

Abstract
Endocannabinoid signaling has been shown to mediate synaptic plasticity by retrogradely inhibiting presynaptic transmitter release in several systems. We found that endocannabinoids act tonically to regulate AMPA miniature postsynaptic current (mPSC) frequency in embryonic motor circuits of the chick spinal cord. Further, strong postsynaptic depolarizations also induced a short-lived endocannabinoid-mediated suppression of mEPSC frequency. Unlike many previous studies, endocannabinoid signaling was not found to influence evoked transmitter release. The results suggest a special role for spontaneous glutamatergic mPSCs and their control by endocannabinoids in the developing spinal cord. We determined that blocking endocannabinoid signaling, which increases spontaneous glutamatergic release, increased spontaneous network activity in vitro and in vivo. Previous work in spinal motoneurons had shown that reducing spontaneous network activity (SNA) chronically in vivo led to homeostatic increases in AMPA and GABA mPSC amplitude (homeostatic synaptic plasticity). Blocking endocannabinoid signaling in vivo, and thus increasing SNA, triggered compensatory decreases of both AMPA and GABA mPSC amplitudes. These findings, combined with previous results, are consistent with the idea that this form of homeostatic synaptic plasticity is a bidirectional process in the living embryo. Together, our results suggest a role for tonic signaling of endocannabinoids as a potential mechanism to regulate the level of SNA, which is known to be critical for synaptic maturation in the embryonic spinal cord.
Introduction
Embryonic movements are produced by spontaneous bursts of spiking activity arising episodically in the developing spinal network. This spontaneous network activity (SNA) results from the highly excitable nature of the synaptic connections in the spinal circuitry, as both GABAergic and AMPAergic currents are excitatory at this stage. SNA is expressed in most, if not all, developing networks, suggesting a fundamental importance for the maturation of these neuronal circuits (Blankenship and Feller, 2010; O'Donovan et al., 1998). In the embryonic spinal cord SNA has been shown to be important for the proper development of the limbs, and motoneuron axon pathfinding (Gonzalez-Islas and Wenner, 2010; O'Donovan et al., 1998). In addition, we have recently shown that SNA is important in the regulation of synaptic strength through a form of homeostatic synaptic plasticity (Gonzalez-Islas and Wenner, 2006). When SNA was blocked in vivo in the chick from embryonic day 8 to 10 (E8–E10), the amplitude of AMPA and GABA miniature postsynaptic currents (mPSCs) was altered in a manner that should compensate for activity blockade. On the other hand it is unknown whether homeostatic plasticity is triggered by chronic increases in the levels of SNA in the embryonic cord.
Additionally, a faster form of SNA-dependent synaptic plasticity also occurs in developing spinal motoneurons. Immediately following an episode of SNA, evoked and spontaneous glutamatergic synaptic responses are depressed, and then recover in the interepisode interval (IEI; Fedirchuk et al., 1999; Tabak et al., 2001; C. Gonzalez-Islas and P. Wenner, unpublished results). This postepisode depression and recovery appear to be at least partially mediated by changes in presynaptic release. The mechanism responsible for the SNA-dependent modulation of AMPAergic synaptic strength is not clear. We considered the possibility that endocannabinoid signaling, implicated in many different forms of synaptic plasticity, was involved. It is widely known that after strong depolarizations and metabotropic signaling, as would occur during episodes of SNA, neurons can synthesize and release endocannabinoids; the released endocannabinoids retrogradely activate a presynaptic G-protein-coupled cannabinoid receptor (CB1), which in turn inhibits neurotransmitter release (Alger, 2009; Heifets and Castillo, 2009; El Manira and Kyriakatos, 2010; Ohno-Shosaku et al., 2012). Further, previous work suggested that hippocampal SNA leads to endocannabinoid liberation, thus inhibiting presynaptic vesicle release (Bernard et al., 2005). We therefore hypothesized that episodes of SNA could trigger the release of endocannabinoids and reduce presynaptic glutamatergic release following the episode.
We show that endocannabinoid signaling did not appear to mediate the postepisode modulation of mEPSC frequency or evoked (action potential-dependent) glutamatergic responses. Despite this, endocannabinoid signaling did mediate a transient depolarization-induced suppression of mEPSCs and had a profound effect on a tonic regulation of mEPSCs, demonstrating an importance of mEPSC regulation. Additionally, altering endocannabinoid signaling also influenced the frequency of SNA in vitro and in vivo, likely because of the changes in glutamatergic spontaneous release. By blocking endocannabinoid receptors in vivo, we also were able to demonstrate a form of compensatory synaptic plasticity following increases in SNA. The results suggest an important role for endocannabinoids in the maturation of motor networks.
Materials and Methods
Dissection.
Experiments were performed on isolated spinal cords of stage 36 (Hamburger and Hamilton, 1951) White Leghorn chicken embryos of either sex. Eggs were incubated for 10 d (E10) (GQF Manufacturing) at 38°C. The lumbosacral spinal cord region, with attached spinal nerves, was dissected under cooled (15°C) oxygenated Tyrode's solution containing the following (in mm): 139 NaCl, 12 d-glucose, 17 NaHCO3, 3 KCl, 1 MgCl2, and 3 CaCl2. Tyrode's solution was constantly bubbled with a mixture of 95% O2-5% CO2 to maintain a pH of ∼7.3. After dissection, the cord was allowed to recover for at least 6 h in Tyrode's at 18°C. The cord was then transferred to a recording chamber and continuously perfused with oxygenated Tyrode's solution that was slowly heated to recording temperature (28°C). Stable spontaneous network activity was established before starting the experiments (constant burst frequency, every 10–12 min).
Immunoblot.
Lumbosacral spinal cords were homogenized in radioimmunoprecipitation assay buffer supplemented with protease and phosphatase inhibitors (Sigma-Aldrich). Samples were then centrifuged at 16,000 g for 5 min to remove cell debris. Protein concentration was quantitated using BCA reagent (Pierce). Samples were separated on 4–15% SDS-PAGE, and blotted to a nitrocellulose membrane. The primary antibody against CB1 receptor was from Alomone Labs (rabbit anti-CB1R ACR-001). The secondary antibody was a horseradish peroxidase-goat anti-rabbit IgG from Sigma-Aldrich. The blot was visualized by ECL chemiluminescence (GE Healthcare). Blots were done in duplicate, and each sample represents a lysate of 2 different cords.
In ovo drug application.
At E8, a window in the shell of the egg was opened to allow monitoring chick embryo movements and drug application, and 5 μm AM 251 (based on a 50 ml egg volume; Tocris Bioscience) in dimethylsulfoxide (DMSO) was applied onto the chorioallantoic membrane of the chick embryo. Modification of the spontaneous activity was monitored by assessing the duration of embryonic movements in a 5 min window at different times during treatment. Only eggs with a cardiac rate >100 beats/min at E10 were used (untreated E10 embryos: 156 ± 3, n = 10; treated E10 embryos: 162 ± 5, n = 10).
Recording of mPSCs.
Whole-cell patch-clamp recordings were made from motoneurons localized between lumbosacral segments 1 and 3 (LS1–LS3) to acquire mPSC. Tetrodotoxin was not used to isolate mPSCs during the IEI, as the vast majority of these events were action potential independent (Chub and O'Donovan, 2001; Gonzalez-Islas and Wenner, 2006). Motoneurons were antidromically identified by stimulating muscle nerves via tight-fitting glass suction electrodes. Patch-clamp tight seals (>2 GΩ) were obtained using electrodes pulled from thin-walled glass (World Precision Instruments) in two stages, using a P-87 Flaming/Brown micropipette puller (Sutter Instruments) to obtain resistances between 5 and 10 MΩ. Series resistance during recording varied from 15 to 20 MΩ among different motoneurons and was not compensated. Recordings were terminated whenever significant increases in series resistance (>20%) occurred. Standard extracellular recording solution for mPSCs contained the following (in mm): 139 NaCl, 5 KCl, 17 NaHCO3, 3 CaCl2, 1 MgCl2, and 12 d-glucose, and pH was adjusted to 7.3 with NaOH. The extracellular solution was constantly bubbled with a mixture of 95% O2-5% CO2 to maintain a pH of ∼7.3. The intracellular patch solution for mPSC recordings contained the following (in mm): 10 NaCl, 100 K-gluconate, 36 KCl, 10 HEPES, 1.1 EGTA, 1 MgCl2, 0.1 CaCl2, 1 Na2ATP, and 0.1 MgGTP; pH was adjusted to 7.3 with KOH. The junction potential was determined for each experiment (approximately −13 mV) and corrected off-line. Motoneuron GABA and AMPA mPSCs were separated based on their kinetics, as shown previously for this preparation (Gonzalez-Islas and Wenner, 2006); thus no antagonists were added to the bath to isolate mPSCs (unless specified in the results). Glycine receptor antagonists were not used since glycinergic mPSCs were not observed in chick embryo motoneurons at the stages studied (Gonzalez-Islas and Wenner, 2006). No NMDA-sensitive currents could be detected when motoneurons were held at −70 mV and with 1 mm MgCl2 in the bath (Gonzalez-Islas and Wenner, 2006). mPSCs were acquired on an EPC 8 patch-clamp amplifier (Heka Elektronics), digitized on-line using PClamp 10 (Molecular Devices), and analyzed off-line with MiniAnalysis (Synaptosoft).
Recording of ventral root potentials.
Tight-fitting glass suction electrodes were used to record and stimulate the ventral roots, dorsal roots, and ventrolateral funiculus (VLF) as described previously (O'Donovan, 1989; O'Donovan and Landmesser, 1987; Xu et al., 2005). These ventral root recordings represent the electrotonically degraded motoneuron population potentials as shown originally in the 1940s (Eccles, 1946; Brooks et al., 1948) and later using glass suction electrodes as sucrose gap recordings (Roberts and Wallis, 1978; Lüscher et al., 1979; Brink et al., 1981), and more recently in the in vitro preparation (O'Donovan, 1987; Wenner and O'Donovan, 1999; Butt and Kiehn, 2003). The signals were amplified (1000×) and filtered (DC to 0.3–5 kHz) by an AC/DC differential amplifier (A-M Systems), and digitally recorded using Axograph acquisition software (Molecular Devices). Further analyses of the data were performed off-line. The ventral root response amplitude was measured following stimulation of the dorsal root or VLF.
Statistics.
Data from averages are expressed as mean ± SEM. Student's t test (α = 0.05) was used to perform the statistical analyses of mPSC amplitudes, frequencies, and decay times. Best-fit analyses were performed for cumulative distributions using a Kolmogorov–Smirnov test (α = 0.05). Statistical analysis of mEPSCs (see Figs. 3D, 7), evoked responses (see Fig. 6), IEI (see Fig. 8), and same cell comparisons of Table 1 were performed using a paired t test.
Figure 3.

Increasing CB1 receptor activation reduces AMPA mPSC frequency. A, Representative traces of AMPA mPSCs from a motoneuron before (black trace) and 20 min after bath addition of the CB1 receptor agonist WIN (5 μm; gray trace). B, Superimposed traces of averaged AMPA mPSCs before (black trace) and after bath addition of WIN (gray trace). C, D, Summary of the effects of WIN (1–10 μm) on AMPA and GABA mPSC amplitude (C) and frequency (D). *p < 0.05. E, Cumulative distribution plot showing the increase in the IEI (reduction in frequency) after addition of 5 μm WIN for the entire distribution of AMPA mPSCs.
Figure 7.

Endocannabinoid signaling and the modulation of AMPA mPSC frequency. A, Whole-cell recording of a motoneuron shows two consecutive episodes and the modulation of mPSCs in the IEI. B, Plot of average AMPA mPSC frequency for consecutive 2 min bins through the IEI. Plots represent values before (black squares) and after (gray diamonds) bath application of 5 μm AM 251 for six different cells. The slopes were obtained from fitting the averaged data to linear functions. C, Normalizing AMPA mPSC frequencies to the frequency in the last 2 min period of the IEI shows that the gradual increase in AMPA mPSC frequency is slightly reduced but is still present after the addition of AM 251. D, Schematic representation of the protocol followed for DSE. AMPA mPSC frequency was measured for 2 min holding the cell at −70 mV, then stepping to 0 mV for 1 min. AMPA mPSC frequency was then measured in the first and second minute after depolarization. E, Summary of the effect of 0 mV step on AMPA mPSC frequency in the first and second minute following depolarization. A significant reduction in AMPA mPSC frequency was observed in the first, but not second minute in control conditions (black bars). *p < 0.04. Bath application of AM 251 (gray bars) prevented this form of DSE.
Figure 6.

CB1 signaling does not influence evoked glutamatergic responses. A, Ventral root (VR) recording showing the alternation of dorsal root (DR) and VLF stimulation. Ventral root recordings expanded from VLF stimulations before and after an episode of SNA are shown below. B, Plot of the amplitudes of VLF-evoked glutamatergic responses in motoneurons, as measured from the ventral root. The responses modulate in an episode-dependent manner, dropping after an episode (vertical lines) and recovering in the IEI. Application of AM 251 or AM 404 had no effect on these responses. C, Dorsal root-evoked responses are not modulated by SNA, and are unaffected following bath application of AM 251 or AM 404. D, Summary of data showing the applied drugs had no effect on VLF- and dorsal root-evoked responses. Twenty dorsal root-evoked values were averaged per preparation in the presence of the drug (20 min after addition of drug) and normalized to last 20 values before drug. VLF-evoked values from two IEIs in the presence of the drug (20 min after addition of drug) were averaged and normalized to the average value of the responses in the last two IEIs before the drug.
Figure 8.
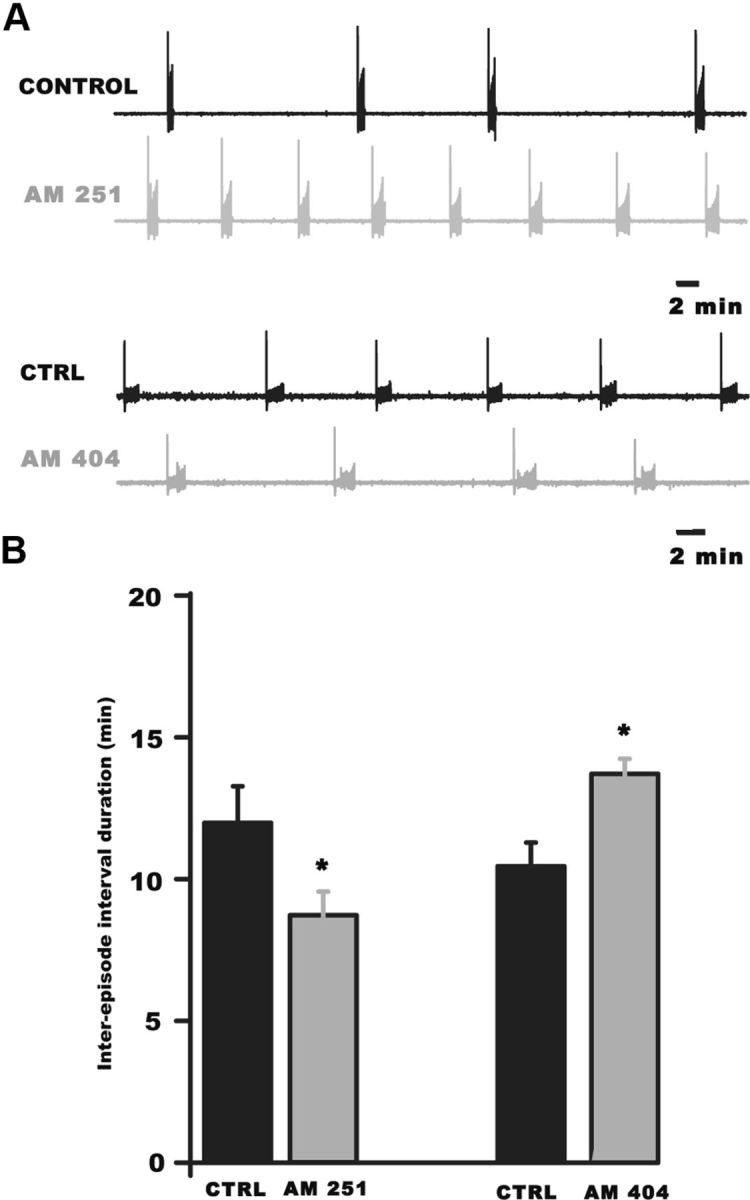
Endocannabinoid signaling influences embryonic spontaneous network activity in the chick spinal cord in vitro. A, Example traces of ventral root recordings showing the expression of SNA before (control, black) and after the addition of endocannabinoid signaling modulators (gray). B, Bar graph summarizing the effects of activation or inhibition of CB1 receptors on the duration of the IEI of SNA in isolated spinal preparations. Antagonizing CB1 receptors with AM 251 (5 μm) increased AMPA SNA frequency (decreased IEI) in vitro. Increasing endocannabinoid signaling with AM 404 significantly decreased SNA frequency (increased IEI) in vitro. *p < 0.005.
Table 1.
Effect of cannabinoid modulators on motoneuron resting membrane properties and on the mPSC parameters
| Parameter | Control (n = 7) | AM 251 (n = 7) | Control (n = 15) | WIN 55,212-2 (n = 17) | Control (n = 9) | ACEA (n = 6) | Control (n = 5) | AM 404 (n = 5) | Control (n = 8) | LY 2183240 (n = 13) |
|---|---|---|---|---|---|---|---|---|---|---|
| Membrane potential (mV) | −63 ± 9.2 | −66 ± 8.7 | −65 ± 5.4 | −71 ± 4.7 | −71 ± 5.3 | −65 ± 4.2 | −59 ± 6.4 | −62 ± 7.1 | −61 ± 3.5 | −60 ± 4.2 |
| Membrane resistance (MΩ) | 519 ± 76 | 487 ± 53 | 582 ± 56 | 573 ± 28 | 576 ± 89 | 521 ± 49 | 448 ± 62 | 472 ± 85 | 519 ± 38 | 530 ± 27 |
| AMPA mPSC mean amplitude (pA) | 8.4 ± 0.2 | 8.5 ± 0.5 | 15.2 ± 1.5 | 15.6 ± 1.4 | 16.8 ± 0.4 | 11.0 ± 0.3 | 8.8 ± 0.5 | 9.1 ± 1.5 | 12.8 ± 0.6 | 12.9 ± 0.7 |
| AMPA mPSC frequency (Hz)‡ | 0.24 ± 0.03 | 1.03 ± 0.03 | 0.26 ± 0.08 | 0.10 ± 0.02 | 0.88 ± 0.24 | 0.4 ± 0.08 | 0.4 ± 0.04 | 0.11 ± 0.02 | 0.45 ± 0.14 | 0.15 ± 0.05 |
| (n = 7) | (n = 7)‡ | (n = 8) | (n = 8)‡ | (n = 4) | (n = 4)‡ | (n = 5) | (n = 5)‡ | (n = 5) | (n = 5)‡ | |
| AMPA mPSC decay time (ms) | 4.8 ± 0.2 | 5.2 ± 0.3 | 4.9 ± 0.7 | 5.1 ± 0.6 | 6.7 ± 0.3 | 7.1 ± 0.4 | 7.4 ± 1.0 | 9.3 ± 1.1 | 5.6 ± 0.3 | 6.6 ± 0.5 |
| GABA mPSC mean amplitude (pA) | 13.3 ± 0.4 | 12.0 ± 0.6 | 23.7 ± 2.1 | 25.3 ± 2 | 24.4 ± 1.1 | 14.5 ± 0.5 | 17.8 ± 1.8 | 11.5 ± 1.4 | n.m. | n.m. |
| GABA mPSC frequency (Hz) | 0.57 ± 0.04 | 0.61 ± 0.04 | 0.28 ± 0.1 | 0.31 ± 0.08 | 0.57 ± 0.06 | 0.54 ± 0.07 | 0.52 ± 0.07 | 0.35 ± 0.11 | n.m. | n.m. |
| GABA mPSC decay time (ms) | 26.3 ± 0.8 | 27.4 ± 1.1 | 32 ± 4 | 33.6 ± 2.7 | 32.8 ± 0.1 | 35.4 ± 0.1 | 30.8 ± 1.3 | 34.7 ± 1.1 | n.m. | n.m. |
Bold numbers indicate significant differences (p < 0.05), n.m. indicates not measured.
‡Denotes that the value was obtained from the same cells before and during drug addition (number of cells shown in parentheses). Averages are expressed as mean ± SEM Student's t test (α = 0.05) was used to perform the statistical analyses of membrane potential, resistance, mPSC amplitude, frequency, and decay time.
Drugs.
AM 251 (N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide); AM 404 (N-(4-hydroxyphenyl)-5Z,8Z,11Z,14Z-eicosatetraenamide); WIN 55,212-2 mesylate ((R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl) pyrrolo [1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate); ACEA (N-(2-chloroethyl)-5Z,8Z,11Z,14Z-eicosatetraenamide); and LY 2183240 (5-[(1,1′-biphenyl]-4-yl)methyl]-N,N-dimethyl-1H-tetrazole-1-carboxamide) were purchased from Tocris Bioscience. KCl was purchased from Fisher Scientific and potassium d-gluconate from Alfa Aesar. All other salts and compounds were purchased from Sigma-Aldrich.
Results
Antagonizing endocannabinoid receptors increased AMPAergic mPSC frequency
Our initial hypothesis was that the large depolarizations produced by episodes of SNA evoke the release of endocannabinoids and activate CB1 receptors, resulting in the weakening of AMPAergic synaptic strength. Thus, we first biochemically verified that CB1 receptors were present in the developing chick spinal cord. Specific CB1 receptor antibodies recognized a single band in Western blots from E10 chick embryo spinal cords (Fig. 1).
Figure 1.

CB1 receptors are present in the developing chick spinal cord. Western blots showing the presence of CB1 in E10 chick embryo spinal cords. Lanes 1 and 2, Total spinal cord lysates incubated with CB1 receptor antibody. Lanes 3 and 4, Total spinal cord lysates incubated first with epitope peptide and then CB1 receptor antibody to show specificity of the antibody.
Next, we tested the possibility that the endocannabinoid signaling would influence AMPA mPSCs. We bath applied an inverse agonist of the CB1 receptor AM 251 (5 μm) while recording whole cell from antidromically identified motoneurons in the isolated E10 spinal cord preparation. Indeed, after adding AM 251 (>20 min), AMPA-mPSC frequency was robustly increased (Fig. 2; Table 1; p = 0.005) without changes in AMPA mPSC amplitude (Fig. 2B,E; Table 1; p = 0.2) or kinetics (Fig. 2B; Table 1; p = 0.13). Further, AM 251 had no effect on GABA mPSC frequency (Fig. 2C), amplitude (Fig. 2B,E; Table 1), or kinetics (Fig. 2B; Table 1). These results suggested that ongoing glutamatergic quantal release was tonically inhibited by CB1 activation.
Figure 2.

Blocking CB1 receptors increased AMPA mPSC frequency. A, Representative traces of mPSCs from an E10 motoneuron before (black trace) and after bath application of the CB1 inverse agonist AM 251 (5 μm; gray trace). B, Superimposed average traces of AMPA mPSC (left) and GABA mPSC (right) before (black trace) and after bath addition of AM 251 (gray trace). No change in mPSC kinetics was found in the presence of AM 251. C, Summary of the effect of AM 251 on mPSC frequencies. Only AMPA mPSC frequency increased significantly after AM 251 addition. *p < 0.01. D, Cumulative distribution plot showing the reduction of the IEI after AM 251 addition. E, Bar graph showing AM 251 had no effect on mPSC amplitudes.
Activation of endocannabinoid signaling decreased AMPAergic mPSC frequency
To determine whether exogenous CB1 activation could reduce the frequency of AMPA mPSCs, we bath applied the CB1 agonist WIN 55,212–2 mesylate (WIN), and recorded mPSC frequency. Surprisingly, AMPA mPSC frequency was no different in control cells and cells exposed to WIN for ≥60 min (1–10 μm, control −0.47 ± 0.06, n = 7; WIN −0.59 ± 0.10, n = 7). However, we found that AMPA mPSC frequency was significantly reduced in individual motoneurons after a 20 min bath application of WIN (Fig. 3D,E; Table 1; p = 0.04). Thus, WIN reduced mEPSC frequency within the same cell after 20–40 min, but not in separate populations of cells that were recorded before and ≥60 min after WIN application. These findings suggest that AMPA mPSC frequency is initially reduced in the presence of WIN, but that mEPSC frequency then recovers in a compensatory manner (see Discussion). WIN did not appear to influence AMPA mPSC amplitude or GABA mPSC amplitude or frequency (Fig. 3; Table 1).
To further confirm that cannabinoid agonists can reduce the frequency of AMPA mPSCs we used another CB1 receptor agonist, ACEA. Bath application of ACEA (5 μm) reduced AMPA mPSC frequency 20–40 min after bath application of the drug (Fig. 4A,E; Table 1; p = 0.0001). In this case, we did not observe the recovery of AMPA mPSC frequency (≥60 min in drug) found with WIN. ACEA had no effect on GABA mPSC frequency (Fig. 4E, left), but did significantly reduce the amplitude of both AMPA and GABA mPSCs (Fig. 4C,E; p = 0.01 AMPA mPSC, p = 0.01 GABA mPSC). We hypothesized that the effect of ACEA on amplitude was not through the CB1 receptor, but rather through some nonspecific interaction. To test this we recorded mEPSCs in the presence of ACEA and AM 251 to block any CB1 receptor contribution. Figure 4E (right) shows that normalized AMPAergic mPSC amplitude was reduced by ACEA to the same extent whether CB1 signaling was blocked or not, suggesting this effect was not mediated by the CB1 receptor. On the other hand, Figure 4E (left) shows that the reduction in AMPAergic mPSC frequency produced by ACEA was blocked when CB1 signaling was blocked by addition of AM 251. This finding suggests that the effect of ACEA on mEPSC frequency was through the CB1 receptor.
Figure 4.
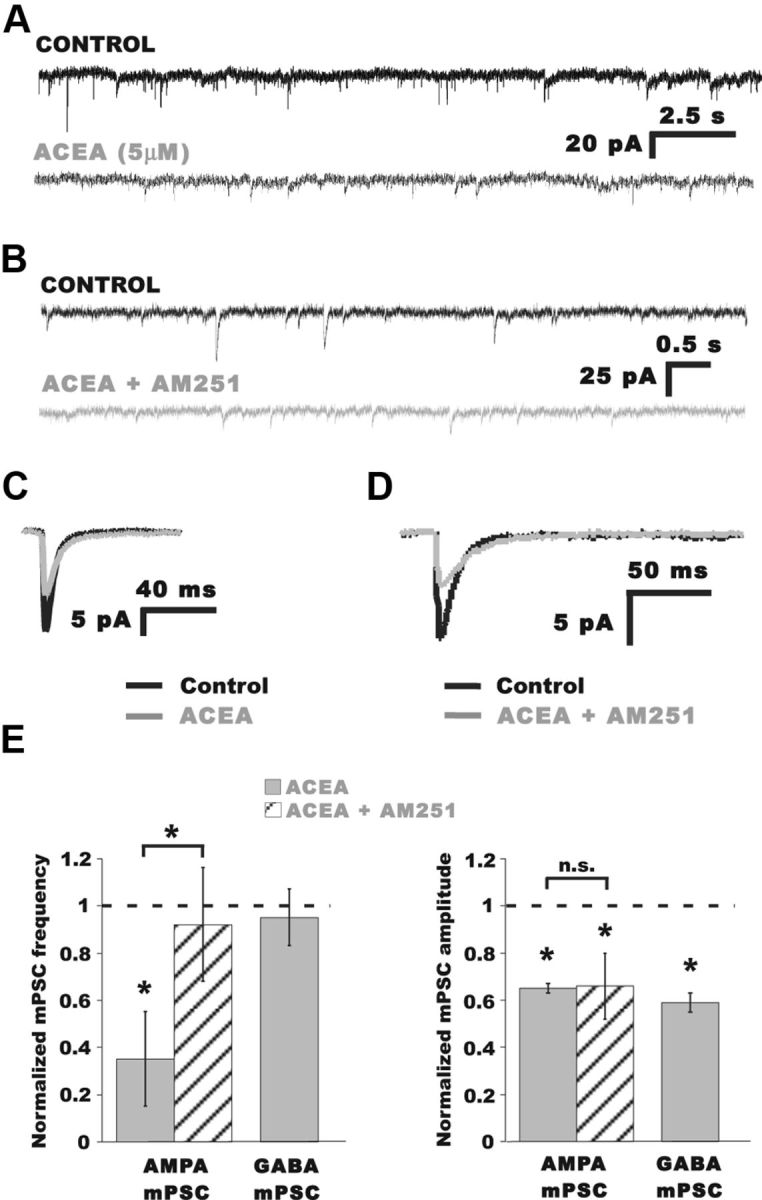
The CB1 agonist ACEA decreased AMPA mPSC frequency. A, Representative traces of mPSCs from a motoneuron before (black trace) and 20 min after bath application of the CB1 receptor agonist ACEA (5 μm; gray trace). B, Representative traces of mPSCs from a motoneuron in normal Tyrode's (black trace) and 20 min after simultaneous bath application of the CB1 receptor agonist ACEA and the CB1 antagonist AM 251 (5 and 5 μm, respectively; gray trace). C, Superimposed traces of averaged AMPA mPSCs before (black trace) and after bath addition of ACEA (gray trace). D, Superimposed traces of averaged AMPA mPSCs before (black trace) and after simultaneous bath addition of ACEA and AM 251 (gray trace). E, Summary of the effects of ACEA and ACEA + AM 251 on normalized AMPA mPSC frequency (left) and amplitude (right). The reduction of AMPA mPSC frequency following addition of ACEA (Table 1) was blocked when CB1 signaling was inhibited with AM 251 (1.10 ± 0.05 Hz control; 0.81 ± 0.22 Hz in ACEA + AM 251; n = 5). A slight reduction in mEPSC frequency was observed in AM 251 + ACEA compared with controls (p = 0.03, paired t test) as would be expected given the dramatic reduction in mEPSC amplitude, dropping some mEPSCs below our ability to detect them. Both AMPA and GABA mPSC amplitudes were significantly reduced after ACEA (Table 1). Unlike frequency, however, mEPSC amplitude was reduced by ACEA to the same extent whether CB1 signaling was blocked by AM 251 or not (12.1 ± 1.4 pA control; 8.0 ± 1.1 pA in ACEA + AM 251; n = 5). *p ≤ 0.05.
The CB1 agonists demonstrated that CB1 activation could reduce AMPA mPSC frequency. Application of the inverse agonist demonstrated that inhibition of CB1 signaling could increase mEPSC frequency; this increase could occur through the blockade of endocannabinoid activation of the CB1 receptor or by blocking a constitutively active CB1 receptor in the absence of endocannabinoids. To determine whether endocannabinoids are released and activate the CB1 receptor in the embryonic spinal cord, we introduced drugs that are thought to elevate endogenously produced endocannabinoids. Therefore, we bath applied 20 μm AM 404 (Pertwee et al., 2010). After at least 20 min in AM 404, AMPA mPSC frequency was significantly reduced (Fig. 5C,D; Table 1; p = 0.001), while amplitude was unaffected (Fig. 5B,E; p = 0.5). AM 404 reduced GABA mPSC amplitude (p = 0.03; Fig. 5B,E) but did not appear to have a significant effect on GABA mPSC frequency (Fig. 5C).
Figure 5.

Increasing endocannabinoid signaling decreased AMPA mPSC frequency. A, Representative traces of mPSCs from a motoneuron before (black trace) and 20 min after bath addition of the endocannabinoid reuptake blocker AM 404, 20 μm (gray trace). B, Superimposed traces of averaged AMPA mPSC (left) and GABA mPSC (right) before (black trace) and after bath addition of AM 404 (gray trace). C, Summary of the effect of AM 404 on mPSC frequency. *p = 0.001. D, Cumulative distribution plot showing the increase in the AMPA mPSC IEI (reduction in frequency) after AM 404 addition. E, Summary of the effect of AM 404 on mPSC amplitude. No effect on AMPA mPSC amplitude was found in AM 404 but GABA mPSC amplitude was significantly reduced. *p = 0.003.
Since it has been previously shown that AM 404 may also activate vanilloid receptors, we used another highly potent blocker of anandamide uptake that additionally inhibits the enzyme responsible for anandamide hydrolysis (fatty acid amide hydrolase), LY 2183240. We found that increasing the endocannabinoid concentration by bath applying 1 μm LY 2183240 significantly reduced AMPA mPSC frequency (p = 0.03), but had no effect on amplitude (Table 1). These experiments were performed in the presence of gabazine (5 μm) to isolate AMPA mPSCs, and the potential effects of LY 2183240 on GABA mPSCs were not evaluated.
Did endocannabinoids modulate glutamatergic transmission following SNA?
Following an episode of SNA, AMPA currents (evoked and mPSC frequency) were reduced, and then recovered in the IEI (Fedirchuk et al., 1999; Tabak et al. 2001; C. Gonzalez-Islas unpublished; Figs. 6A,B, 7A–C). Therefore, we reasoned that during episodes of SNA endocannabinoids could be liberated as a result of strong depolarizations and metabotropic signaling associated with SNA. During the IEI endocannabinoids would be progressively cleared and CB1-mediated suppression of glutamatergic release would be reduced. If probability of release from glutamatergic terminals was modified by endocannabinoids released during episodes, then we would expect that blocking CB1 receptors would significantly increase evoked glutamatergic currents and reduce episode-dependent modulation of these currents. Thus, we measured ventral root potentials while stimulating interneurons projecting through the VLF every 1 or 2 min. Ventral root potentials represent subthreshold electrotonically degraded population potentials from motoneurons (Fig. 6A; see Materials and Methods). The VLF contains predominately glutamatergic and GABAergic interneurons, many of which make direct synaptic connections onto motoneurons. We measured the strength of the glutamatergic connections by blocking GABAergic synaptic transmission (10–20 μm gabazine), while stimulating the VLF, and recording short latency potentials in motoneurons through ventral root recordings (Fig. 6A,B). The episode-dependent modulation of glutamatergic synaptic strength was clearly observed. When we added AM 251 to block CB1 receptors there was no change in the average amplitude of the potential (97.7%; p = 0.75, n = 5; Fig. 6B,D), or its episode-dependent modulation (amplitude after episode normalized to amplitude before episode—46.5 ± 9.3% before AM 251, 43.3 ± 13.5% after AM 251; p = 0.69, paired t test). We also looked at a glutamatergic pathway that does not modulate in the IEI (Fedirchuk et al., 1999) by stimulating sensory neurons that travel through the dorsal roots, while recording the response in the homonymous ventral root (also in the presence of gabazine). Again, there was no change in the evoked response (Fig. 6C,D; 102.8%, p = 0.56). In addition, we saw no changes when we increased CB1 signaling with AM 404 while assessing the average amplitude of the VLF-evoked potential (Fig. 6B,D; 87.6%, p = 0.16), its episode-dependent modulation (amplitude after episode normalized to amplitude before episode; 28.0 ± 4.9% before AM 251, 25.7 ± 4.7% after AM 251; p = 0.27, paired t test; Fig. 6B), or the average dorsal root-evoked response (Fig. 6C,D; 129.3%, p = 0.29). Together, the results suggest that while endocannabinoid signaling strongly regulated AMPAergic mPSC frequency, it did not play a significant role in the regulation of evoked glutamatergic responses, either at baseline or through episode-dependent modulation.
Although we did not observe an endocannabinoid-dependent effect on the modulation of evoked glutamatergic responses, it was still possible that there could be an effect on the modulation of AMPAergic mPSCs. We tested this possibility by recording mEPSCs using whole-cell electrodes; we anticipated that endocannabinoids could mediate the suppression of mEPSCs even though we were holding the cells at −70 mV due to the fact that metabotropic signaling was still intact, and because voltage-clamp will not be perfect when dendrites are involved. To test this idea, we divided the IEI into six consecutive 2 min bins and measured AMPA mPSC frequency in each bin. In Figure 7B we plotted AMPA mPSC frequency obtained at each interval for cords before and after bath application of 5 μm AM 251 to block CB1 activation. We then assessed the rate of recovery of AMPA mPSC frequency through the slope of the frequency interval plot. We found that overall AMPA mPSC frequency was increased; however, there was still a very clear SNA-dependent modulation of AMPA mPSC frequency (Fig. 7B; control: m = 0.03 Hz/min, r = 0.94; following AM 251: m = 0.08 Hz/min, r = 0.93, n = 6). When we normalized mEPSC frequency values to the last interval before an episode it was again clear that the modulation was observed when CB1 signaling was blocked, although there may have been a slight, but not significant inhibition of the rate of recovery in the presence of AM 251 (Fig. 7C; slopes were not significantly different, m = 0.09 Hz/min, r = 0.94, in control and m = 0.06 Hz/min; r = 0.93 in AM 251). Further, if we compared the suppression of mEPSC frequency in the first 2 min period (normalized to last period before an episode) before and after AM 251, the values were still not significantly different (0.50 ± 0.09 vs 0.65 ± 0.21 Hz, p = 0.52). The results suggested that the activation of CB1 by an SNA-dependent release of endocannabinoids could not explain the observed depression and recovery of AMPA mPSC frequency during the IEI, although we cannot rule out a small contribution from CB1 signaling.
Next we considered the possibility that the depolarization associated with SNA could produce endocannabinoid signaling, but that the depression of spontaneous release might be too short lived to effect mEPSCs in the IEI. Large depolarizations have been shown to induce the production and release of endocannabinoids in other systems, which then act to reduce presynaptic glutamate release through CB1 receptors (depolarization-induced suppression of excitation; DSE). Therefore, we tested if strong depolarizations could suppress mEPSC frequency right after the depolarization (Fig. 7D). We found that following a depolarizing step to 0 mV (60 s, in TTX), mEPSC frequency in motoneurons was significantly reduced in the first, but not the second minute after the step (Fig. 7E; 64.6% first minute, p = 0.037; 96.7% second minutes, p = 0.63, n = 6). Consistent with DSE being mediated by endocannabinoid signaling, we found that no such depression of mEPSC frequency occurred in the presence of AM 251 (Fig. 7E; 104.0% first minutes, p = 0.75; 87.7% second minutes, p = 0.30, n = 6). The results suggested that there were two forms of endocannabinoid signaling. The first was likely associated with SNA, but only transiently expressed. The second form of endocannabinoid signaling appeared to be independent of SNA, acting to tonically suppress spontaneous, but not evoked, vesicle release.
It is possible that the terminals giving rise to mEPSCs and the stimulated terminals that underlie evoked release could be separate populations. However, we are stimulating many different classes of glutamatergic interneurons and glutamatergic sensory neurons and we would expect some overlap with the spontaneous population. Rather, we considered the possibility that CB1 activation could influence spontaneous release independently of evoked release in the same terminal. CB1 activation typically suppresses evoked release by inhibiting voltage-gated calcium channels in the terminal. We tested the calcium dependence of VLF- and dorsal root-evoked responses in the ventral root by comparing ventral root responses in normal (3 mm) then low (1 mm) external calcium. As expected, evoked release was calcium-dependent; the VLF-evoked response was depressed in low calcium (17.4 ± 0.9, 3 mm/10.0 ± 0.5, 1 mm/15.8 ± 1.6 μV, back to 3 mm; n = 7, p < 0.0001). The dorsal root-evoked response was also depressed in low calcium (13.4 ± 0.7, 3 mm/8.2 ± 0.3, 1 mm/15.3 ± 1.6 μV, back to 3 mm; n = 7, p = 0.002). On the other hand, low calcium had no effect on mEPSC frequency (0.43 ± 0.3 Hz, 3 mm; n = 8/0.40 ± 0.4 Hz, 1 mm; n = 6, p = 0.59). Since spontaneous release is not altered by lowered calcium, endocannabinoid-mediated suppression of mEPSC frequency must occur through a calcium-independent pathway. These results suggested that evoked and spontaneous release could be regulated through distinct pathways.
Endocannabinoid signaling influenced the frequency of SNA in the isolated spinal cord
The finding that SNA-dependent modulation of AMPA mPSC frequency and evoked glutamatergic responses were unaffected by endocannabinoid signaling was surprising, but suggested endocannabinoids were important for the regulation of AMPAergic mPSCs, specifically. The function of endocannabinoid signaling in the embryonic spinal cord might then be to set the levels of miniature AMPAergic transmission tonically by reducing the probability of spontaneously released glutamatergic vesicles. We considered the possibility that the frequency of AMPAergic mPSCs might influence the frequency of SNA. We tested this idea by altering AMPAergic mPSC frequency by applying modulators of the CB1 receptor while monitoring the frequency of SNA in the isolated in vitro spinal preparation. Blocking CB1 receptors with AM 251 significantly increased SNA frequency (Fig. 8, reduced the IEI, 11.8 ± 1.2 min control, 9.4 ± 0.8 min AM 251, n = 7, p = 0.004). Moreover, increasing endocannabinoid tone with AM 404 produced an increase in the IEI (Fig. 8; 10.4 ± 0.9 min control, 13.7 ± 0.5 min AM 404, n = 5, p = 0.002).
In ovo blockade of CB1 receptors increased SNA and caused homeostatic increases in AMPA and GABA mPSC amplitude
We tested whether these increases in SNA observed in vitro could be extended to the embryo in vivo. We injected AM 251 onto the chorioallantoic membrane at E8 and measured embryonic movements until E10 (Fig. 9). Like the results in the in vitro cord, embryonic movements were increased after AM 251 was added to the egg; however, this did not occur until 12 h after the in ovo injection. This delay is likely to occur due to the immature development of the glutamatergic system at E8 (Milner and Landmesser, 1999). We have previously shown that blocking SNA in ovo from E8–E10 through lidocaine infusion leads to a homeostatic increase in GABA and AMPA mPSC amplitude. The observation that embryonic movements, and therefore SNA, were increased with AM 251 provided us with the opportunity to test if this increase in SNA in vivo homeostatically reduced AMPAergic and GABAergic mPSC amplitudes. At E8, we again injected AM 251 (5 μm) into the egg to increase SNA in ovo. AM 251 was allowed to act for 2 d and at E10, spinal cords from AM 251-treated embryos were isolated and mPSCs amplitude in spinal motoneurons were measured. After chronic AM 251 treatment, AMPA-mPSC amplitude was significantly reduced compared with untreated controls (Fig. 10; control, 13.8 ± 2.1 pA; AM 251, 11.9 ± 0.7 pA; p = 0.04, n = 6). GABA mPSC amplitude was similarly reduced after the levels of activity were increased with AM 251 (Fig. 10; control, 27.4 ± 2.8 pA; AM 251, 16.9 ± 1.2 pA; p = 0.03, n = 6). No significant effect was found in AMPA mPSC frequency after AM 251 treatment (Fig. 10; control, 0.17 ± 0.04 Hz; AM 251, 0.23 ± 0.05 Hz; p = 0.3, n = 6). In contrast, GABA mPSC frequency was significantly increased compared with controls (Fig. 10; control, 0.44 ± 0.08 Hz; AM 251, 0.79 ± 0.04 Hz; n = 6, p = 0.02). While both AMPA and GABA mPSC amplitude were reduced following increases in SNA in ovo, the amplitude distributions did not undergo synaptic scaling (see Fig. 10 legend; see Discussion).
Figure 9.

Chronic blockade of CB1 receptors increased embryonic limb movements in ovo. Plot of the average embryonic kicking time in ovo in 5 min observation periods during the 2 d treatment with vehicle (DMSO black) and 5 μm AM 251 dissolved in DMSO (gray). Limb movements started to increase in AM 251-treated embryos 12 h after injection and remained more active than controls during the rest of the 2 d long treatment.
Figure 10.

Chronic blockade of CB1 receptors induce compensatory reductions of AMPA and GABA mPSC amplitude in embryonic spinal motoneurons. A, Summary of the effect on mPSCs of 2 d blockade of CB1 receptors in ovo with AM 251. AMPA and GABA mPSC amplitude is decreased compared with controls. *p < 0.05. B, Bar graph showing that GABA mPSC frequency is significantly increased after chronic treatment with AM 251. *p = 0.02. AMPA mPSC frequency was unaffected by the treatment. C, D, Cumulative distribution of mPSC amplitudes for control (treated with DMSO for 2 d; black line) and for AM 251-treated embryos (gray line). AMPA and GABA mPSC amplitudes did not scale across their distributions following AM 251 treatment (Kolmogorov–Smirnov Z = 4.44; p < 0.001 for AMPA mPSC amplitude distribution; Kolmogorov–Smirnov Z = 5.18; p < 0.001 for GABA mPSC amplitude distribution).
Discussion
Embryonic cannabis exposure has been associated with hyperactivity and cognitive impairment in animal models and humans (Huizink and Mulder, 2006; Fried, 2002; Campolongo et al., 2009). This is consistent with the widespread nature of endocannabinoid signaling in several developing systems (Middleton and Protti, 2011; Penzo and Peña, 2009; Jiang et al., 2010; Bernard et al., 2005; Li et al., 2009). Here, we demonstrate that the endocannabinoid signaling system does not play a role in the modulation of glutamatergic synapses during the IEI. However, CB1 signaling does mediate a transient depolarization-induced inhibition of mEPSC frequency, along with a tonic suppression of glutamatergic spontaneous events. Further, we show that endocannabinoid signaling alters the frequency of SNA in vitro and in vivo. Blockade of CB1 in vivo chronically increases the frequency of SNA leading to a compensatory reduction in excitatory GABA and AMPA mPSC amplitude.
Endocannabinoid signaling mediates a transient glutamatergic plasticity, but this does not contribute to the IEI plasticity
Following an episode of SNA, evoked glutamatergic currents and AMPA mPSC frequency were depressed, but then recovered in the following IEI (Figs. 6, 7) (Fedirchuk et al., 1999; Tabak et al., 2001). We hypothesized that the spiking activity and neurotransmission associated with SNA would enhance endocannabinoid signaling and reduce glutamatergic probability of release until endocannabinoid signaling was weakened in the IEI as endocannabinoids were cleared from synapses. This hypothesis was not supported by our findings, since the SNA-dependent modulation of glutamatergic currents was unaffected by blocking CB1 receptors. It is unlikely that other cannabinoid receptors mediate this form of glutamatergic plasticity, as increasing endocannabinoid levels with AM 404 similarly had no effect on glutamatergic modulation. We did find that strong depolarizations, as would occur during episodes of SNA, did lead to an endocannabinoid-mediated depression of mEPSC frequency, but this effect was only observed within 60 s of the depolarization. Therefore, endocannabinoids may not contribute to the glutamatergic modulation observed in the IEI due to their transient nature, possibly resulting from a rapid endocannabinoid clearance. Thus, other mechanisms must exist that mediate the SNA-dependent modulation of glutamatergic transmission, such as nitric oxide (Kyriakatos and El Manira, 2007).
Endocannabinoid signaling tonically inhibits AMPAergic mPSCs while having no observable effect on evoked glutamatergic currents
Our results suggest that endocannabinoid signaling tonically inhibits glutamatergic mPSCs. Blocking endocannabinoid signaling dramatically increased mEPSC frequency, while activating the CB1 receptor suppressed mEPSCs. These results favor the possibility that endocannabinoids are constitutively released at some base level in an SNA-independent manner, and thus tonically inhibit spontaneous glutamatergic vesicle release, thereby lowering AMPAergic tone. While several other studies have demonstrated a tonic endocannabinoid inhibition of presynaptic release of GABAergic transmission (Losonczy et al., 2004; Oliet et al., 2007; Zhu and Lovinger, 2010; Inada et al., 2010; Neu et al., 2007; Hentges et al., 2005; Farkas et al., 2010; Kim and Alger, 2010), this is the first demonstration of tonic inhibition of glutamatergic release (Alger, 2012).
In addition to an influence on AMPAergic mPSC frequency, different endocannabinoid signaling modulators also had effects on mPSC amplitude. For instance, AM 404 produced a significant reduction of GABA mPSC amplitude, and ACEA reduced AMPAergic and GABAergic mPSC amplitude. These effects appear to be due to the nonspecific effects of the drugs (Fig. 4E). AM 404 has been reported to activate the vanilloid receptor TRPV1 (Zygmunt et al., 2000) and ACEA can inhibit voltage-gated calcium channels (Pertwee et al., 2010). Further, the transient effect of WIN, compared with ACEA, may be due to the rapid internalization/desensitization of CB1 receptors, which has been reported following WIN application (Hsieh et al., 1999; Jin et al., 1999; Coutts et al., 2001; Kouznetsova et al., 2002). We cannot speak with certainty about the influence of nonspecific effects, as the results are mixed. However, we are confident in the interpretation that endocannabinoid signaling reduces the base level of AMPAergic mPSCs, because all of the results with different modulators were consistent in this regard.
While endocannabinoid signaling had a clear effect on glutamatergic spontaneous release, it had no detectable effect on evoked responses of two different glutamatergic inputs to motoneurons. When spontaneous and evoked currents are assessed in the same study, they are typically influenced by endocannabinoid signaling in similar ways (Wilson and Nicoll, 2002; Wamsteeker et al., 2010; Chevaleyre et al., 2007; Hentges et al., 2005). To our knowledge, this is the first direct demonstration that endocannabinoids inhibit presynaptic spontaneous release, while having no effect on evoked release. One recent study in the embryonic tectum demonstrated that mEPSC frequency, but not evoked currents, was suppressed in response to postsynaptic depolarization, but this was not mediated by endocannabinoid signaling (Penzo and Peña, 2011). Several studies suggest that spontaneous and evoked vesicle release can be regulated independently through separate pathways (Chung et al., 2010; Geppert et al., 1994; Groffen et al., 2010), although it is not clear whether this is accomplished through distinct pools for spontaneous and evoked release (Denker and Rizzoli, 2010). Our study suggests that endocannabinoid signaling regulates spontaneous release in a calcium-independent manner. It appears that spontaneous and evoked release have distinct regulatory pathways in the embryonic spinal cord, as evoked glutamate release is not influenced by endocannabinoid signaling but is calcium dependent.
The functional role of endocannabinoid signaling in the embryonic spinal cord
Tonic suppression of spontaneous glutamatergic release is quite different from the more established role of endocannabinoid signaling in presynaptic short and long-term plasticity (Alger, 2009; Heifets and Castillo, 2009; El Manira and Kyriakatos, 2010). Thus, endocannabinoid signaling could have a unique function in the developing spinal network. In the current study we have shown that endocannabinoid signaling regulates the frequency of spinal SNA in vitro and in vivo, likely by inhibiting spontaneous glutamatergic release. We have previously shown that episodes are triggered by motoneuron discharge, which recruits spinal interneurons (Wenner and O'Donovan, 2001). Motoneuron discharge typically arises from minis, so an increase in mEPSCs (AM 251) could reduce the IEI by initiating more spikes and triggering the episode earlier. In the developing hippocampus, endocannabinoid signaling reduced the frequency of giant depolarizing potentials (a hippocampal form of SNA) by CB1 inhibition of excitatory GABAergic transmission presynaptically (Bernard et al., 2005). In the motor circuitry of the adult lamprey spinal cord, endocannabinoid signaling increased the frequency of locomotor bouts of activity by presynaptically reducing inhibitory transmission onto motor and interneurons (Kettunen et al., 2005; Kyriakatos and El Manira, 2007; El Manira and Kyriakatos, 2010). It will be important to understand how spinal circuit CB1 signaling transitions from the embryo (suppresses SNA via tonic mEPSC inhibition) to adult (enhances rhythms through evoked IPSC inhibition). SNA in the embryonic cord has been shown to be important in the development of limbs, motoneuron axon pathfinding, and synaptic maturation (O'Donovan et al., 1998; Hanson and Landmesser, 2004; Gonzalez-Islas and Wenner, 2006; Blankenship and Feller, 2010). Therefore, these SNA-dependent developmental features could be regulated by endocannabinoid signaling via its effect on SNA frequency.
In addition to a role in setting SNA frequency, endocannabinoids may have functional significance by selectively controlling glutamatergic spontaneous release. Glutamatergic mPSCs appear to have important functional roles themselves (Sutton and Schuman, 2009). Spontaneous mPSCs can influence firing rates in electrically compact neurons (Carter and Regehr, 2002), and stabilize excitatory synaptic function (Sutton et al., 2006; Jakawich et al., 2010; Frank et al., 2006). Finally, mPSCs can regulate the activity of postsynaptic signaling pathways differently than that controlled by action potential-triggered neurotransmission (Sutton et al., 2007).
Increasing SNA through chronic blockade of endocannabinoid signaling reduces AMPA and GABA mPSC amplitude
Synaptic scaling is a form of homeostatic plasticity that increases or decreases the strength of all synapses onto a neuron by a scaling factor to stabilize firing rates (Turrigiano, 2012). As discussed above, reductions of SNA in ovo led to homeostatic increases in depolarizing GABAergic and AMPAergic mPSC amplitude in embryonic chick motoneurons (Gonzalez-Islas and Wenner, 2006). Studies in culture have suggested that this synaptic scaling process is bidirectional, responding to both decreases and increases in activity (Turrigiano et al., 1998). Because we were able to increase activity in ovo through application of a CB1 receptor antagonist, we were able to show that chronic increases in SNA in vivo reduce AMPAergic and GABAergic mPSC amplitude in a compensatory direction. This demonstrates a synaptic plasticity in the living embryo that appears to be homeostatic, although we do not actually demonstrate homeostasis in this study. Regardless, we show a compensatory plasticity that is bidirectional [changes in synaptic strength to both increases, this study, and decreases (Gonzalez-Islas and Wenner, 2006) in network activity]. On the other hand, the mPSC amplitude distributions did not scale (mPSC amplitude distributions from control and endocannabinoid-treated motoneurons did not appear to be multiplicatively related). The lack of scaling in this process suggests that the compensatory synaptic responses to increases versus decreases in SNA likely involve distinct mechanisms. As discussed above, the finding that endocannabinoid signaling regulates SNA levels, and therefore synaptic maturation, highlights the potential impact of perturbations to endocannabinoid signaling during embryonic development.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grant R01 NS065992 and the University Research Committee Award, Emory University (P. W.). We thank Dr. Shawn Hochman and the reviewers of our manuscript as their suggestions improved the paper significantly.
References
- Alger BE. Endocannabinoids at the synapse a decade after the dies mirabilis (29 March 2001): what we still do not know. J Physiol. 2012;590:2203–2212. doi: 10.1113/jphysiol.2011.220855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alger BE. Endocannabinoid signaling in neural plasticity. Curr Top Behav Neurosci. 2009;1:141–172. doi: 10.1007/978-3-540-88955-7_6. [DOI] [PubMed] [Google Scholar]
- Bernard C, Milh M, Morozov YM, Ben-Ari Y, Freund TF, Gozlan H. Altering cannabinoid signaling during development disrupts neuronal activity. Proc Natl Acad Sci U S A. 2005;102:9388–9393. doi: 10.1073/pnas.0409641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenship AG, Feller MB. Mechanisms underlying spontaneous patterned activity in developing neural circuits. Nat Rev Neurosci. 2010;11:18–29. doi: 10.1038/nrn2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brink E, Jankowska E, McCrea D, Skoog B. Use of sucrose gap for recording postsynaptic population potentials evoked by single interneurones in spinal motoneurones. Brain Res. 1981;223:165–169. doi: 10.1016/0006-8993(81)90817-9. [DOI] [PubMed] [Google Scholar]
- Brooks CM, Eccles JC, Malcolm JL. Synaptic potentials of inhibited motoneurones. J Neurophysiol. 1948;11:417–430. doi: 10.1152/jn.1948.11.5.417. [DOI] [PubMed] [Google Scholar]
- Butt SJ, Kiehn O. Functional identification of interneurons responsible for left-right coordination of hindlimbs in mammals. Neuron. 2003;38:953–963. doi: 10.1016/s0896-6273(03)00353-2. [DOI] [PubMed] [Google Scholar]
- Campolongo P, Trezza V, Palmery M, Trabace L, Cuomo V. Developmental exposure to cannabinoids causes subtle and enduring neurofunctional alterations. Int Rev Neurobiol. 2009;85:117–133. doi: 10.1016/S0074-7742(09)85009-5. [DOI] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Quantal events shape cerebellar interneuron firing. Nat Neurosci. 2002;5:1309–1318. doi: 10.1038/nn970. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Heifets BD, Kaeser PS, Südhof TC, Purpura DP, Castillo PE. Endocannabinoid-mediated long-term plasticity requires cAMP/PKA signaling and RIM1alpha. Neuron. 2007;54:801–812. doi: 10.1016/j.neuron.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chub N, O'Donovan MJ. Post-episode depression of GABAergic transmission in spinal neurons of the chick embryo. J Neurophysiol. 2001;85:2166–2176. doi: 10.1152/jn.2001.85.5.2166. [DOI] [PubMed] [Google Scholar]
- Chung C, Barylko B, Leitz J, Liu X, Kavalali ET. Acute dynamin inhibition dissects synaptic vesicle recycling pathways that drive spontaneous and evoked neurotransmission. J Neurosci. 2010;30:1363–1376. doi: 10.1523/JNEUROSCI.3427-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutts AA, Anavi-Goffer S, Ross RA, MacEwan DJ, Mackie K, Pertwee RG, Irving AJ. Agonist-induced internalization and trafficking of cannabinoid CB1 receptors in hippocampal neurons. J Neurosci. 2001;21:2425–2433. doi: 10.1523/JNEUROSCI.21-07-02425.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denker A, Rizzoli SO. Synaptic vesicle pools: an update. Front Synaptic Neurosci. 2010;2:135. doi: 10.3389/fnsyn.2010.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC. Synaptic potentials of motoneurones. J Neurophysiol. 1946;9:87–120. doi: 10.1152/jn.1946.9.2.87. [DOI] [PubMed] [Google Scholar]
- El Manira A, Kyriakatos A. The role of endocannabinoid signaling in motor control. Physiology. 2010;25:230–238. doi: 10.1152/physiol.00007.2010. [DOI] [PubMed] [Google Scholar]
- Farkas I, Kalló I, Deli L, Vida B, Hrabovszky E, Fekete C, Moenter SM, Watanabe M, Liposits Z. Retrograde endocannabinoid signaling reduces GABAergic synaptic transmission to gonadotropin-releasing hormone neurons. Endocrinology. 2010;151:5818–5829. doi: 10.1210/en.2010-0638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedirchuk B, Wenner P, Whelan PJ, Ho S, Tabak J, O'Donovan MJ. Spontaneous network activity transiently depresses synaptic transmission in the embryonic chick spinal cord. J Neurosci. 1999;19:2102–2112. doi: 10.1523/JNEUROSCI.19-06-02102.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Kennedy MJ, Goold CP, Marek KW, Davis GW. Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron. 2006;52:663–677. doi: 10.1016/j.neuron.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried PA. Conceptual issues in behavioral teratology and their application in determining long-term sequelae of prenatal marihuana exposure. J Child Psychol Psychiatry. 2002;43:81–102. doi: 10.1111/1469-7610.00005. [DOI] [PubMed] [Google Scholar]
- Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Südhof TC. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell. 1994;79:717–727. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Islas C, Wenner P. Developmental plasticity of inhibitory circuitry. New York: Springer; 2010. Role of spontaneous activity in the maturation of GABAergic synapses in spinal circuits. [Google Scholar]
- Gonzalez-Islas C, Wenner P. Spontaneous network activity in the embryonic spinal cord regulates AMPAergic and GABAergic synaptic strength. Neuron. 2006;49:563–575. doi: 10.1016/j.neuron.2006.01.017. [DOI] [PubMed] [Google Scholar]
- Groffen AJ, Martens S, Díez Arazola R, Cornelisse LN, Lozovaya N, de Jong AP, Goriounova NA, Habets RL, Takai Y, Borst JG, Brose N, McMahon HT, Verhage M. Doc2b is a high-affinity Ca2+ sensor for spontaneous neurotransmitter release. Science. 2010;327:1614–1618. doi: 10.1126/science.1183765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger V, Hamilton HL. A series of normal stages in the developing chick embryo. Dev Dyn. 1951;195:231–272. doi: 10.1002/aja.1001950404. [DOI] [PubMed] [Google Scholar]
- Hanson MG, Landmesser LT. Normal patterns of spontaneous activity are required for correct motor axon guidance and the expression of specific guidance molecules. Neuron. 2004;43:687–701. doi: 10.1016/j.neuron.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Heifets BD, Castillo PE. Endocannabinoid signaling and long-term synaptic plasticity. Annu Rev Physiol. 2009;71:283–306. doi: 10.1146/annurev.physiol.010908.163149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentges ST, Low MJ, Williams JT. Differential regulation of synaptic inputs by constitutively released endocannabinoids and exogenous cannabinoids. J Neurosci. 2005;25:9746–9751. doi: 10.1523/JNEUROSCI.2769-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh C, Brown S, Derleth C, Mackie K. Internalization and recycling of the CB1 cannabinoid receptor. J Neurochem. 1999;73:493–501. doi: 10.1046/j.1471-4159.1999.0730493.x. [DOI] [PubMed] [Google Scholar]
- Huizink AC, Mulder EJ. Maternal smoking, drinking or cannabis use during pregnancy and neurobehavioral and cognitive functioning in human offspring. Neurosci Biobehav Rev. 2006;30:24–41. doi: 10.1016/j.neubiorev.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Inada H, Maejima T, Nakahata Y, Yamaguchi J, Nabekura J, Ishibashi H. Endocannabinoids contribute to metabotropic glutamate receptor-mediated inhibition of GABA release onto hippocampal CA3 pyramidal neurons in an isolated neuron/bouton preparation. Neuroscience. 2010;165:1377–1389. doi: 10.1016/j.neuroscience.2009.11.054. [DOI] [PubMed] [Google Scholar]
- Jakawich SK, Nasser HB, Strong MJ, McCartney AJ, Perez AS, Rakesh N, Carruthers CJ, Sutton MA. Local presynaptic activity gates homeostatic changes in presynaptic function driven by dendritic BDNF synthesis. Neuron. 2010;68:1143–1158. doi: 10.1016/j.neuron.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B, Huang S, de Pasquale R, Millman D, Song L, Lee HK, Tsumoto T, Kirkwood A. The maturation of GABAergic transmission in visual cortex requires endocannabinoid-mediated LTD of inhibitory inputs during a critical period. Neuron. 2010;66:248–259. doi: 10.1016/j.neuron.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Brown S, Roche JP, Hsieh C, Celver JP, Kovoor A, Chavkin C, Mackie K. Distinct domains of the CB1 cannabinoid receptor mediate desensitization and internalization. J Neurosci. 1999;19:3773–3780. doi: 10.1523/JNEUROSCI.19-10-03773.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettunen P, Kyriakatos A, Hallén K, El Manira A. Neuromodulation via conditional release of endocannabinoids in the spinal locomotor network. Neuron. 2005;45:95–104. doi: 10.1016/j.neuron.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Kim J, Alger BE. Reduction in endocannabinoid tone is a homeostatic mechanism for specific inhibitory synapses. Nat Neurosci. 2010;13:592–600. doi: 10.1038/nn.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouznetsova M, Kelley B, Shen M, Thayer SA. Desensitization of cannabinoid-mediated presynaptic inhibition of neurotransmission between rat hippocampal neurons in culture. Mol Pharmacol. 2002;61:477–485. doi: 10.1124/mol.61.3.477. [DOI] [PubMed] [Google Scholar]
- Kyriakatos A, El Manira A. Long-term plasticity of the spinal locomotor circuitry mediated by endocannabinoid and nitric oxide signaling. J Neurosci. 2007;27:12664–12674. doi: 10.1523/JNEUROSCI.3174-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Bender KJ, Drew PJ, Jadhav SP, Sylwestrak E, Feldman DE. Endocannabinoid signaling is required for development and critical period plasticity of the whisker map in somatosensory cortex. Neuron. 2009;64:537–549. doi: 10.1016/j.neuron.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losonczy A, Biró AA, Nusser Z. Persistently active cannabinoid receptors mute a subpopulation of hippocampal interneurons. Proc Natl Acad Sci U S A. 2004;101:1362–1367. doi: 10.1073/pnas.0304752101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher HR, Ruenzel P, Fetz E, Henneman E. Postsynaptic population potentials recorded from ventral roots perfused with isotonic sucrose: connections of groups Ia and II spindle afferent fibers with large populations of motoneurons. J Neurophysiol. 1979;42:1146–1164. doi: 10.1152/jn.1979.42.4.1146. [DOI] [PubMed] [Google Scholar]
- Middleton TP, Protti DA. Cannabinoids modulate spontaneous synaptic activity in retinal ganglion cells. Vis Neurosci. 2011;28:393–402. doi: 10.1017/S0952523811000198. [DOI] [PubMed] [Google Scholar]
- Milner LD, Landmesser LT. Cholinergic and GABAergic inputs drive patterned spontaneous motoneuron activity before target contact. J Neurosci. 1999;19:3007–3022. doi: 10.1523/JNEUROSCI.19-08-03007.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu A, Földy C, Soltesz I. Postsynaptic origin of CB1-dependent tonic inhibition of GABA release at cholecystokinin-positive basket cell to pyramidal cell synapses in the CA1 region of the rat hippocampus. J Physiol. 2007;578:233–247. doi: 10.1113/jphysiol.2006.115691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donovan MJ. Developmental approaches to the analysis of vertebrate central pattern generators. J Neurosci Methods. 1987;21:275–286. doi: 10.1016/0165-0270(87)90122-1. [DOI] [PubMed] [Google Scholar]
- O'Donovan MJ. Motor activity in the isolated spinal cord of the chick embryo: synaptic drive and firing pattern of single motoneurons. J Neurosci. 1989;9:943–958. doi: 10.1523/JNEUROSCI.09-03-00943.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donovan MJ, Landmesser L. The development of hindlimb motor activity studied in the isolated spinal cord of the chick embryo. J Neurosci. 1987;7:3256–3264. doi: 10.1523/JNEUROSCI.07-10-03256.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donovan MJ, Chub N, Wenner P. Mechanisms of spontaneous activity in developing spinal networks. J Neurobiol. 1998;37:131–145. doi: 10.1002/(sici)1097-4695(199810)37:1<131::aid-neu10>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Tanimura A, Hashimotodani Y, Kano M. Endocannabinoids and retrograde modulation of synaptic transmission. Neuroscientist. 2012;18:119–132. doi: 10.1177/1073858410397377. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Baimoukhametova DV, Piet R, Bains JS. Retrograde regulation of GABA transmission by the tonic release of oxytocin and endocannabinoids governs postsynaptic firing. J Neurosci. 2007;27:1325–1333. doi: 10.1523/JNEUROSCI.2676-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzo MA, Peña JL. Endocannabinoid-mediated long-term depression in the avian midbrain expressed presynaptically and postsynaptically. J Neurosci. 2009;29:4131–4139. doi: 10.1523/JNEUROSCI.5466-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzo MA, Peña JL. Depolarization-induced suppression of spontaneous release in the avian midbrain. J Neurosci. 2011;31:3602–3609. doi: 10.1523/JNEUROSCI.6388-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, Greasley PJ, Hansen HS, Kunos G, Mackie K, Mechoulam R, Ross RA. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB and CB. Pharmacol Rev. 2010;62:588–631. doi: 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts MH, Wallis DI. Dorsal and ventral root potentials recorded in vivo by the sucrose-gap method [proceedings] J Physiol. 1978;277:42P–43P. [PubMed] [Google Scholar]
- Sutton MA, Schuman EM. Partitioning the synaptic landscape: distinct microdomains for spontaneous and spike-triggered neurotransmission. Sci Signal. 2009;2:pe19. doi: 10.1126/scisignal.265pe19. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Taylor AM, Ito HT, Pham A, Schuman EM. Postsynaptic decoding of neural activity: eEF2 as a biochemical sensor coupling miniature synaptic transmission to local protein synthesis. Neuron. 2007;55:648–661. doi: 10.1016/j.neuron.2007.07.030. [DOI] [PubMed] [Google Scholar]
- Tabak J, Rinzel J, O'Donovan MJ. The role of activity-dependent network depression in the expression and self-regulation of spontaneous activity in the developing spinal cord. J Neurosci. 2001;21:8966–8978. doi: 10.1523/JNEUROSCI.21-22-08966.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol. 2012;4:a005736. doi: 10.1101/cshperspect.a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Wamsteeker JI, Kuzmiski JB, Bains JS. Repeated stress impairs endocannabinoid signaling in the paraventricular nucleus of the hypothalamus. J Neurosci. 2010;30:11188–11196. doi: 10.1523/JNEUROSCI.1046-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenner P, O'Donovan MJ. Identification of an interneuronal population that mediates recurrent inhibition of motoneurons in the developing chick spinal cord. J Neurosci. 1999;19:7557–7567. doi: 10.1523/JNEUROSCI.19-17-07557.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenner P, O'Donovan MJ. Mechanisms that initiate spontaneous network activity in the developing chick spinal cord. J Neurophysiol. 2001;86:1481–1498. doi: 10.1152/jn.2001.86.3.1481. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endocannabinoid signaling in the brain. Science. 2002;296:678–682. doi: 10.1126/science.1063545. [DOI] [PubMed] [Google Scholar]
- Xu H, Whelan PJ, Wenner P. Development of an inhibitory interneuronal circuit in the embryonic spinal cord. J Neurophysiol. 2005;93:2922–2933. doi: 10.1152/jn.01091.2004. [DOI] [PubMed] [Google Scholar]
- Zhu PJ, Lovinger DM. Developmental alteration of endocannabinoid retrograde signaling in the hippocampus. J Neurophysiol. 2010;103:1123–1129. doi: 10.1152/jn.00327.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zygmunt PM, Chuang H, Movahed P, Julius D, Högestätt ED. The anandamide transport inhibitor AM 404 activates vanilloid receptors. Eur J Pharmacol. 2000;396:39–42. doi: 10.1016/s0014-2999(00)00207-7. [DOI] [PubMed] [Google Scholar]