

Abstract
Objectives:
Patients on anti-tuberculosis therapy (ATT) are more prone to drug interactions in the presence of coexisting illnesses which warrant drug therapy. Rifampicin is a strong CYP enzyme inducer while isoniazid is a potent CYP inhibitor. The objective of the study was to find the net effect of one month ATT on CYP2C9 enzyme and to correlate it with respect to the CYP2C9 genetic polymorphisms.
Materials and Methods:
Forty eight newly diagnosed tuberculosis patients were included in the study based on the inclusion-exclusion criteria. Before commencing ATT, they were given a single dose of phenytoin 300 mg as a probe drug for CYP2C9. Blood sample was collected after three hours to carry out CYP2C9 genotyping by PCR-RFLP method. Phenotyping for CYP2C9 enzyme was done by measuring the ratio of phenytoin and its metabolite p-HPPH (para hydroxy phenyl hydantoin) by reverse phase HPLC (high performance liquid chromatography) method before and after one month of ATT.
Results:
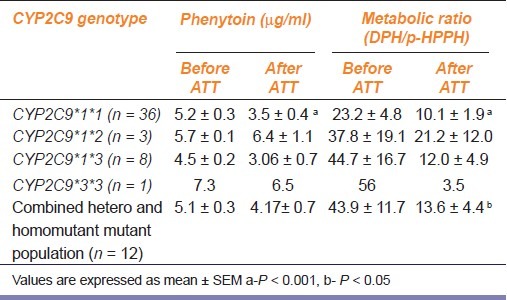
In the CYP2C9*1*1 genotype, the mean plasma concentrations of phenytoin before and after one month of ATT were 5.2 ± 0.3 μg/ml and 3.5 ± 0.4 μg/ml respectively, a reduction by 33% showing significant induction (P < 0.001). There was also significant decrease in the metabolic ratio after one month of ATT from 23.2 ± 4.8 to 10.1 ± 1.9 (P < 0.001). The metabolic ratio was also observed to reduce significantly (P < 0.05) when the CYP2C9*1*2, CYP2C9*1*3, and CYP2C9*3*3 data were pooled together.
Conclusion:
The presence of polymorphisms in the CYP2C9 gene does not affect the induction potential of ATT.
KEY WORDS: Anti-tuberculosis therapy, CYP2C9, induction, inhibition, pharmacogenetics
Introduction
Tuberculosis is one of the leading infectious causes of death in the world. According to the WHO estimates, 1.7 million people died of tuberculosis in the developing world in 2009.[1] Some patients with tuberculosis may also have other coexisting diseases like HIV, diabetes, osteoarthritis, hypertension which will also require drug therapy for a sustained period. The standard treatment for tuberculosis is a combination of rifampicin, isoniazid, pyrazinamide and ethambutol. Since antituberculosis drug therapy (ATT) is given for a minimum period of six months, patients having other coexisting diseases will be taking other additional drugs along with ATT. This can make the patient predisposed to drug interactions.
Rifampicin is known to induce both phase I and phase II drug metabolizing enzymes. It has been described to be a pleiotropic inducer as several enzymes are induced by it, some of them being CYP1A, CYP2A, CYP2C, CYP2D6, CYP2E1 and CYP11B1.[2] Enzyme induction leads to the production of more enzymes, usually after three or more days of drug treatment. Increased enzyme levels will lead to increased metabolic activity and to a quicker degradation of drugs used for the other diseases. This might lead to a therapeutic failure of the drugs used. A classic example of the above situation is the combination of oral contraceptives (OCP) with ATT where rifampicin leads to OCP failure. Isoniazid is known to inhibit CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2E1 and CYP3A4.[3] So when drugs metabolized by the above enzymes are given to a patient on ATT, there is a risk of drug toxicity due to impaired metabolism. However when an inducer and an inhibitor is given together for a patient with tuberculosis the net effect on the drug metabolizing enzyme has not been widely studied at therapeutic doses in vivo. So the combined effect of ATT on one of the drug metabolizing enzyme CYP2C9 is attempted.
CYP2C9 is one of the major drug metabolizing enzymes known to metabolize many of the drugs such as NSAIDs, sulfonylureas, oral anticoagulants, diuretics, angiotensin II blockers, phenytoin, cyclophosphamide, fluoxetine, etc.[4] The gene coding for this enzyme CYP2C9 is mapped on chromosome 10 between q23 and q24. Two inherited amino acid substitutions in CYP2C9 namely, Arg144Cys (*2 allele) and Ile359Leu (*3 allele) are known to affect catalytic function of the enzyme CYP2C9.[5] The genotype frequency of CYP2C9 in Tamil Nadu population done in an earlier study was CYP2C9 *1/*1 – 82%, CYP2C9 *1/*2 – 4% and CYP2C9 *1/*3 – 12%.[6] So the aim of the study was to find the effect of anti-tuberculosis therapy on polymorphic drug metabolizing enzyme CYP2C9.
Materials and Methods
The study was conducted at the Tuberculosis Clinic of the Department of Pulmonary Medicine, Jawaharlal Institute of Postgraduate Medical Education and Research [JIPMER], Pondicherry. The study was approved by the Institutional Ethics Committee. After explaining the study procedure, informed consent was obtained from each participant The patients were diagnosed based on the sputum smear acid fast bacteria (AFB) positivity or a histopathological examination of the specimen showing features suggestive of tuberculosis or a pleural fluid adenosine deaminase titers >60. Unrelated newly diagnosed patients with pulmonary and extra-pulmonary tuberculosis, of either sex or age above 18 years residing in Tamil Nadu and Pondicherry for more than three generations and speaking Tamil as mother tongue were included in the study. All patients with coexisting severe illnesses which warrant hospitalization, pregnant women and patients taking any other medications which induce or inhibit CYP2C9 were excluded from the study. After explaining the procedure and taking a written informed consent from the TB patients, they were administered tab phenytoin 300 mg in an empty stomach, as a probe drug for CYP2C9. Three hours later, 5 ml of blood was collected in EDTA tubes. The patients were asked to return after one month of anti-tuberculosis therapy as prescribed by the chest physicians (rifampicin 10 mg/kg/, isoniazid 5 mg/kg, pyrazinamide 25 mg/kg and ethambutol 15 mg/kg per day). The phenotyping procedure was repeated again after one month of ATT to look for the change in the CYP2C9 enzyme activity.
After centrifugation of the sample, the cellular part (leucocytes) was used for DNA extraction by phenol chloroform method and the CYP2C9 genotyping by PCR-RFLP method.[7] The remnant plasma was used for estimation of phenytoin and its metabolite p-HPPH (Para hydroxyl phenyl hydantoin) by reverse phase high performance liquid chromatography according to the method described earlier.[8] Carbamazepine was used as the internal standard. Pure powders of phenytoin and the metabolite 5-(para-hydroxyphenylhydantoin (p-HPPH) were procured from Aldrich Chem Co. Wisconsin, USA.
Statistical Analysis
All data are presented as mean ± SEM. The data are analysed using GraphPad Instat statistical software (GraphPad Software Inc, San Diego, CA, USA) version 5.02. The mean values of phenytoin, p-HPPH and phenytoin metabolic ratio were tested for deviation from the normality using Kolmogorov-Smirnov test. The concentration of phenytoin and metabolic ratio were compared before and after ATT using paired t test. The concentration of phenytoin and metabolic ratio were compared between the different CYP2C9 genotype groups using Kruskal Wallis test. The level of statistical significance was set at P < 0.05.
Results
A total of 48 tuberculosis patients participated in this study. The demographic characteristics of the study population are shown in Table 1. There was no significant difference in the demographic parameters observed among the CYP2C9 genotype groups. In the CYP2C9*1*1 genotype, the mean plasma concentrations of phenytoin before and after one month of ATT were 5.2 ± 0.3 μg/ml and 3.5 ± 0.4 μg/ml respectively, a reduction by 33% showing significant induction. (P < 0.001) [Table 2]. The range of phenytoin concentration was 3.27 – 7.07 μg/ml. This difference in phenytoin concentration showed significant change only in the wild type population of CYP2C9 but was absent in the hetero mutant population [Table 2]. The mean concentration of phenytoin after one month of anti-tuberculosis treatment was 3.5 μg/ml and the range was 0.2-8.1 μg/ml. The metabolic ratio was also observed to reduce significantly (P < 0.05) when the CYP2C9*1*2, CYP2C9*1*3, and CYP2C9*3*3 data were pooled together. (P < 0.05) as shown in Table 2.
Table 1.
Demographic parameters of the study patients
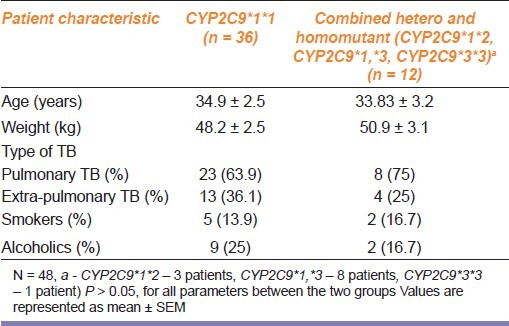
Table 2.
Change in phenytoin concentration and metabolic ratio before and after 1 month of ATT in different CYP2C9 genotypes
The most common adverse effects with ATT observed was dizziness (9.2%), the others being myalgia, pruritus, nausea, vomiting, dyspepsia, headache, macular rash, diplopia and alopecia. These adverse reactions occurred mainly during the first two weeks of therapy with ATT and subsided spontaneously. There was no correlation between the adverse effects observed with respect to the genotype.
Discussion
To the best of our knowledge, this is the first human study to examine the relationship between impact of genotype and induction/inhibition potential in tuberculosis patients. Phenytoin was used as a probe drug to evaluate the activity of CYP2C9 enzyme, since it is metabolised predominantly by this enzyme and its use as a probe drug for CYP2C9 enzyme had earlier been validated in an earlier publication using the ratio of phenytoin and its metabolite concentration.[9] It would have been more informative to study the effect of ATT on multiple enzymes simultaneously but considering the fact that giving a cocktail of probe drugs would make it cumbersome to a patient already receiving multiple drugs as part of his treatment, we restricted ourselves to studying CYP2C9. We chose to limit the study period to 30 days mainly for logistic reasons because after 30 days patients would anyway have to visit the hospital to receive the ATT drugs for the follow up. After the second month of treatment most patients were referred to the local primary health centres to receive the drugs by DOTS therapy. So it would be difficult to expect patients to visit the hospital solely for the study purpose after 120 days. Additionally, the induction and inhibition potential of ATT can be reasonably gauged with one month of ATT.
The mean plasma concentration of phenytoin observed after 3 hours of oral intake of a single dose of phenytoin (300 mg) was 5.1 μg/ml. There was no significant difference in the plasma concentration of phenytoin or its metabolite between different CYP2C9 genotype groups at baseline. This finding was similar to that observed in a study that was done on healthy volunteers in Tamilian population done in our lab.[9] In epileptic patients with polymorphic CYP2C9 enzyme such as CYP2C9*1*3 it has been observed that the phenytoin requirement is considerably reduced due to the diminished enzyme activity and the dose requirement was still lower in the homomutant population.[10] Such variation is not observed here probably because the CYP2C9 enzyme has still not reached saturation kinetics as it is a single dose study.
The mean concentration of phenytoin after one month of anti-tuberculosis treatment was 3.5 μg/ml and the range was 0.2-8.1 μg/ml. The significant decrease (33%) in the phenytoin concentration after one month of anti-tuberculosis treatment implies that there is an enhancement of the metabolism of the probe drug phenytoin by the enzyme CYP2C9. The metabolic ratio (DPH/p-HPPH) has also shown a significant decrease after one month of ATT. All these results are in conformity with previous studies that showed that rifampicin is a strong pleiotropic inducer of drug metabolizing enzymes including CYP2C9 (2). Several clinical studies have shown that rifampicin increases the clearance of co-administered drugs such as fluconazole, pioglitazone, oral contraceptives, gliclazide, sulfasalazine, atazanavir, saquinavir, etc.[11,12] Although rifabutin is known to have a lesser induction potential as compared to rifampicin, a literature search did not show any study highlighting the genotype impact on the induction and inhibition potential of rifabutin.
Although isoniazid is a weak inhibitor of CYP2C9 enzyme, when it is given in combination with rifampicin, ethambutol and pyrazinamide for 30 days, the net effect that has been observed in this study is enhanced induction. This is in conformity to an earlier study done in tuberculosis patients which showed that the inducing effect of rifampicin is much more pronounced than the inhibitory effects of isoniazid when both the drugs are given in combination.[13] However it has also been observed that isoniazid can cause phenytoin toxicity when both the drugs are administered together due to the predominant inhibitory effect of isoniazid.[14,15]
The average decrease in phenytoin concentration and the decrease in metabolic ratio after one month of ATT was observed in both the groups. This indicates that anti-tuberculosis treatment has produced a significant induction in the mutant groups. Despite the low amount of enzyme levels present in patients of heterozygous and homozygous variant genotype, induction of enzymes was detected in this population. This would mean that the genotype of the individual does not play a major role in determining the degree of induction of CYP2C9. These findings are similar to a study done in healthy volunteers who were given rifampicin 450 mg for four days to see if it could change the kinetics of the probe drug tolbutamide. Tolbutamide clearance was similar across all the CYP2C9 genotypes.[16] This is in contrast to an earlier study using mephenytoin as a probe drug, which showed that induction of the CYP enzymes by rifampicin was only observed in extensive metabolisers but not in poor metabolisers.[17] We however could not find any reason for the sixteen fold increase in induction that was observed in the patient with CYP2C9*3*3.
In addition to the polymorphisms of P450 genes, genetic variations of nuclear receptors and regulatory proteins that modulate the transcriptional processes of P450 expression, intracellular and tissue concentration of inducers, physiological factors such as hormones, development and disease and other environmental elements could play a significant role in the variability of P450 expression.[18] In this study, all patients were given the anti-tuberculosis drugs based on their body weight. The extent of enzyme induction is also a dose dependent process. Thus even individual differences in the intracellular concentration of inducers such as rifampicin could cause differences in the degree of induction. The intracellular concentration of the inducers can be influenced by a variety of drug transporters such as P- glycoprotein which can cause efflux of the drug. P-glycoprotein is encoded by the human MDR1 gene. It has been observed that in MDR1 overexpressed human colon carcinoma cells, the induction of CYP3A4 by rifampicin was reduced when compared to the parental cells.[19] Oral treatment with rifampicin in increasing doses was also shown to result in elevated drug levels in the livers of MDR1a (-/-) when compared to of MDR1a (+/+) mice.[20] A similar mechanism could also be operating in these patients with impaired induction. A polymorphism in the MDR1 gene may also influence enzyme induction by rifampicin.
The human liver-specific organic anion-transporting polypeptide C (OATP-C) mediates the hepatocellular uptake of rifampicin. Variation of OATP can also reduce the intake of rifampicin and thereby weaken rifampicin mediated pregnane X receptor activation.[21,22] There are no published data on the frequencies of OATP-C polymorphisms in Indian population. Interleukin - 6 decreases both rifampicin and phenobarbital mediated induction of CYP2B6, CYP2C8, CYP2C9 and CYP3A4.[23] Mycobacterium tuberculosis has shown to activate interleukin 6 and thereby decrease the induction of CYP2C9.[24] If there is a variation in interleukin 6 activation it can also partly explain the variability in enzyme induction.
Limitations of the study
The lack of adequate sample size in the homozygous and heterozygous population of individuals with CYP2C9 polymorphisms has made it difficult to give conclusive evidence on the influence of genetic polymorphisms on enzyme induction. Nevertheless considering the fact that the frequency of the CYP2C9 variants is reduced in the general population, it is not surprising to find a lower sample size in this study among TB patients. The results of this study may offer sufficient ground for elucidating further, the interaction of genotype and the induction inhibition potential of ATT therapy. The dose of phenytoin that was given to the patients was a fixed dose and not one based on the individual patient's body weight. This could in addition contribute to the inter-individual variation in the metabolism.
Our study has shown that induction of CYP2C9 occurs with one month of ATT. However the induction was detected appreciably in both the heterozygous population of CYP2C9*1*2 and CYP2C9*1*3 thereby showing that the genotype of the individual may not significantly influence the degree of induction. The role of other genetic factors such as mutation in promoter region of OATP and MDR1 polymorphisms in induction needs to be studied to have a better understanding on the influence of genotype on induction of the CYP2C9 enzyme.
Conclusions
Our study has shown that one month of anti-tuberculosis treatment can cause a significant decrease in the metabolic ratio of phenytoin implying increased induction of CYP2C9.The concentration of phenytoin after one month of anti-tuberculosis therapy was also shown to reduce significantly which could be due to the induction of CYP2C9. The maximum induction observed was not greater than two fold. However the induction of CYP2C9 was also found to be significant in individuals with deficient enzyme activity when pooled together as in CYP2C9*1*2 and CYP2C9*1*3 thereby showing that the CYP2C9 genotype of the individual may not influence the degree of induction caused by ATT.
Acknowledgments
We gratefully acknowledge the Indian Council of Medical Research for funding this project (Sanction number: 53/17/2003 - BMS dated 12-3-2007). We thank Mr. Rajan, Ms. Kayathri, Ms. Anuradha, Ms. Indumathi, and Ms. Priya for their technical assistance in this project.
Footnotes
Source of Support: Indian Council of Medical Research.
Conflict of Interest: No.
References
- 1.Global tuberculosis control 2010 fact sheet (2010) WHO web site. [Last accessed on 2011 June 9]. Available from: www.who.int .
- 2.Rae JM, Johnson MD, Lippman ME, Flockhart DA. Rifampin is a selective, pleiotropic inducer of drug metabolism genes in human hepatocytes: Studies with cDNA and oligonucleotide expression arrays. J Pharmacol Exp Ther. 2001;299:849–57. [PubMed] [Google Scholar]
- 3.Nishimura Y, Kurata N, Sakurai E, Yasuhara H. Inhibitory effect of anti-tuberculosis drugs on human cytochrome P450-mediated activities. J Pharmacol Sci. 2004;96:293–300. doi: 10.1254/jphs.fp0040296. [DOI] [PubMed] [Google Scholar]
- 4.Kirchheiner J, Brockmoller J. Clinical consequences of cytochrome P450 2C9 polymorphisms. Clin Pharmacol Ther. 2005;77:1–16. doi: 10.1016/j.clpt.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 5.Miners JO, Birkett DJ. Cytochrome P4502C9: An enzyme of major importance in human drug metabolism. Br J Clin Pharmacol. 1998;45:25–38. doi: 10.1046/j.1365-2125.1998.00721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adithan C, Gerard N, Vasu S, Balakrishnan R, Shashindran CH, Krishnamoorthy R. Allele and genotype frequency of CYP2C9 in Tamil Nadu population. Eur J Clin Pharmacol. 2003;59:707–9. doi: 10.1007/s00228-003-0666-3. [DOI] [PubMed] [Google Scholar]
- 7.Sullivan-Klose TH, Ghanayem BI, Bell DA, Zhang ZY, Kaminsky LS, Shenfield GM, et al. The role of the CYP2C9-Leu359 allelic variant in the tolbutamide polymorphism. Pharmacogenetics. 1996;4:341–9. doi: 10.1097/00008571-199608000-00007. [DOI] [PubMed] [Google Scholar]
- 8.Gerson B, Bell F, Chan S. Antiepileptic agents--primidone, phenobarbital, phenytoin, and carbamazepine by reversed-phase liquid chromatography. Clin Chem. 1984;30:105–8. [PubMed] [Google Scholar]
- 9.Rosemary J, Surendiran A, Rajan S, Shashindran CH, Adithan C. Influence of the CYP2C9 AND CYP2C19 polymorphisms on phenytoin hydroxylation in healthy individuals from south India. Indian J Med Res. 2006;123:665–70. [PubMed] [Google Scholar]
- 10.Hung CC, Lin CJ, Chen CC, Chang CJ, Liou HH. Dosage recommendation of phenytoin for patients with epilepsy with different CYP2C9/CYP2C19 polymorphisms. Ther Drug Monit. 2004;26:534–40. doi: 10.1097/00007691-200410000-00012. [DOI] [PubMed] [Google Scholar]
- 11.Acosta EP, Kendall MA, Gerber JG, Ston-Smith B, Koletar SL, Zolopa AR, et al. Effect of concomitantly administered rifampin on the pharmacokinetics and safety of atazanavir administered twice daily. Anti-microb Agents Chemother. 2007;51:104–10. doi: 10.1128/AAC.00341-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ament PW, Bertolino JG, Liszewski JL. Clinically significant drug interactions. Am Fam Physician. 2011;61:1745–54. [PubMed] [Google Scholar]
- 13.Kay L, Kampmann JP, Svendsen TL, Vergman B, Hansen JE, Skovsted L, et al. Influence of rifampicin and isoniazid on the kinetics of phenytoin. Br J Clin Pharmacol. 1985;20:323–6. doi: 10.1111/j.1365-2125.1985.tb05071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller RR, Porter J, Greenblatt DJ. Clinical importance of the interaction of phenytoin and isoniazid: A report from the Boston collaborative drug surveillance program. Chest. 1979;75:356–8. doi: 10.1378/chest.75.3.356. [DOI] [PubMed] [Google Scholar]
- 15.Walubo A, Aboo A. Phenytoin toxicity due to concomitant anti-tuberculosis therapy. S Afr Med J. 1995;85:1175–6. [PubMed] [Google Scholar]
- 16.Vormfelde SV, Brockmoller J, Bauer S, Herchenhein P, Kuon J, Meineke I, et al. Relative impact of genotype and enzyme induction on the metabolic capacity of CYP2C9 in healthy volunteers. Clin Pharmacol Ther. 2009;86:54–61. doi: 10.1038/clpt.2009.40. [DOI] [PubMed] [Google Scholar]
- 17.Zhou HH, Anthony LB, Wood AJ, Wilkinson GR. Induction of polymorphic 4′-hydroxylation of S-mephenytoin by rifampicin. Br J Clin Pharmacol. 1990;30:471–5. doi: 10.1111/j.1365-2125.1990.tb03799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang C, Lin JH, Lu AY. Metabolism-based drug-drug interactions: What determines individual variability in cytochrome P450 induction? Drug Metab Dispos. 2005;33:603–13. doi: 10.1124/dmd.104.003236. [DOI] [PubMed] [Google Scholar]
- 19.Mills JB, Rose KA, Sadagopan N, Sahi J, De Morais SM. Induction of drug metabolism enzymes and MDR1 using a novel human hepatocyte cell line. J Pharmacol Exp Ther. 2004;309:303–9. doi: 10.1124/jpet.103.061713. [DOI] [PubMed] [Google Scholar]
- 20.Schuetz EG, Schinkel AH, Relling MV, Schuetz JD. P-glycoprotein: A major determinant of rifampicin-inducible expression of cytochrome P4503A in mice and humans. Proc Natl Acad Sci U S A. 1996;93:4001–5. doi: 10.1073/pnas.93.9.4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tirona RG, Leake BF, Wolkoff AW, Kim RB. Human organic anion transporting polypeptide-C (SLC21A6) is a major determinant of rifampin-mediated pregnane X receptor activation. J Pharmacol Exp Ther. 2003;304:223–8. doi: 10.1124/jpet.102.043026. [DOI] [PubMed] [Google Scholar]
- 22.Zaher H, Zu Schwabedissen HE, Tirona RG, Cox ML, Obert LA, Agrawal N, et al. Targeted disruption of murine organic anion-transporting polypeptide 1b2 (Oatp1b2/Slco1b2) significantly alters disposition of prototypical drug substrates pravastatin and rifampin. Mol Pharmacol. 2008;74:320–9. doi: 10.1124/mol.108.046458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pascussi JM, Gerbal-Chaloin S, Pichard-Garcia L, Daujat M, Fabre JM, Maurel P, et al. Interleukin-6 negatively regulates the expression of pregnane X receptor and constitutively activated receptor in primary human hepatocytes. Biochem Biophys Res Commun. 2000;274:707–13. doi: 10.1006/bbrc.2000.3219. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Broser M, Rom WN. Activation of the interleukin 6 gene by mycobacterium tuberculosis or lipopolysaccharide is mediated by nuclear factors NF-IL6 and NF-kappa B. Proc Natl Acad Sci U S A. 1994;15:2225–9. doi: 10.1073/pnas.91.6.2225. [DOI] [PMC free article] [PubMed] [Google Scholar]