

Abstract
The title compound, C17H16N2O3, has an E conformation about the azobenzene (–N=N–) linkage. The benzene rings are twisted slightly with respect to each other [6.79 (9)°], while the dihedral angle between the plane through the carboxy group and the attached benzene ring is 3.2 (2)°. In the crystal, molecules are oriented with the carboxy groups head-to-head, forming O—H⋯O hydrogen-bonded inversion dimers. These dimers are connected by C—H⋯O hydrogen-bonds into layers lying parallel to the (013) plane.
Related literature
For the physical properties of compounds containing an azobenzene (–N=N–) linkage, see: Chigrinov (2005 ▶); Hegde (2007 ▶). For related structures, see: Yu & Liu (2009 ▶); Lai et al. (2002 ▶); Centore & Tuzi (2003 ▶). For standard bond lengths, see Allen et al. (1987 ▶).
Experimental
Crystal data
C17H16N2O3
M r = 296.33
Triclinic,
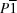
a = 7.0937 (7) Å
b = 9.8687 (10) Å
c = 11.2490 (11) Å
α = 87.334 (8)°
β = 73.475 (8)°
γ = 75.174 (8)°
V = 729.54 (13) Å3
Z = 2
Cu Kα radiation
μ = 0.77 mm−1
T = 100 K
0.26 × 0.16 × 0.04 mm
Data collection
Oxford Diffraction Gemini diffractometer
Absorption correction: multi-scan (CrysAlis RED; Oxford Diffraction, 2006 ▶) T min = 0.85, T max = 0.97
9940 measured reflections
2783 independent reflections
2298 reflections with I > 2.0σ(I)
R int = 0.024
Refinement
R[F 2 > 2σ(F 2)] = 0.054
wR(F 2) = 0.146
S = 1.00
2773 reflections
199 parameters
H-atom parameters constrained
Δρmax = 0.51 e Å−3
Δρmin = −0.33 e Å−3
Data collection: CrysAlis CCD (Oxford Diffraction, 2006 ▶); cell refinement: CrysAlis CCD; data reduction: CrysAlis RED (Oxford Diffraction, 2006 ▶); program(s) used to solve structure: Superflip (Palatinus & Chapuis, 2007 ▶); program(s) used to refine structure: CRYSTALS (Betteridge et al., 2003 ▶); molecular graphics: Mercury (Macrae et al., 2006 ▶); software used to prepare material for publication: CRYSTALS.
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536812038718/su2476sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812038718/su2476Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812038718/su2476Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O3—H2⋯O1i | 0.87 | 1.76 | 2.612 (3) | 166 (1) |
| C21—H211⋯O1ii | 0.95 | 2.50 | 3.275 (3) | 139 (1) |
Symmetry codes: (i) 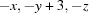 ; (ii)
; (ii) 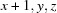 .
.
Acknowledgments
This research was supported by a UMP research grant (No. RDU100338).
supplementary crystallographic information
Comment
An in a molecule introduces the possibility of photochromism and photoisomerization (Chigrinov, 2005). Photonics, in which the light can be controlled by light as stimulus has been exploited (Hegde, 2007). Upon absorption of UV light (~ 365 nm) the energetically more stable E conformation, transforms into the Z conformation. The reverse transformation of the Z isomer into the E isomer can be brought about by irradiation with visible light (in the range of 400–500 nm). The latter can also occur in the "dark" by a process known as "thermal back relaxation" in a period ranging from minutes to tens of hours depending on the system. In this case molecules again transform from the metastable cis-conformation to the energetically stable trans-conformation. In conclusion, the present investigation on rod-shaped azo dyes is very useful for a variety of photonic applications. Excellent quality, cost effective, easy to prepare, are properties which make these devices very attractive for future generations. Detailed investigations on the physics of these azo dyes is under intense consideration.
The bond lengths (Allen et al.,1987) and bond angles in the titled compound (Fig. 1) are normal. The carbonyl group (C2/O1/O3) is almost coplanar with the attached benzene ring (C4-C7/C21/C22) with a dihedral angle of only 3.2 (2)°. The length of N8═N9 bond is 1.263 (2) Å and the torsion angle for the azo unit (C7—N8—N9—C10) is -177.75 (16)° rather than ca. ±180° as observed elsewhere: For example: 4,4-Azinodibenzoic acid (Yu and Liu, 2009) and (E)-ethyl 4-((4-(decanoyloxy)phenyl)diazenyl)benzoate (Lai et al.,2002). However, it is comparable with the value of 175.10° observed for (E)-4-((4-((2-hydroxyethyl)(methyl)amino)phenyl)diazenyl)benzoic acid (Centore & Tuzi, 2003). The benzene rings (C4-C7/C21/C22 and C10-C3/C19/C20) lie at a mutual dihedral angles of 6.79 (9)°, compared to 16.69° in (E)-4-((4-((2-hydroxyethyl)(methyl)amino)phenyl)diazenyl)benzoic acid (Centore & Tuzi, 2003). The C15—C16—C17—C18 torsion angle in the butyl group is 126.1 (3)°.
In the crystal, the carboxyl groups are oriented head-to-head forming hydrogen bonded inversion dimers (Table 1 and Fig. 2). These dimers are further linked by C—H···O hydrogen bonds to a generate a layer parallel to the (013) plane (Table 1 and Fig. 2).
Experimental
The title compound was prepared from ethyl 4-aminobenzoate. Firstly the diazonuim salt was prepared using one equivalent of sodium nitrite to one equivalent of ethyl 4-aminobenzoate in methanol - water mixture at 275 K, in the presence of 3 equivalents of aqueous hydrochloric acid, which was coupled with phenol to yield ethyl 4-[(4-hydroxyphenyl)diazenyl]benzoate. This compound was then alkylated with 4-bromo-1-butene in the presence of potassium carbonate as base to give the ester, ethyl 4-{[4-(but-3-en-1-yloxy)phenyl]diazenyl}benzoate. This compound was then hydrolyzed under basic conditions to yield the title benzoic acid. Brown plate-like crystals of the title compound were obtained by slow evaporation of a solution in methanol.
Refinement
The H atoms were all located in a difference Fourier map, but those attached to carbon atoms were repositioned geometrically. They were all initially refined with soft restraints on the bond lengths and angles to regularize their geometry: C—H = 0.93 (2)–0.98 (2) Å and O—H = 0.82 (2) Å with Uiso(H) = k × Ueq(O,C) where k = 1.5 for the OH H atom and = 1.2 for the C-bound H atoms. In the final cycles or refinement they were allowed to ride on their parent atom.
Figures
Fig. 1.

The molecular structure of the title molecule with the atom numbering and displacement ellipsoids drawn at the 50% probability level.
Fig. 2.

A view along the c axis of the crystal packing of the title compound, with the hydrogen bonds shown as dashed lines [the C-bound H atoms have been omitted for clarity].
Crystal data
| C17H16N2O3 | Z = 2 |
| Mr = 296.33 | F(000) = 312 |
| Triclinic, P1 | Dx = 1.349 Mg m−3 |
| Hall symbol: -P 1 | Cu Kα radiation, λ = 1.54180 Å |
| a = 7.0937 (7) Å | Cell parameters from 3768 reflections |
| b = 9.8687 (10) Å | θ = 4–71° |
| c = 11.2490 (11) Å | µ = 0.77 mm−1 |
| α = 87.334 (8)° | T = 100 K |
| β = 73.475 (8)° | Plate, brown |
| γ = 75.174 (8)° | 0.26 × 0.16 × 0.04 mm |
| V = 729.54 (13) Å3 |
Data collection
| Oxford Diffraction Gemini diffractometer | 2783 independent reflections |
| Radiation source: sealed x-ray tube | 2298 reflections with I > 2.0σ(I) |
| Graphite monochromator | Rint = 0.024 |
| ω scans | θmax = 71.5°, θmin = 4.1° |
| Absorption correction: multi-scan (CrysAlis RED; Oxford Diffraction, 2006) | h = −8→8 |
| Tmin = 0.85, Tmax = 0.97 | k = −11→11 |
| 9940 measured reflections | l = −13→13 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Hydrogen site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.054 | H-atom parameters constrained |
| wR(F2) = 0.146 | Method = Modified Sheldrick w = 1/[σ2(F2) + ( 0.06P)2 + 0.76P], where P = (max(Fo2,0) + 2Fc2)/3 |
| S = 1.00 | (Δ/σ)max = 0.0003776 |
| 2773 reflections | Δρmax = 0.51 e Å−3 |
| 199 parameters | Δρmin = −0.33 e Å−3 |
| 0 restraints |
Special details
| Refinement. This compound, 9940 numbers of reflections were collected and measured during the refinement. Symmetry related reflections were measured more than once and after merging the symmetry equivalent reflections there were only 2783 reflection left. 10 more reflections were filtered, as sigma cutoff was set as 3 and (sinθ/x)set to>0.01 (to eliminate reflection measured near the vicinity of beam stop) therefore numbers of reflection reduced to 2773. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.0122 (2) | 1.34005 (15) | 0.07329 (14) | 0.0276 | |
| C2 | 0.1938 (3) | 1.3516 (2) | 0.04699 (18) | 0.0221 | |
| O3 | 0.2517 (2) | 1.45464 (14) | −0.00910 (14) | 0.0281 | |
| C4 | 0.3484 (3) | 1.2387 (2) | 0.08338 (17) | 0.0215 | |
| C5 | 0.2990 (3) | 1.1180 (2) | 0.14000 (18) | 0.0231 | |
| C6 | 0.4449 (3) | 1.0125 (2) | 0.17190 (18) | 0.0233 | |
| C7 | 0.6447 (3) | 1.0260 (2) | 0.14525 (18) | 0.0220 | |
| N8 | 0.8099 (2) | 0.92608 (18) | 0.17206 (15) | 0.0238 | |
| N9 | 0.7751 (2) | 0.81065 (17) | 0.21222 (15) | 0.0235 | |
| C10 | 0.9450 (3) | 0.7184 (2) | 0.24132 (18) | 0.0243 | |
| C11 | 1.1313 (3) | 0.7536 (2) | 0.2247 (2) | 0.0293 | |
| C12 | 1.2896 (3) | 0.6624 (2) | 0.2565 (2) | 0.0321 | |
| C13 | 1.2665 (3) | 0.5349 (2) | 0.30679 (19) | 0.0302 | |
| O14 | 1.4341 (2) | 0.45519 (16) | 0.33669 (15) | 0.0364 | |
| C15 | 1.4290 (4) | 0.3232 (2) | 0.3930 (2) | 0.0349 | |
| C16 | 1.6307 (4) | 0.2701 (3) | 0.4247 (2) | 0.0399 | |
| C17 | 1.6430 (4) | 0.1366 (3) | 0.4911 (2) | 0.0388 | |
| C18 | 1.7890 (4) | 0.0213 (3) | 0.4581 (3) | 0.0455 | |
| C19 | 1.0850 (3) | 0.4962 (2) | 0.32338 (19) | 0.0310 | |
| C20 | 0.9240 (3) | 0.5897 (2) | 0.28909 (19) | 0.0287 | |
| C21 | 0.6931 (3) | 1.1467 (2) | 0.09065 (19) | 0.0252 | |
| C22 | 0.5462 (3) | 1.2529 (2) | 0.06039 (18) | 0.0238 | |
| H51 | 0.1631 | 1.1098 | 0.1566 | 0.0298* | |
| H61 | 0.4112 | 0.9298 | 0.2120 | 0.0302* | |
| H111 | 1.1483 | 0.8433 | 0.1888 | 0.0379* | |
| H121 | 1.4168 | 0.6860 | 0.2448 | 0.0406* | |
| H151 | 1.3110 | 0.3369 | 0.4683 | 0.0453* | |
| H152 | 1.4146 | 0.2584 | 0.3328 | 0.0447* | |
| H161 | 1.6418 | 0.3468 | 0.4751 | 0.0512* | |
| H162 | 1.7478 | 0.2531 | 0.3482 | 0.0518* | |
| H171 | 1.5325 | 0.1356 | 0.5657 | 0.0520* | |
| H182 | 1.9017 | 0.0237 | 0.3829 | 0.0605* | |
| H181 | 1.7859 | −0.0632 | 0.5068 | 0.0604* | |
| H191 | 1.0658 | 0.4083 | 0.3577 | 0.0394* | |
| H201 | 0.8003 | 0.5652 | 0.2975 | 0.0369* | |
| H211 | 0.8278 | 1.1552 | 0.0740 | 0.0331* | |
| H221 | 0.5794 | 1.3355 | 0.0213 | 0.0322* | |
| H2 | 0.1704 | 1.5151 | −0.0423 | 0.0500* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0212 (7) | 0.0239 (8) | 0.0399 (8) | −0.0047 (6) | −0.0132 (6) | 0.0029 (6) |
| C2 | 0.0228 (10) | 0.0191 (10) | 0.0252 (10) | −0.0034 (8) | −0.0092 (8) | −0.0018 (8) |
| O3 | 0.0270 (8) | 0.0210 (8) | 0.0385 (8) | −0.0048 (6) | −0.0154 (6) | 0.0082 (6) |
| C4 | 0.0213 (10) | 0.0195 (10) | 0.0243 (9) | −0.0033 (8) | −0.0087 (8) | −0.0015 (8) |
| C5 | 0.0183 (9) | 0.0230 (11) | 0.0295 (10) | −0.0048 (8) | −0.0094 (8) | 0.0016 (8) |
| C6 | 0.0236 (10) | 0.0203 (10) | 0.0279 (10) | −0.0064 (8) | −0.0098 (8) | 0.0030 (8) |
| C7 | 0.0213 (10) | 0.0198 (10) | 0.0248 (10) | −0.0009 (8) | −0.0100 (8) | −0.0005 (8) |
| N8 | 0.0217 (8) | 0.0225 (9) | 0.0272 (9) | −0.0028 (7) | −0.0094 (7) | 0.0016 (7) |
| N9 | 0.0207 (8) | 0.0218 (9) | 0.0258 (8) | −0.0003 (7) | −0.0073 (7) | −0.0008 (7) |
| C10 | 0.0235 (10) | 0.0231 (11) | 0.0228 (10) | 0.0018 (8) | −0.0079 (8) | −0.0014 (8) |
| C11 | 0.0256 (11) | 0.0273 (11) | 0.0338 (11) | −0.0009 (9) | −0.0116 (9) | −0.0001 (9) |
| C12 | 0.0260 (11) | 0.0317 (12) | 0.0391 (12) | −0.0018 (9) | −0.0145 (9) | −0.0008 (10) |
| C13 | 0.0281 (11) | 0.0311 (12) | 0.0276 (10) | 0.0047 (9) | −0.0119 (9) | −0.0046 (9) |
| O14 | 0.0356 (9) | 0.0287 (8) | 0.0461 (9) | 0.0007 (7) | −0.0216 (7) | 0.0028 (7) |
| C15 | 0.0410 (13) | 0.0270 (12) | 0.0348 (12) | −0.0002 (10) | −0.0149 (10) | −0.0006 (9) |
| C16 | 0.0456 (14) | 0.0359 (14) | 0.0420 (13) | −0.0055 (11) | −0.0228 (11) | 0.0019 (10) |
| C17 | 0.0383 (13) | 0.0374 (13) | 0.0408 (13) | −0.0019 (10) | −0.0187 (10) | 0.0032 (10) |
| C18 | 0.0463 (15) | 0.0372 (14) | 0.0563 (16) | −0.0033 (11) | −0.0264 (13) | 0.0022 (12) |
| C19 | 0.0390 (12) | 0.0218 (11) | 0.0273 (10) | 0.0000 (9) | −0.0086 (9) | 0.0013 (8) |
| C20 | 0.0272 (11) | 0.0270 (11) | 0.0284 (11) | −0.0022 (9) | −0.0064 (8) | −0.0009 (8) |
| C21 | 0.0196 (9) | 0.0265 (11) | 0.0310 (10) | −0.0066 (8) | −0.0087 (8) | 0.0002 (8) |
| C22 | 0.0237 (10) | 0.0196 (10) | 0.0294 (10) | −0.0055 (8) | −0.0101 (8) | 0.0031 (8) |
Geometric parameters (Å, º)
| O1—C2 | 1.271 (2) | C13—O14 | 1.367 (3) |
| C2—O3 | 1.268 (2) | C13—C19 | 1.395 (3) |
| C2—C4 | 1.483 (3) | O14—C15 | 1.426 (3) |
| O3—H2 | 0.868 | C15—C16 | 1.529 (3) |
| C4—C5 | 1.401 (3) | C15—H151 | 0.993 |
| C4—C22 | 1.395 (3) | C15—H152 | 0.996 |
| C5—C6 | 1.382 (3) | C16—C17 | 1.479 (3) |
| C5—H51 | 0.952 | C16—H161 | 0.998 |
| C6—C7 | 1.403 (3) | C16—H162 | 0.997 |
| C6—H61 | 0.965 | C17—C18 | 1.312 (4) |
| C7—N8 | 1.421 (3) | C17—H171 | 0.974 |
| C7—C21 | 1.391 (3) | C18—H182 | 0.989 |
| N8—N9 | 1.263 (2) | C18—H181 | 0.978 |
| N9—C10 | 1.423 (3) | C19—C20 | 1.408 (3) |
| C10—C11 | 1.411 (3) | C19—H191 | 0.959 |
| C10—C20 | 1.382 (3) | C20—H201 | 0.947 |
| C11—C12 | 1.371 (3) | C21—C22 | 1.382 (3) |
| C11—H111 | 0.977 | C21—H211 | 0.945 |
| C12—C13 | 1.384 (3) | C22—H221 | 0.959 |
| C12—H121 | 0.960 | ||
| O1—C2—O3 | 123.76 (18) | O14—C15—C16 | 106.04 (19) |
| O1—C2—C4 | 118.68 (17) | O14—C15—H151 | 109.0 |
| O3—C2—C4 | 117.56 (17) | C16—C15—H151 | 111.8 |
| C2—O3—H2 | 119.6 | O14—C15—H152 | 108.8 |
| C2—C4—C5 | 121.02 (17) | C16—C15—H152 | 111.3 |
| C2—C4—C22 | 119.37 (17) | H151—C15—H152 | 109.7 |
| C5—C4—C22 | 119.61 (18) | C15—C16—C17 | 112.4 (2) |
| C4—C5—C6 | 120.55 (18) | C15—C16—H161 | 106.1 |
| C4—C5—H51 | 119.1 | C17—C16—H161 | 112.0 |
| C6—C5—H51 | 120.4 | C15—C16—H162 | 111.1 |
| C5—C6—C7 | 119.42 (18) | C17—C16—H162 | 107.0 |
| C5—C6—H61 | 120.9 | H161—C16—H162 | 108.3 |
| C7—C6—H61 | 119.7 | C16—C17—C18 | 125.6 (3) |
| C6—C7—N8 | 125.56 (18) | C16—C17—H171 | 116.3 |
| C6—C7—C21 | 119.98 (18) | C18—C17—H171 | 118.0 |
| N8—C7—C21 | 114.44 (17) | C17—C18—H182 | 117.5 |
| C7—N8—N9 | 115.89 (16) | C17—C18—H181 | 120.8 |
| N8—N9—C10 | 112.56 (16) | H182—C18—H181 | 121.7 |
| N9—C10—C11 | 122.76 (18) | C13—C19—C20 | 118.9 (2) |
| N9—C10—C20 | 118.03 (18) | C13—C19—H191 | 122.3 |
| C11—C10—C20 | 119.21 (19) | C20—C19—H191 | 118.8 |
| C10—C11—C12 | 120.7 (2) | C19—C20—C10 | 120.4 (2) |
| C10—C11—H111 | 119.5 | C19—C20—H201 | 120.6 |
| C12—C11—H111 | 119.8 | C10—C20—H201 | 118.9 |
| C11—C12—C13 | 119.9 (2) | C7—C21—C22 | 120.46 (18) |
| C11—C12—H121 | 120.8 | C7—C21—H211 | 119.3 |
| C13—C12—H121 | 119.3 | C22—C21—H211 | 120.3 |
| C12—C13—O14 | 114.02 (19) | C4—C22—C21 | 119.94 (18) |
| C12—C13—C19 | 120.84 (19) | C4—C22—H221 | 119.3 |
| O14—C13—C19 | 125.1 (2) | C21—C22—H221 | 120.7 |
| C13—O14—C15 | 119.93 (18) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O3—H2···O1i | 0.87 | 1.76 | 2.612 (3) | 166 (1) |
| C21—H211···O1ii | 0.95 | 2.50 | 3.275 (3) | 139 (1) |
Symmetry codes: (i) −x, −y+3, −z; (ii) x+1, y, z.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: SU2476).
References
- Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987). J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
- Betteridge, P. W., Carruthers, J. R., Cooper, R. I., Prout, K. & Watkin, D. J. (2003). J. Appl. Cryst. 36, 1487.
- Centore, R. & Tuzi, A. (2003). Cryst. Eng. 6, 87–97.
- Chigrinov, V. G. (2005). ICOCN 2005 Digest, p. 285.
- Hegde, G. (2007). PhD thesis, University Malaysia Pahang, Malaysia.
- Lai, L.-L., Su, F.-Y., Lin, Y.-J., Ho, C.-H., Wang, E., Hung, C.-H., Liu, Y.-H. & Wang, Y. (2002). Helv. Chim. Acta, 85, 1517–1522.
- Macrae, C. F., Edgington, P. R., McCabe, P., Pidcock, E., Shields, G. P., Taylor, R., Towler, M. & van de Streek, J. (2006). J. Appl. Cryst. 39, 453–457.
- Oxford Diffraction (2006). CrysAlis CCD and CrysAlis RED Oxford Diffraction Ltd, Abingdon, England.
- Palatinus, L. & Chapuis, G. (2007). J. Appl. Cryst. 40, 786–790.
- Yu, Q.-D. & Liu, Y.-Y. (2009). Acta Cryst. E65, o2326. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536812038718/su2476sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812038718/su2476Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812038718/su2476Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report