Fischer KA, Nakahara K, Carthew RW, Ruohola-Baker H. Stem cell division is regulated by the micro- RNA pathway. Nature. 2005;435:974–8. doi: 10.1038/nature03816. [DOI] [PubMed] [Google Scholar]
- 11.Choi S, Myers JN. Molecular pathogenesis of oral squamous cell carcinoma: Implications for therapy. J Dent Res. 2008;87:14–23. doi: 10.1177/154405910808700104. [DOI] [PubMed] [Google Scholar]
- 12.Jensen KB, Jones J, Wat FM. A stem cell gene expression profile of human squamous cell carcinomas. Cancer Lett. 2008;272:23–31. doi: 10.1016/j.canlet.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Imanguli MM, Pavletic SZ, Guadagnini JP, Brahim JS, Atkinson JC. Chronic graft versus host disease of oral mucosa: Review of available therapies. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2006;101:175–83. doi: 10.1016/j.tripleo.2005.08.028. [DOI] [PubMed] [Google Scholar]
- 14.Demarosi F, Lodi G, Carrassi A, Soligo D, Sardella A. Oral malignancies following HSCT: Graft versus host disease and other risk factors. Oral Oncol. 2005;41:865–77. doi: 10.1016/j.oraloncology.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Otsubo H, Yokoe H, Miya T, Atsuta F, Miura N, Tanzawa H, et al. Gingival squamous cell carcinoma in a patient with chronic graft-versus-host disease. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;84:171–4. doi: 10.1016/s1079-2104(97)90065-2. [DOI] [PubMed] [Google Scholar]
- 16.Monroe MM, Anderson EC, Clayburgh DR, Wong MH. Cancer stem cells in head and neck squamous cell carcinoma- review. J Oncol. 2011:1–8. doi: 10.1155/2011/762780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 18.Chen YC, Chang CJ, Hsu HS, Chen YW, Tai LK, Tseng LM, et al. Inhibition of tumorigenicity and enhancement of radiochemosensitivity in head and neck squamous cell cancer-derived ALDH1-positive cells by knockdown of Bmi-1. Oral Oncol. 2010;46:158–65. doi: 10.1016/j.oraloncology.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 19.Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–7. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]