

Abstract
This review summarizes our current understanding of exocrine pancreas development, including the formation of acinar, ductal and centroacinar cells. We discuss the transcription factors associated with various stages of exocrine differentiation, from multipotent progenitor cells to fully differentiated acinar and ductal cells. Within the branching epithelial tree of the embryonic pancreas, this involves the progressive restriction of multipotent pancreatic progenitor cells to either a central “trunk” domain giving rise to the islet and ductal lineages, or a peripheral “tip” domain giving rise to acinar cells. This review also discusses the soluble morphogens and other signaling pathways that influence these events. Finally, we examine centroacinar cells as an enigmatic pancreatic cell type whose lineage remains uncertain, and whose possible progenitor capacities continue to be explored.
Introduction
Metazoans have developed highly specialized exocrine cell types dedicated to the synthesis and secretion of proteolytic and other digestive enzymes. Examples across multiple phyla include zymogen gland cells in hydra endoderm [1], F-cells in the crustacean hepatopancreas [2], hatching gland cells in teleost and amphibian embryos [3, 4], and the acinar cells of the vertebrate pancreas. In most vertebrates, pancreatic acinar cells develop and function in close spatial proximity to their endocrine counterparts, and are presumed to be derived from a common multi-lineage progenitor cell (MPC).
Driven by the need to develop treatments for diabetes, much of the research in pancreatic developmental biology has historically focused on the endocrine compartment. Recently, studies focusing on mechanisms of exocrine pancreas development have become increasingly common. These studies reflect the important role of acinar and ductal cells in pancreatic disease, as well as an increasing awareness that adult exocrine cell types might serve as effective sources for beta cell neogenesis[5, 6]. These studies have also demonstrated that endocrine and exocrine ontogenies are highly intertwined, and further suggested an unanticipated plasticity between lineages. In addition, emerging evidence suggests that the islet and ductal lineages share a common immediate progenitor, and are more closely related than the ductal and acinar lineages. Given this interplay, it may be somewhat disingenuous to consider exocrine pancreas development as an isolated topic. Nevertheless, in this review we summarize current knowledge regarding development of the exocrine pancreas, including the specification, differentiation and function of acinar, centroacinar and ductal cell types. In its focus on exocrine development, this review neglects many other areas of pancreatic developmental biology, including foregut patterning, early morphogenesis, endocrine differentiation and developmental plasticity. Outstanding summaries of these topics can be found in a number of recent reviews, including several published in the current issue [7-13]. While this effort clearly emphasizes principles generated from studies of pancreas development in the mouse, where appropriate we also attempt to incorporate relevant observations from other vertebrate species.
Cell types of the exocrine pancreas
Pancreatic Acinar Cells
Acinar cells in the vertebrate pancreas are frequently considered to be the paradigmatic polarized secretory cell; they were used in the Nobel Prize-winning initial demonstration of the vectorial sequence of protein trafficking from endoplasmic reticulum (RER), to Golgi, to condensing vacuole, to secretory granule [14]. Pancreatic acinar cells are pyramidal in shape, and quite large, reaching up to 30 microns in apical-to-basal height. The acinar cell’s extreme dedication to the synthesis and secretion of digestive zymogens is demonstrated by a remarkably dense accumulation of rough endoplasmic reticulum, as well as an apical cytoplasm laden with secretory granules. This high degree of specialization is also evident on a molecular level, where a remarkable fraction of total acinar cell mRNA is devoted to transcripts encoding digestive zymogens [15].
In addition to multiple well known proteolytic enzymes, pancreatic acinar cells synthesize and secrete glycoside hydrolases such as Amylase, as well as ribonucleases, lipases and phospholipases [16, 17]). Among these enzymes, Amylase, Trypsin(ogen), Carboxypeptidase A (CPA), and Elastase are most frequently employed as acinar cell-specific markers. Other commonly utilized acinar cell markers include the plant lectin Peanut Agglutinin (PNA) [18] and the transcription factors Ptf1a, Mist1 and Rbpjl [19, 20]. Acinar cell-specific gene expression is frequently driven by the heterotrimeric PTF1 transcriptional complex, which binds to tandem E- and TC-box elements found in enhancer/promoter elements of many zymogen genes [21]. While the expression levels of different digestive zymogens are frequently assumed to co-vary, there is evidence of differential regulation of different zymogen classes, as well as non-synchronous activation of zymogen gene expression during acinar cell differentiation [15, 22, 23].
Centroacinar Cells
The acinus consists of an organized cluster of acinar cells which secrete digestive enzymes into a central lumen, from which the enzymes flow into ducts. Perhaps the most enigmatic of all pancreatic cell types, the centroacinar cell lies at the junction of the secretory acinus and its associated terminal ductal epithelium. These cells are variably depicted as an extension of the most terminal ductal epithelium as it invaginates into the secretory acinus [24], or alternatively as providing a fenestrated “cap” to the apical surface of acinar cells [25]. At this point, it remains uncertain whether centroacinar and terminal duct cells represent two different cell types or are functionally equivalent, and the possibility exists that multiple cell types may be located in a centroacinar position. In contrast to the much larger pancreatic acinar cells, the main body of centroacinar cells is typically less than 10 microns in diameter, with minimal cytoplasm and a high nuclear-to-cytoplasmic ratio. Work in both mammalian systems and zebrafish has suggested that these cells also extend long cytoplasmic processes, providing apparent contact with other centroacinar cells as well as adjacent acinar and islet cell types [24, 26]. While this unique feature is quite distinctive, the functional significance of these extensions remains unknown.
In addition to their unique location and morphology, many studies have also called attention to centroacinar cells as a candidate multipotent progenitor cell in adult pancreas [27-31]. Centroacinar cells have been shown to rapidly proliferate following either partial pancreatectomy [29, 32], streptozotocin induced destruction of the insulin-producing β-cells [28, 33], and acute or chronic administration of caerulein [30]. As discussed more extensively below, our group and others have also identified centroacinar and terminal duct cells as the exclusive domain of active Notch-signaling in adult mouse and zebrafish pancreas; this is evidenced by the expression of Hes1, a Notch target gene, or by the activation of a Notch-responsive fluorescent reporter in these cells [27]. In addition to ongoing Notch-pathway activation, at least some centroacinar cells also express Sox9, another marker of progenitor cells in the developing pancreas [28]. We have also demonstrated that a centroacinar cell population is characterized by high-level ALDH1 enzymatic activity. Furthermore, these cells display a unique in vitro progenitor capacity, including a markedly heightened ability to form pancreatospheres in suspension culture, as well as a unique ability to contribute to embryonic exocrine and endocrine lineages. Nevertheless, the ability of ALDH1-expressing centroacinar cells to act as in vivo progenitors has never been documented, and while the degree of overlap between the ALDH1, Sox9 and Hes1 lineages remains to be established, in vivo analyses of the adult Sox9 and Hes1 lineages have failed to detect multilineage progenitor activity[34, 35].
Developmentally, the origin of centroacinar cells has not been clearly established, in part due to a paucity of centroacinar cell-specific markers, as well as an incomplete understanding of possible cellular heterogeneity among cells residing in the centroacinar position. In zebrafish, adult centroacinar cells arise from progenitor cells within the larval pancreatic duct [36]. In the mouse, rare cells expressing Sox9 along with markers of both nascent ducts (e.g. Nkx6.1) and nascent acini (e.g. Ptf1a) have been identified at E14.5 [37]. As discussed below, these cells may represent an early manifestation of the centroacinar lineage.
Ductal Epithelial Cells
The adult pancreatic ductal epithelial tree serves two critical physiologic functions: first, to secrete bicarbonate-rich fluid to dilute and pH-optimize the protein concentrate secreted by acinar cells; and second, to convey this mix to the intestinal lumen. Ducts are typically classified by size and position within the ductal epithelial tree, with the most terminal/intercalated ducts draining into intralobular ducts, followed by interlobular ducts, and finally the main pancreatic duct, which drains into the intestine [38]. Classical conceptions of ductal morphogenesis have typically invoked a direct extension of the gut lumen into an ever more highly branched epithelial tree. However, recent highly detailed examinations of duct morphogenesis [39-41] suggest a more complex mechanism. This process involves initial epithelial stratification and the formation of multiple small lumens that subsequently remodel through changes in cell shape and position to ultimately fuse and form a ramifying, single-lumen ductal system.
Classical markers used to label and identify ductal epithelium include the functional markers Carbonic Anhydrase II, Mucin 1 (also known to label acinar cells), Cystic Fibrosis Transmembrane Receptor (CFTR), and various Cytokeratins (many of which display species-specific expression). Murine pancreatic ducts are also selectively marked by expression of CD133 and Osteopontin [37, 42, 43], as well as binding by the Dolichos biflorus (DBA) and Wisteria fluoribunda (WFA) plant lectins [18, 44]. Although many studies continue to consider the ductal epithelial tree as an essentially uniform structure along its peripheral-to-central axis, this likely represents a drastic oversimplification, and the detailed characterization of inter-segment heterogeneity represents an important challenge for the field. Considerable evidence already exists supporting such segment-specific identities. Morphologically, the most terminal ductal epithelial cells exhibit a somewhat flattened or squamous shape and minimal cytoplasm, while more central stereotypical ductal epithelial cells are cuboidal and rich in mitochondria. At the level of protein expression, there also exists extensive evidence of selective marker expression along different segments of the ductal epithelial tree, including primarily peripheral expression of CFTR, Muc1, and Aquaporins 1 and 5 [45, 46]. In addition, different “duct-specific” transcription factors, such as Hnf1β, Hnf6 and Sox9, are expressed heterogeneously throughout the ductal epithelial tree (Figure 1); the significance of this heterogeneity has not yet been explored. Evidence also suggests that the normal morphogenesis of different segments of the ductal epithelial tree may be dependent upon different transcriptional programs. Mice lacking the transcription factor Hnf6 have normal terminal/intercalated ducts but exhibit cystic dilation of inter- and intra-lobular ducts [47]. Many additional aspects of pancreatic ductal biology have been nicely summarized in a recent detailed review [38].
Figure 1. Transcription factor expression in adult ductal epithelium.
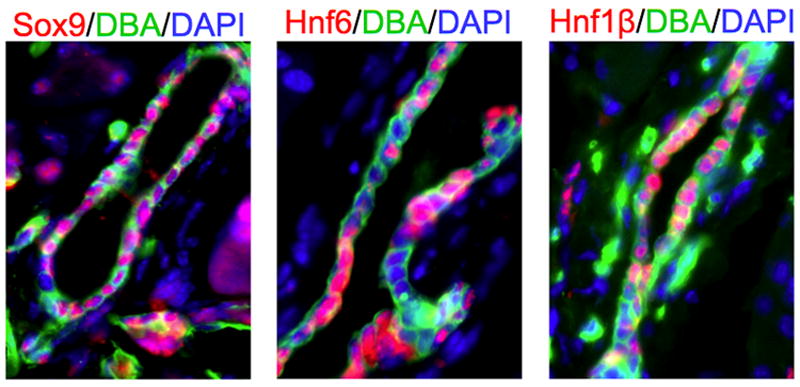
Immunofluorescent labeling for Sox9, Hnf6 and Hnf1β in adult mouse pancreas. Note heterogeneous expression, with neighboring cells often displaying different patterns of transcription factor expression.
In addition to the ductal epithelial segments discussed above, an important paper recently called attention to a previously noted but neglected ductal epithelial compartment referred to as the pancreatic duct gland; a compartment that had been previously noted, but which has been neglected in more recent work [48-51]. These glands are comprised of blind outpouches of the main pancreatic duct, and cells within these glands show a unique columnar morphology and express genes not found in normal cuboidal duct cells, including Shh, Pdx1, Hes1 and gastric-type mucins. In response to chronic epithelial injury, these glands undergo selective expansion and may contribute to formation of mucinous metaplasia and pancreatic intraepithelial neoplasia [48].
Regulation of exocrine differentiation by soluble morphogens
In mammals, the pancreas forms from separate dorsal and ventral pancreatic buds. The buds arise, proliferate and ramify within the adjacent dorsal and ventral pancreatic mesenchyme. Historically, initial insights into the regulation of exocrine pancreatic development focused on the influence of mesenchyme-derived soluble morphogens, including their influences on endocrine vs. exocrine lineage selection (reviewed in Serup, this issue). The first evidence that mesenchymal tissue was required for the normal development of the pancreatic epithelium was provided by studies involving in vitro culture of microdissected E11.5 dorsal pancreatic buds [52]. Associated dorsal mesenchyme was demonstrated to be required for normal growth of the epithelial bud, as well as the development of exocrine acini, but not the expression of endocrine genes [52, 53]. These studies defined endocrine differentiation as the effective “default path” for isolated pancreatic epithelium [54]. Furthermore, these early experiments demonstrated that while mesenchymal signals were required for normal bud growth and eventual exocrine differentiation, subsequent to E10.5 the mesenchymal influence was permissive rather than instructive, with non-pancreatic mesenchyme from other segments of the foregut equally able to support pancreatic epithelial growth.
It is now apparent that these results reflect the fact that mesenchymal tissues associated with budding foregut derivatives frequently express a common set of soluble morphogens. For example, FGF10, produced by the dorsal pancreatic mesenchyme [55], is also expressed in mesenchymal tissues associated with the nascent lung buds [55, 56]. Mesenchyme-derived FGF family members indeed play a critical role in exocrine pancreatic development [57-59] (reviewed in Serup, this issue). Specifically, FGFR-2 IIIb and its ligands FGF-1, FGF-7, and FGF-10 are expressed throughout pancreatic development and promote the growth, morphogenesis, and differentiation of exocrine cells [57]. Subsequent studies have demonstrated that a significant component of the FGF10 influence involves the activation of epithelial Notch-signaling, thereby maintaining a pool of undifferentiated progenitor cells capable of supporting ongoing epithelial growth [58, 60, 61]. In addition to allowing progenitor cells to avoid early default endocrine differentiation, and thereby promoting later exocrine fates, the delay in progenitor differentiation induced by FGF10 effectively increases the window during which progenitor cells activate Ngn3 and contribute to the endocrine lineage. In so doing, FGF10 also effectively increases the ultimate number of endocrine cells [55, 62].
Other soluble morphogens capable of influencing exocrine differentiation include EGF, which promotes proliferative growth at the expense of both exocrine and endocrine differentiation [63], and members of the TGF-β/BMP superfamily (reviewed in Serup, this issue). One such mesenchyme-derived factor, the TGF-β antagonist Follistatin, was found to mimic both inductive and repressive effects of the mesenchyme, promoting development of exocrine tissue while limiting endocrine differentiation [64]. Conversely, the addition of exogenous TGF-β to isolated pancreatic epithelium was found to promote endocrine at the expense of exocrine differentiation [65], and transgenic expression of a dominant negative TGF-β type II receptor was found to promote expansion of exocrine cells [66]. Together, these data suggest that pancreatic mesenchyme limits default endocrine differentiation through the combined effects of FGF signaling activation and TGF-β inactivation within the developing pancreatic epithelium.
While most studies examining the effect of soluble morphogens on pancreatic exocrine development invoke a mesenchyme-to-epithelium signaling paradigm, additional recent studies have identified autonomous mesenchymal signaling pathways that exert potent influences on the development of associated pancreatic epithelium. For example, analysis of phospho-Smads 1, 5 and 8 have suggested selective activation of BMP signaling in E11.5 mouse pancreatic mesenchyme, as well as in chick pancreatic mesenchyme at a similar developmental stage [67]. In the chick, this was associated with mesenchymal expression of BMP4 and BMP7, suggesting a mesenchyme-autonomous BMP signaling network. In both mouse and chick, inhibition of mesenchymal signaling following electroporation of the BMP antagonist Noggin resulted in pancreatic epithelial hypoplasia, reduced branching, excessive endocrine differentiation and impaired exocrine differentiation. Electroporation of a dominant negative Alk3 receptor into E11.5 mouse pancreatic mesenchyme effectively phenocopied the effect of Noggin [67]. These studies suggest that TGF-β type II receptor signaling in the epithelium and Alk3 receptor signaling in the mesenchyme exert opposing effects on exocrine lineage commitment and/or differentiation (reviewed in Serup, this issue).
While many soluble morphogens appear to modulate competing progenitor commitment to the endocrine and exocrine lineages, recent studies suggest that multipotent pancreatic progenitor cells (MPCs) are allocated to spatially distinct trunk and tip domains, with trunk progenitors giving rise to primarily endocrine and ductal cells, and tip progenitors giving rise to primarily acinar cells (this concept is discussed in detail in the following section) (discussed extensively in Serup; Rieck et al., this issue). It is therefore not surprising that specific soluble morphogens can also influence the relative allocation of progenitor cells to ductal and acinar fates. Indeed, this influence appears to be mediated by retinoid signaling, which has been shown to promote ductal differentiation through up-regulation of mesenchymal laminin-1 [68-70]. Specifically, 9-cis retinoic acid (9cRA) inhibits acinar differentiation in the developing pancreas, in favor of ducts, and 9cRA does not induce ductal differentiation in the absence of mesenchyme or following inhibition of laminin signaling [69]. Another mesenchyme-derived soluble factor selectively promoting pancreatic ductal proliferation and differentiation is epimorphin (syntaxin 2), which is also known to induce epithelial branching in a variety of tissues [71, 72].
Recently, through the use of a mesenchymal-specific Nkx3.2 (Bapx1):Cre driver, the mesenchymal contribution to pancreas development was probed in vivo [73]. Using this Cre driver to accomplish selective ablation of the pancreatic mesenchyme using either Cre-activated Diphtheria toxin or Diphtheria toxin receptor alleles, this study demonstrated that mesenchymal cells are required to support normal pancreatic growth and branching at both early and late developmental stages. This effect was conveyed by promoting the proliferation of both differentiated and undifferentiated cell types, with minimal direct effects on endocrine and exocrine differentiation. Furthermore, mesenchyme-specific deletion of β-catenin largely phenocopied the effect of Diphtheria toxin-mediated mesenchymal ablation, implicating β-catenin as an essential mediator of mesenchymal expansion and survival [73].
While many of the influences of the pancreatic mesenchyme on pancreatic exocrine differentiation are presumed to emanate from mesenchymal fibroblasts, endothelial cells are also known to be potent modulators of both early and late pancreatic development [44, 74-77] (reviewed in Villasenor and Cleaver, this issue). With respect to exocrine differentiation, recent studies have demonstrated that vascular endothelial cells normally associate with ductal and islet progenitors in the trunk domain of the pancreatic epithelial bud, and specifically inhibit allocation of MPCs to the tip domain, thereby inhibiting acinar cell differentiation [74, 76]
Progenitor commitment to endocrine and exocrine fates: transcription factor topology in trunks and tips
Ultimately, the multiple soluble morphogens directing MPCs to adopt endocrine and exocrine cell fates convey their effects through altered expression and/or activity of lineage-specifying transcription factors. As in the case of progressive endocrine differentiation (as reviewed in Rieck, Bankaitis and Wright, this issue), the differentiation of exocrine pancreatic cell types requires specific temporal and spatial cascades of transcription factor activation. In the case of acinar cell differentiation, the basic helix-loop-helix transcription factor Ptf1a is required not only for the commitment of progenitor cells to an acinar cell fate, but also for maintenance of acinar cell differentiation in adult pancreas. In this regard, the role played by Ptf1a differs from the role played by the bHLH transcription factor Ngn3, which is required for initiation but not maintenance of endocrine differentiation. As discussed below, this difference is reflected in the contrasting manner in which these two bHLH proteins interact with the Notch-signaling pathway.
Over the past 15 years, it has become apparent that frequently reported reciprocal changes in endocrine vs. exocrine cell fates reflect the fact that all pancreatic epithelial cells types share a common origin from MPCs. These progenitors are defined by a unique gene expression signature involving at least partially overlapping expression of Pdx1, Ptf1a, Sox9, Hes1, Hnf1β, Nkx6.1 and Nkx6.2 [35, 37, 78-82]. While the common origin of both endocrine and exocrine cell types from pools of progenitor cells expressing these transcription factors has been indisputably established by Cre/lox-based lineage tracing strategies [35, 37, 81, 83, 84], it should be stressed that unlike imaging- and transplantation-based lineage-tracing techniques often used in invertebrates, Cre/lox-based lineage tracing in mice and other vertebrates establishes the lineage of collective and potentially heterogeneous cell populations rather than individual progenitor cells. However, direct evidence of a specific MPC cell type contributing to both endocrine and exocrine lineages has been confirmed by clonal analysis of developing pancreatic epithelium. Single-cell labeling of E11.5 murine dorsal pancreatic buds using limiting dilutions of replication-incompetent retrovirus leads to subsequent clusters of cells with both endocrine and exocrine components [85]. ERT2 allows for temporal control of Cre-activity and more recent studies that used limiting dilutions of tamoxifen to activate cell type-specific CreER lineage labels in a highly mosaic manner have generated similar results [35, 79]. For the Hes1 lineage, low density and presumably clonal activation of a Hes1:CreERT2 lineage marker on E9.5 leads to expanded clusters of labeled islet, acinar and ductal cells at E17.5, indicating apparent clonal derivation [35]. Similar observations have been made following clonal activation of a Carboxypeptidase A lineage label at E11.5 [79].
While each of the above transcription factors appears to play a role in the specification or maintenance of the pancreatic progenitor pool, the commitment of MPCs to either the endocrine or exocrine lineages involves the progressive restriction of their initially overlapping patterns of expression. Spatially, this involves the progressive organization and patterning of the developing pancreas into distinct central and peripheral fields. In the setting of ongoing branching morphogenesis, these become resolved into central “trunks” and peripheral “tips”; with trunk domains becoming progressively restricted to islet and ductal fates, and tip domains becoming progressively restricted to the acinar lineage [78, 79, 81]. Thus the progressive restriction of MPC fates does not respect a simple endocrine vs. exocrine paradigm, but instead involves selection between competing islet/ductal and acinar cell fates. In this discussion, developmental timepoints refer primarily to events in the dorsal pancreatic bud and to the onset of detectable protein expression or the timing of tamoxifen administration to activate lineage-specific CreER labeling.
Between E9.5 and E10.5, Pdx1, Ptf1a and Sox9 are co-expressed in the vast majority of MPCs [28, 82]. By E10.5, most Pdx1+, Ptf1+, Sox9+ MPCs also express Hes1 and Nkx6.1 [78, 86] Between E11.5 and E12.5, the stratified pancreatic buds begin to organize into a branched epithelial tree. By E11.5, the majority of cells have activated expression of Hnf1β and CPA, in combination with ongoing expression of Pdx1, Ptf1a, Sox9, Hes1, Nkx6.1, and Nkx6.2. At this stage, Cre/lox-based lineage tracing confirms that MPCs expressing Pdx1, Sox9, Hnf1β, Hes1 and CPA/Ptf1a remain tri-potent, effectively contributing to the future islet, ductal and acinar lineages [35, 37, 79, 81, 83].
By E12.5, overlapping expression of these transcription factors begins to diminish, and the bud becomes spatially resolved into trunk and tip domains. As this occurs, Ptf1a and CPA become progressively restricted to peripheral tips of the epithelial branches, while Hnf1β, Nkx6.1 and Nkx6.2 become progressively restricted to trunks [78, 79, 81]. In contrast, at E12.5, Pdx1 and Sox9 continue to be broadly expressed. Examples of trunk- and tip-restricted transcription factor expression are provided in Figure 2. Notably, the appearance of cells expressing only trunk- or tip-associated genes occurs even before the onset of branching morphogenesis. For example, scattered cells expressing either Nkx6.1 or Ptf1a (but not both) can be detected as early E10.5 [78]; whether or not these cells are already lineage-restricted and subsequently spatially sort into appropriate trunk and tip domains, or remain plastic and modify trunk and tip gene expression based on location, remains unknown.
Figure 2. Transcription factor topologies in developing mouse pancreas.
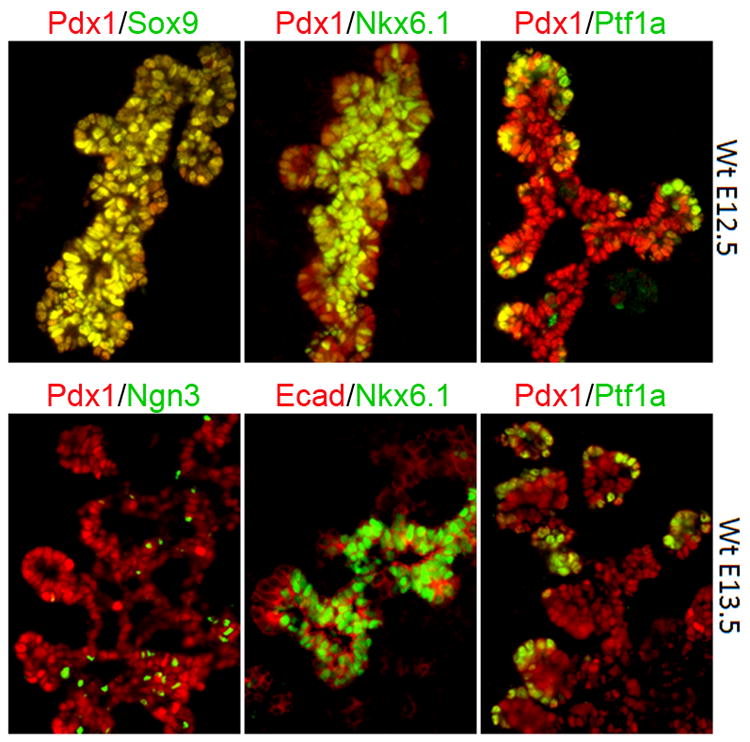
Immunofluorescent labeling for Pdx1, Sox9, Nkx6.1, Ptf1a, Ngn3 and E-cadherin (Ecad) in emerging trunk and tip domains of developing mouse pancreas. Hnf1β and Nkx6.1 become progressively restricted to the central trunk region of the branching pancreatic epithelium, from which Ngn+ cells begin to emerge. In contrast, Ptf1a is restricted to peripheral tips. As late as E13.5, Pdx1 and Sox9 are expressed in both central trunk and and peripheral tip domains.
Associated with the establishment of discrete Ptf1a+, CPA+, Hnf1β-, Nkx6.1-, Nkx6.2-, Sox9+ tip domains and Ptf1a-, CPA-, Hnf1β+, Nkx6.1+, Nkx6.2+, Sox9+ trunk domains, MPCs become progressively lineage restricted. Studies using a tamoxifen-inducible Hnf1β:CreER allele have revealed that E11.5 and E12.5 Hnf1β+ progenitor cells contribute to the islet, ductal and acinar lineages, although the proportion of acinar cells originating from Hnf1β+ progenitors is measurably decreased following tamoxifen administration at E12.5 compared to E11.5 [81]. Conversely, lineage labeling using a CPA:CreER has demonstrated that E10.5, E11.5 and E12.5 CPA+ progenitors contribute to islet, ductal and acinar cells, but CPA+ cells labeled by tamoxifen injection on E13.5 contribute exclusively to the acinar lineage. Together, these studies demonstrate the progressive restriction of trunk progenitors to the islet and ductal fates, and a corresponding restriction of tip progenitors to the acinar lineage. In contrast, the pool of progenitor cells expressing Sox9 remains tri-potent up until birth, reflecting ongoing expression not only in trunk progenitors but also in a subset of cells at the junction of the tip and trunk domains [28, 34, 37]. In this regard, detailed studies of the tip/trunk junction at E14.5 have demonstrated a tip population of differentiating acinar cells expressing Ptf1a but not Hnf1β or Sox9, a trunk population expressing Hnf1β and Sox9, but not Ptf1a, and an intermediate population expressing Ptf1a and Sox9 but not Hnf1β [78]. A subset of these Ptf1a+, Sox9+ cells also appear to express Hes1 [28]. As discussed below, these cells may represent the initial manifestation of the centroacinar lineage. In adult mouse and human pancreas, Sox9 expression is restricted to centroacinar and terminal duct cells, with negligible ongoing contribution to the endocrine lineage in the absence of injury [34, 37]. Following the induction of chronic pancreatitis by way of pancreatic duct ligation, Sox9+ centroacinar and terminal duct cells are capable of generating duct-associated cells expressing Ngn3, but these cells fail to complete an endocrine differentiation program [37]. While conflicting data exist regarding the normal contribution of adult Sox9+ cells to the exocrine lineage [34, 37], demonstration of a postnatal contribution of the adult Sox9+ cells to the acinar lineage appears to require activation of a Sox9:CreER lineage label with very high doses of tamoxifen. This raises the possibility of leaky Cre activity in acinar cells themselves, even in the absence of Sox9 protein expression.
While these studies have clearly defined the progressive fates of tip and trunk progenitor cells, a detailed mechanistic understanding of how each of these transcription factors contributes to the determination of acinar, ductal and centroacinar fates remains incomplete. Nevertheless, certain functional interactions have been clearly established. In particular, recent studies have demonstrated specific and opposing roles for Nkx6.1/6.2 and Ptf1a in promoting trunk vs. tip cell fates. Reflecting their initial co-expression but subsequent partitioning to trunk and tip domains [78, 80], recent functional studies have demonstrated antagonistic interaction between these factors. Specifically, the Nkx6 factors, which by E12.5 are largely restricted to trunk progenitors, have been demonstrated to be both required and sufficient to promote endocrine and antagonize acinar cell fate. Tip-restricted Ptf1a exerts the opposite influence [78]. Combined deletion of Nkx6.1 and Nkx6.2 results in extension of Ptf1a expression into the E12.5 trunk region, coupled with an increase in the number of Amylase-positive cells and a corresponding decrease in the number of cells expressing Ngn3. In contrast, forced expression of either Nkx6.1 or Nkx6.2 throughout the pancreatic epithelium promotes endocrine differentiation, while repressing the acinar cell fate. Notably, the number of ductal epithelial cells is unaffected by ectopic Nkx6 factor expression, reflecting the shared origin of islet and ductal cells from the trunk domain [78]
Notch signaling negatively regulates exocrine differentiation
Superimposed on the resolution of the branching epithelial tree into central trunk and peripheral tip domains is the influence of the Notch-signaling pathway, known to be a critical regulator of both endocrine and exocrine differentiation in both mouse and zebrafish pancreas [26, 35, 36, 61, 87-94]; (reviewed in Serup, this issue). As described above, Notch pathway activation (as imperfectly assessed by Hes1 expression) appears to be widespread in early MPCs [35, 86, 90], and subsequently becomes progressively silenced coincident with the onset at E13.5 of burst of endocrine differentiation known as the secondary transition. Specifically, Hes1 expression is lost in endocrine progenitors as they activate Ngn3 and delaminate from trunk epithelium [95], as well as in Ptf1a- and CPA-expressing tip progenitors as they begin to initiate acinar cell differentiation [35, 90]. By E14.5, Hes1 expression is largely confined to Ngn3-negative, CPA-negative trunk epithelium. This expression likely includes cells in the emerging centroacinar compartment, and by birth Hes1 expression is entirely restricted to centroacinar and terminal duct cells, most of which also express Sox9 [28, 31, 35, 90]. Centroacinar localization of cells with active Notch-signaling has also been observed in developing and adult zebrafish pancreas, and evidence suggests that these cells may also function as MPCs [26, 36].
This progressive restriction of Notch-signaling is associated with progressive restriction in the fate of the Hes1 lineage [35]. When labeled by tamoxifen injection at either E9.5 or E11.5, Hes1+ MPCs are tri-potent, contributing to the islet, ductal and acinar lineages. However, at the onset of the secondary transition at E13.5, these cells begin to progressively lose their potential to contribute to the endocrine lineage, with tamoxifen injection on E15.5 failing to label future alpha- or beta-cells. These data suggest that, during the secondary transition, the emergence of Ngn3+ cells from Hes1-expressing progenitors in the trunk domain occurs during a discrete temporal window between E13.5 and E15.5. In contrast, Hes1+ trunk cells, which become progressively restricted to the terminal ductal and centroacinar positions, continue to contribute to the ductal and acinar cell lineages throughout development, while serving as apparent lineage-restricted duct cell progenitors after birth [35]. Thus in addition to cells expressing Pdx1 and Sox9, Hes1+ centroacinar cells, perhaps related to the Ptf1a+, Sox9+, Hnf1β-negative cell population described on E14.5 [78], may represent the only other progenitor cell type capable of crossing the trunk/tip divide.
Functionally, Notch appears to effectively block both endocrine and exocrine differentiation [90-94] (reviewed in Serup, this issue). In the case of endocrine differentiation, this occurs by direct repression of Ngn3 expression [92, 94-96], while in the case of exocrine differentiation, it appears to occur through selective inhibition of Ptf1a’s late induction of acinar cell-specific gene expression [90, 97]. In contrast, the early pancreatic specification and morphogenetic influences of Ptf1a are permitted in the context of activated Notch-signaling, and Ptf1a itself appears to play a role in early Notch pathway activation, through induction of Dll1 expression [86]. With respect to regulation of ductal differentiation, several studies have demonstrated that Notch promotes embryonic MPC’s to adopt a trunk progenitor phenotype [78] and ultimately a ductal fate [35, 98]. Thus, the influence of Notch on pancreatic development may transcend a simple inhibitory effect on endocrine and acinar cell differentiation, and include an effect on lineage selection mediated through an influence on trunk/tip patterning. These concepts are presented schematically in Figure 3.
Figure 3. Emergence of ductal, islet and acinar lineages from progressively restricted trunk and tip progenitor cells.
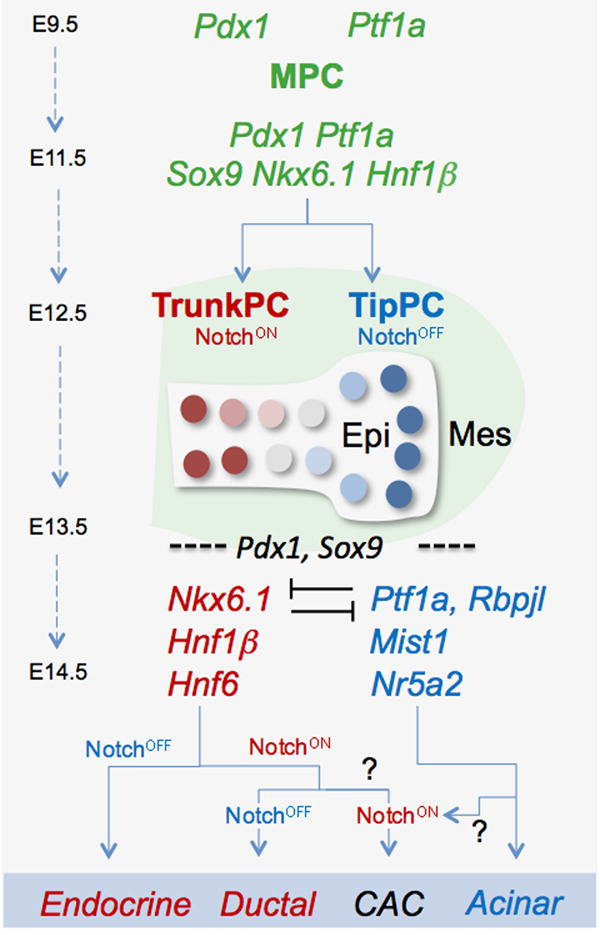
Initially tri-potent multil-ineage progenitor cells (MPCs) expressing both Pdx1 and Ptf1a ultimately give rise to ductal, islet and and acinar cells. On E11.5 most MPCs also co-express Sox9, Hnf1 β, Hnf6, Nkx6.1, and Nkx6.2. By E12.5, Hnf1 β, Hnf6, Nkx6.1, and Nkx6.2 become largely restricted to trunk progenitor cells (TrunkPCs), while Ptf1a becomes restricted to tip progenitors (TipPCs), with Notch signaling promoting the TrunkPC identity. By E13.5, acinar and lslet differentiation have been initiated, associated with the activation of additional lineage-specific transcription factors. Among TrunkPCs, Notch activation promotes a ductal fate. By the time of gestation, active Notch signaling is largely confined to centroacinar cells (CAC), whose derivation from Trunk vs. TipPCs remains unknown. CACs may be derived from either trunk progenitors maintaining active Notch signaling or from tip progenitors reactivating Notch.
In adult pancreas, continued Notch activation in centroacinar cells appears to be required for ongoing contribution of these cells to the ductal lineage; selective inactivation of Notch-signaling in Hes1+ centroacinar cells leads to their rapid differentiation into acinar cells, eliminating their ability to serve as adult ductal progenitors [87]. The pro-ductal influence of Notch may be suggested by evidence of abnormal Notch pathway activation in the majority of human pancreatic ductal cancers [99].
Induction of “terminal” exocrine differentiation
While little is known regarding transcriptional regulation of ductal and centroacinar differentiation, the functions of specific transcription factors in islet and acinar differentiation have been well characterized. Associated with the onset of the secondary transition on or about E13.5, endocrine differentiation is initiated by loss of Hes1 and activation of Ngn3 in cells derived from trunk progenitors, followed by the activation of additional endocrine-specific transcription factors and the generation of individual islet cell types [12].
Similarly, acinar cell differentiation is initiated within the tip domain, characterized by ongoing expression of Ptf1a and additional activation of the mammalian Supressor of Hairless homologue Rbpjl [21, 100]. Co-expression of Ptf1a and Rbpjl appears to activate a self-reinforcing gene expression module, effectively locking cells into a fixed state of acinar cell differentiation [100]. However, even this state of apparent terminal differentiation can be reversed by either reactivation of Notch-signaling [99, 101] or by reprogramming with combinations of endocrine-specific transcription factors [6]. Another transcription factor required for generation of the mature acinar phenotype is the bHLH transcription factor Mist1. Expression of Mist1 appears to be a common feature of many differentiated exocrine cell types, including parotid acinar cells, gastric chief cells, and cells of the zebrafish hatching gland [102-104]. In pancreatic acinar cells, Mist1 is required for establishment of the mature secretory phenotype and further acts to limit the proliferation of mature acinar cells [20, 103, 105, 106]. Differentiating and differentiated acinar cells also express the nuclear receptor LRH1/Nr5a2 [107], which has been shown to both physically interact with Ptf1a and independently activate expression of genes encoding digestive zymogens and other acinar cell-specific genes. Conditional deletion of LRH1 in adult pancreatic tissue results in decreased expression of several acinar cell-specific genes, as well as altered concentrations of acinar cell proteases and lipases in secreted pancreatic fluid. In addition, LRH1-deficient mice displayed impaired secretagogue-induced pancreatic fluid secretion. This raises the possibility of an additional influence of LRH1 on adult pancreatic duct cell function, although it remains unknown whether or not this represents a duct cell autonomous effect [107].
As discussed above, multiple transcription factors appear to contribute to patterning of the central trunk domain from which ductal epithelial cells are subsequently derived. However, at present, no specific transcription factors have been implicated in the initiation and maintenance of duct-specific gene expression, although ProxHnf1β and Hnf6 likely play instructive roles. In the normal pancreas, Prox1 is widely expressed at E13.5, but is low or absent in acinar cells in the adult[108, 109]. In a condititonal knockout of Prox1, there is relatively normal expression of both Hnf6 and Hnf1β, while Prox1 expression is decreased in the Hnf6 deficient mouse, suggesting that Hnf6 is upstream of Prox1 [109, 110]. Prox1-deficient mice are viable, but have dilated ductal lumens and progressively decreasing amounts of acinar tissue [109], suggesting an possible role for Prox1 in maintaining normal duct function.
In the absence of identified positive regulators of ductal differentiation, it is tempting to consider that ductal epithelial cells represent the progeny of “left behind” trunk progenitors that have avoided an endocrine fate by virtue of active Notch-signaling and failure to activate high levels of Ngn3. While evidence exists supporting this hypothesis [98, 111, 112], it remains likely that additional positive regulators of ductal differentiation remain to be identified.
Acknowledgments
The authors wish to thanks Charles Murtaugh and Michael Parsons for helpful discussions. This work was supported by the Chicago Diabetes Project and NIH grant DK61215 (to SDL).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chera S, et al. Silencing of the hydra serine protease inhibitor Kazal1 gene mimics the human SPINK1 pancreatic phenotype. J Cell Sci. 2006;119(Pt 5):846–57. doi: 10.1242/jcs.02807. [DOI] [PubMed] [Google Scholar]
- 2.Mohrlen F, et al. Activation of pro-astacin. Immunological and model peptide studies on the processing of immature astacin, a zinc-endopeptidase from the crayfish Astacus astacus. Eur J Biochem. 2001;268(9):2540–6. doi: 10.1046/j.1432-1327.2001.02136.x. [DOI] [PubMed] [Google Scholar]
- 3.Nokhbatolfoghahai M, Downie JR. Amphibian hatching gland cells: pattern and distribution in anurans. Tissue Cell. 2007;39(4):225–40. doi: 10.1016/j.tice.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 4.Inohaya K, et al. Temporal and spatial patterns of gene expression for the hatching enzyme in the teleost embryo, Oryzias latipes. Dev Biol. 1995;171(2):374–85. doi: 10.1006/dbio.1995.1289. [DOI] [PubMed] [Google Scholar]
- 5.Bonner-Weir S, et al. The pancreatic ductal epithelium serves as a potential pool of progenitor cells. Pediatr Diabetes. 2004;5(Suppl 2):16–22. doi: 10.1111/j.1399-543X.2004.00075.x. [DOI] [PubMed] [Google Scholar]
- 6.Zhou Q, et al. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455(7213):627–32. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seymour PA, Sander M. Historical perspective: beginnings of the beta-cell: current perspectives in beta-cell development. Diabetes. 2011;60(2):364–76. doi: 10.2337/db10-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pan FC, Wright C. Pancreas organogenesis: from bud to plexus to gland. Dev Dyn. 2011;240(3):530–65. doi: 10.1002/dvdy.22584. [DOI] [PubMed] [Google Scholar]
- 9.Gittes GK. Developmental biology of the pancreas: a comprehensive review. Dev Biol. 2009;326(1):4–35. doi: 10.1016/j.ydbio.2008.10.024. [DOI] [PubMed] [Google Scholar]
- 10.Murtaugh LC. Pancreas and beta-cell development: from the actual to the possible. Development. 2007;134(3):427–38. doi: 10.1242/dev.02770. [DOI] [PubMed] [Google Scholar]
- 11.Jensen J. Gene regulatory factors in pancreatic development. Dev Dyn. 2004;229(1):176–200. doi: 10.1002/dvdy.10460. [DOI] [PubMed] [Google Scholar]
- 12.Oliver-Krasinski JM, Stoffers DA. On the origin of the beta cell. Genes Dev. 2008;22(15):1998–2021. doi: 10.1101/gad.1670808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Puri S, Hebrok M. Cellular plasticity within the pancreas--lessons learned from development. Dev Cell. 2010;18(3):342–56. doi: 10.1016/j.devcel.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jamieson JD, Palade GE. Intracellular transport of secretory proteins in the pancreatic exocrine cell. I. Role of the peripheral elements of the Golgi complex. J Cell Biol. 1967;34(2):577–96. doi: 10.1083/jcb.34.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han JH, Rall L, Rutter WJ. Selective expression of rat pancreatic genes during embryonic development. Proc Natl Acad Sci U S A. 1986;83(1):110–4. doi: 10.1073/pnas.83.1.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rutter WJ, K JD, Bradshaw WS, Clark WR, Ronzio RA, Sanders TG. Regulation of Specific Protein Synthesis in Cytodifferentiation. J Cell Physiol. 1968;72(S1):1. doi: 10.1002/jcp.1040720403. [DOI] [PubMed] [Google Scholar]
- 17.Jorgensen M, et al. An Illustrated Review of Early Pancreas Development in Mouse. Endocrine Reviews. 2007;28(6):685–705. doi: 10.1210/er.2007-0016. [DOI] [PubMed] [Google Scholar]
- 18.Scoggins CR, et al. p53-dependent acinar cell apoptosis triggers epithelial proliferation in duct-ligated murine pancreas. Am J Physiol Gastrointest Liver Physiol. 2000;279(4):G827–36. doi: 10.1152/ajpgi.2000.279.4.G827. [DOI] [PubMed] [Google Scholar]
- 19.Masui T, et al. Replacement of Rbpj with Rbpjl in the PTF1 complex controls the final maturation of pancreatic acinar cells. Gastroenterology. 2010;139(1):270–80. doi: 10.1053/j.gastro.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pin CL, et al. The bHLH transcription factor Mist1 is required to maintain exocrine pancreas cell organization and acinar cell identity. J Cell Biol. 2001;155(4):519–30. doi: 10.1083/jcb.200105060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beres TM, et al. PTF1 is an organ-specific and Notch-independent basic helix-loop-helix complex containing the mammalian Suppressor of Hairless (RBP-J) or its paralogue, RBP-L. Mol Cell Biol. 2006;26(1):117–30. doi: 10.1128/MCB.26.1.117-130.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rovira M, et al. Murine embryonic stem cell-derived pancreatic acinar cells recapitulate features of early pancreatic differentiation. Gastroenterology. 2008;135(4):1301–1310. 1310e1–5. doi: 10.1053/j.gastro.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schick J, Kern H, Scheele G. Hormonal stimulation in the exocrine pancreas results in coordinate and anticoordinate regulation of protein synthesis. J Cell Biol. 1984;99(5):1569–74. doi: 10.1083/jcb.99.5.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leeson TS, Leeson R. Close association of centroacinar/ductular and insular cells in the rat pancreas. Histol Histopathol. 1986;1(1):33–42. [PubMed] [Google Scholar]
- 25.Pour PM. Pancreatic Centroacinar Cells. Int J Pancreatol. 1994;15(1):51–64. [PubMed] [Google Scholar]
- 26.Parsons MJ, et al. Notch-responsive cells initiate the secondary transition in larval zebrafish pancreas. Mech Dev. 2009;126(10):898–912. doi: 10.1016/j.mod.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miyamoto Y, et al. Notch mediates TGFalpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell. 2003;3(6):565–76. doi: 10.1016/s1535-6108(03)00140-5. [DOI] [PubMed] [Google Scholar]
- 28.Seymour PA, et al. SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proc Natl Acad Sci U S A. 2007;104(6):1865–70. doi: 10.1073/pnas.0609217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hayashi K, et al. Regional differences in the cellular proliferation activity of the regenerating rat pancreas after partial pancreatectomy. Arch Histol Cytol. 1999;62(4):337–46. doi: 10.1679/aohc.62.337. [DOI] [PubMed] [Google Scholar]
- 30.Gasslander T, Ihse I, Smeds S. The importance of the centroacinar region in cerulein-induced mouse pancreatic growth. Scand J Gastroenterol. 1992;27(7):564–70. doi: 10.3109/00365529209000120. [DOI] [PubMed] [Google Scholar]
- 31.Stanger BZ, et al. Pten constrains centroacinar cell expansion and malignant transformation in the pancreas. Cancer Cell. 2005;8(3):185–95. doi: 10.1016/j.ccr.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 32.Hayashi KY, et al. Differentiation and proliferation of endocrine cells in the regenerating rat pancreas after 90% pancreatectomy. Arch Histol Cytol. 2003;66(2):163–74. doi: 10.1679/aohc.66.163. [DOI] [PubMed] [Google Scholar]
- 33.Nagasao J, et al. Morphological changes in the rat endocrine pancreas within 12 h of intravenous streptozotocin administration. Anat Histol Embryol. 2005;34(1):42–7. doi: 10.1111/j.1439-0264.2004.00566.x. [DOI] [PubMed] [Google Scholar]
- 34.Furuyama K, et al. Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat Genet. 2011;43(1):34–41. doi: 10.1038/ng.722. [DOI] [PubMed] [Google Scholar]
- 35.Kopinke D, et al. Lineage tracing reveals the dynamic contribution of Hes1+ cells to the developing and adult pancreas. Development. 2011;138(3):431–41. doi: 10.1242/dev.053843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, et al. Genetic inducible fate mapping in larval zebrafish reveals origins of adult insulin-producing beta-cells. Development. 2011;138(4):609–17. doi: 10.1242/dev.059097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kopp JL, et al. Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development. 2011;138(4):653–65. doi: 10.1242/dev.056499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reichert M, Rustgi AK. Pancreatic ductal cells in development, regeneration, and neoplasia. J Clin Invest. 2011;121(12):4572–8. doi: 10.1172/JCI57131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Villasenor A, et al. Epithelial dynamics of pancreatic branching morphogenesis. Development. 2010;137(24):4295–305. doi: 10.1242/dev.052993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hick AC, et al. Mechanism of primitive duct formation in the pancreas and submandibular glands: a role for SDF-1. BMC Dev Biol. 2009;9:66. doi: 10.1186/1471-213X-9-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kesavan G, et al. Cdc42-mediated tubulogenesis controls cell specification. Cell. 2009;139(4):791–801. doi: 10.1016/j.cell.2009.08.049. [DOI] [PubMed] [Google Scholar]
- 42.Kilic G, Wang J, Sosa-Pineda B. Osteopontin is a novel marker of pancreatic ductal tissues and of undifferentiated pancreatic precursors in mice. Dev Dyn. 2006;235(6):1659–67. doi: 10.1002/dvdy.20729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lardon J, et al. Stem cell marker prominin-1/AC133 is expressed in duct cells of the adult human pancreas. Pancreas. 2008;36(1):e1–6. doi: 10.1097/mpa.0b013e318149f2dc. [DOI] [PubMed] [Google Scholar]
- 44.Lammert E, Cleaver O, Melton D. Induction of pancreatic differentiation by signals from blood vessels. Science. 2001;294(5542):564–7. doi: 10.1126/science.1064344. [DOI] [PubMed] [Google Scholar]
- 45.Burghardt B, et al. Distribution of aquaporin water channels AQP1 and AQP5 in the ductal system of the human pancreas. Gut. 2003;52(7):1008–16. doi: 10.1136/gut.52.7.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kopinke D, Murtaugh LC. Exocrine-to-endocrine differentiation is detectable only prior to birth in the uninjured mouse pancreas. BMC Dev Biol. 2010;10:38. doi: 10.1186/1471-213X-10-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pierreux CE, et al. The transcription factor hepatocyte nuclear factor-6 controls the development of pancreatic ducts in the mouse. Gastroenterology. 2006;130(2):532–41. doi: 10.1053/j.gastro.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 48.Strobel O, et al. Pancreatic duct glands are distinct ductal compartments that react to chronic injury and mediate Shh-induced metaplasia. Gastroenterology. 2010;138(3):1166–77. doi: 10.1053/j.gastro.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Geleff S, Bock P. Pancreatic duct glands. II. Lectin binding affinities of ductular epithelium, ductular glands, and Brunner glands. Histochemistry. 1984;80(1):31–8. doi: 10.1007/BF00492768. [DOI] [PubMed] [Google Scholar]
- 50.Bock P, Geleff S. Pancreatic duct glands. III. Morphology of secretory epithelium and endoepithelial glands. Z Mikrosk Anat Forsch. 1984;98(6):857–72. [PubMed] [Google Scholar]
- 51.Bock P. Pancreatic duct glands. I. Staining reactions of acid glycoprotein secret. Acta Histochem. 1978;61(1):118–26. doi: 10.1016/S0065-1281(78)80055-5. [DOI] [PubMed] [Google Scholar]
- 52.Golosow N, Grobstein C. Epitheliomesenchymal interaction in pancreatic morphogenesis. Dev Biol. 1962;4:242–55. doi: 10.1016/0012-1606(62)90042-8. [DOI] [PubMed] [Google Scholar]
- 53.Wessels NK, Cohen JH. Early pancreas organogenesis: morphogenesis, tissue interactions, and mass effects. Dev Biol. 1967;15:237–270. doi: 10.1016/0012-1606(67)90042-5. [DOI] [PubMed] [Google Scholar]
- 54.Gittes GK, et al. Lineage-specific morphogenesis in the developing pancreas: role of mesenchymal factors. Development. 1996;122(2):439–47. doi: 10.1242/dev.122.2.439. [DOI] [PubMed] [Google Scholar]
- 55.Bhushan A, et al. Fgf10 is essential for maintaining the proliferative capacity of epithelial progenitor cells during early pancreatic organogenesis. Development. 2001;128(24):5109–17. doi: 10.1242/dev.128.24.5109. [DOI] [PubMed] [Google Scholar]
- 56.Bellusci S, et al. Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development. 1997;124(23):4867–78. doi: 10.1242/dev.124.23.4867. [DOI] [PubMed] [Google Scholar]
- 57.Miralles F, et al. Signaling through fibroblast growth factor receptor 2b plays a key role in the development of the exocrine pancreas. Proc Natl Acad Sci U S A. 1999;96(11):6267–72. doi: 10.1073/pnas.96.11.6267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miralles F, et al. Interplay between FGF10 and Notch signalling is required for the self-renewal of pancreatic progenitors. Int J Dev Biol. 2006;50(1):17–26. doi: 10.1387/ijdb.052080fm. [DOI] [PubMed] [Google Scholar]
- 59.Le Bras S, et al. Fibroblast growth factor 2 promotes pancreatic epithelial cell proliferation via functional fibroblast growth factor receptors during embryonic life. Diabetes. 1998;47(8):1236–42. [PubMed] [Google Scholar]
- 60.Hart A, Papadopoulou S, Edlund H. Fgf10 maintains notch activation, stimulates proliferation, and blocks differentiation of pancreatic epithelial cells. Dev Dyn. 2003;228(2):185–93. doi: 10.1002/dvdy.10368. [DOI] [PubMed] [Google Scholar]
- 61.Norgaard GA, Jensen JN, Jensen J. FGF10 signaling maintains the pancreatic progenitor cell state revealing a novel role of Notch in organ development. Dev Biol. 2003;264(2):323–38. doi: 10.1016/j.ydbio.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 62.Attali M, et al. Control of beta-cell differentiation by the pancreatic mesenchyme. Diabetes. 2007;56(5):1248–58. doi: 10.2337/db06-1307. [DOI] [PubMed] [Google Scholar]
- 63.Cras-Meneur C, et al. Epidermal growth factor increases undifferentiated pancreatic embryonic cells in vitro: a balance between proliferation and differentiation. Diabetes. 2001;50(7):1571–9. doi: 10.2337/diabetes.50.7.1571. [DOI] [PubMed] [Google Scholar]
- 64.Miralles F, Czernichow P, Scharfmann R. Follistatin regulates the relative proportions of endocrine versus exocrine tissue during pancreatic development. Development. 1998;125(6):1017–24. doi: 10.1242/dev.125.6.1017. [DOI] [PubMed] [Google Scholar]
- 65.Sanvito F, et al. TGF-beta 1 influences the relative development of the exocrine and endocrine pancreas in vitro. Development. 1994;120(12):3451–62. doi: 10.1242/dev.120.12.3451. [DOI] [PubMed] [Google Scholar]
- 66.Bottinger EP, et al. Expression of a dominant-negative mutant TGF-beta type II receptor in transgenic mice reveals essential roles for TGF-beta in regulation of growth and differentiation in the exocrine pancreas. EMBO J. 1997;16(10):2621–33. doi: 10.1093/emboj/16.10.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ahnfelt-Ronne J, et al. Mesenchymal bone morphogenetic protein signaling is required for normal pancreas development. Diabetes. 2010;59(8):1948–56. doi: 10.2337/db09-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kadison A, et al. Retinoid signaling directs secondary lineage selection in pancreatic organogenesis. J Pediatr Surg. 2001;36(8):1150–6. doi: 10.1053/jpsu.2001.25734. [DOI] [PubMed] [Google Scholar]
- 69.Kobayashi H, et al. Retinoid signaling controls mouse pancreatic exocrine lineage selection through epithelial-mesenchymal interactions. Gastroenterology. 2002;123(4):1331–40. doi: 10.1053/gast.2002.35949. [DOI] [PubMed] [Google Scholar]
- 70.Crisera CA, et al. Expression and role of laminin-1 in mouse pancreatic organogenesis. Diabetes. 2000;49(6):936–44. doi: 10.2337/diabetes.49.6.936. [DOI] [PubMed] [Google Scholar]
- 71.Gumbiner BM. Epithelial morphogenesis. Cell. 1992;69(3):385–7. doi: 10.1016/0092-8674(92)90440-n. [DOI] [PubMed] [Google Scholar]
- 72.Tulachan SS, et al. Mesenchymal epimorphin is important for pancreatic duct morphogenesis. Dev Growth Differ. 2006;48(2):65–72. doi: 10.1111/j.1440-169X.2006.00846.x. [DOI] [PubMed] [Google Scholar]
- 73.Landsman L, et al. Pancreatic mesenchyme regulates epithelial organogenesis throughout development. PLoS Biol. 2011;9(9):e1001143. doi: 10.1371/journal.pbio.1001143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Magenheim J, et al. Blood vessels restrain pancreas branching, differentiation and growth. Development. 2011;138(21):4743–52. doi: 10.1242/dev.066548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lammert E, Cleaver O, Melton D. Role of endothelial cells in early pancreas and liver development. Mech Dev. 2003;120(1):59–64. doi: 10.1016/s0925-4773(02)00332-5. [DOI] [PubMed] [Google Scholar]
- 76.Pierreux CE, et al. Epithelial: Endothelial cross-talk regulates exocrine differentiation in developing pancreas. Dev Biol. 2010;347(1):216–27. doi: 10.1016/j.ydbio.2010.08.024. [DOI] [PubMed] [Google Scholar]
- 77.Jacquemin P, et al. An endothelial-mesenchymal relay pathway regulates early phases of pancreas development. Dev Biol. 2006;290(1):189–99. doi: 10.1016/j.ydbio.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 78.Schaffer AE, et al. Nkx6 transcription factors and Ptf1a function as antagonistic lineage determinants in multipotent pancreatic progenitors. Dev Cell. 2010;18(6):1022–9. doi: 10.1016/j.devcel.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhou Q, et al. A multipotent progenitor domain guides pancreatic organogenesis. Dev Cell. 2007;13(1):103–14. doi: 10.1016/j.devcel.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 80.Hald J, et al. Generation and characterization of Ptf1a antiserum and localization of Ptf1a in relation to Nkx6.1 and Pdx1 during the earliest stages of mouse pancreas development. J Histochem Cytochem. 2008;56(6):587–95. doi: 10.1369/jhc.2008.950675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Solar M, et al. Pancreatic exocrine duct cells give rise to insulin-producing beta cells during embryogenesis but not after birth. Dev Cell. 2009;17(6):849–60. doi: 10.1016/j.devcel.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 82.Burlison JS, et al. Pdx-1 and Ptf1a concurrently determine fate specification of pancreatic multipotent progenitor cells. Dev Biol. 2008;316(1):74–86. doi: 10.1016/j.ydbio.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129(10):2447–57. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- 84.Kawaguchi Y, et al. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet. 2002;32(1):128–34. doi: 10.1038/ng959. [DOI] [PubMed] [Google Scholar]
- 85.Fishman MP, Melton DA. Pancreatic lineage analysis using a retroviral vector in embryonic mice demonstrates a common progenitor for endocrine and exocrine cells. Int J Dev Biol. 2002;46(2):201–7. [PubMed] [Google Scholar]
- 86.Ahnfelt-Ronne J, et al. Ptf1a-mediated control of Dll1 reveals an alternative to the lateral inhibition mechanism. Development. 2012;139(1):33–45. doi: 10.1242/dev.071761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kopinke D, et al. Ongoing Notch signaling maintains phenotypic fidelity in the adult exocrine pancreas. Dev Biol. 2011 doi: 10.1016/j.ydbio.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ahnfelt-Ronne J, et al. Preservation of proliferating pancreatic progenitor cells by Delta-Notch signaling in the embryonic chicken pancreas. BMC Dev Biol. 2007;7:63. doi: 10.1186/1471-213X-7-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fujikura J, et al. Notch/Rbp-j signaling prevents premature endocrine and ductal cell differentiation in the pancreas. Cell Metab. 2006;3(1):59–65. doi: 10.1016/j.cmet.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 90.Esni F, et al. Notch inhibits Ptf1 function and acinar cell differentiation in developing mouse and zebrafish pancreas. Development. 2004;131(17):4213–24. doi: 10.1242/dev.01280. [DOI] [PubMed] [Google Scholar]
- 91.Murtaugh LC, et al. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci U S A. 2003;100(25):14920–5. doi: 10.1073/pnas.2436557100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hald J, et al. Activated Notch1 prevents differentiation of pancreatic acinar cells and attenuate endocrine development. Dev Biol. 2003;260(2):426–37. doi: 10.1016/s0012-1606(03)00326-9. [DOI] [PubMed] [Google Scholar]
- 93.Jensen J, et al. Control of endodermal endocrine development by Hes-1. Nat Genet. 2000;24(1):36–44. doi: 10.1038/71657. [DOI] [PubMed] [Google Scholar]
- 94.Apelqvist A, et al. Notch signalling controls pancreatic cell differentiation. Nature. 1999;400(6747):877–81. doi: 10.1038/23716. [DOI] [PubMed] [Google Scholar]
- 95.Jensen J, et al. Independent development of pancreatic alpha- and beta-cells from neurogenin3-expressing precursors: a role for the notch pathway in repression of premature differentiation. Diabetes. 2000;49(2):163–76. doi: 10.2337/diabetes.49.2.163. [DOI] [PubMed] [Google Scholar]
- 96.Lee JC, et al. Regulation of the pancreatic pro-endocrine gene neurogenin3. Diabetes. 2001;50(5):928–36. doi: 10.2337/diabetes.50.5.928. [DOI] [PubMed] [Google Scholar]
- 97.Ghosh B, Leach SD. Interactions between hairy/enhancer of split-related proteins and the pancreatic transcription factor Ptf1-p48 modulate function of the PTF1 transcriptional complex. Biochem J. 2006;393(Pt 3):679–85. doi: 10.1042/BJ20051063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Greenwood AL, et al. Notch signaling reveals developmental plasticity of Pax4(+) pancreatic endocrine progenitors and shunts them to a duct fate. Mech Dev. 2007;124(2):97–107. doi: 10.1016/j.mod.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 99.Miyamoto Y, et al. Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell. 2003;3(6):565–76. doi: 10.1016/s1535-6108(03)00140-5. [DOI] [PubMed] [Google Scholar]
- 100.Masui T, et al. Early pancreatic development requires the vertebrate Suppressor of Hairless (RBPJ) in the PTF1 bHLH complex. Genes Dev. 2007;21(20):2629–43. doi: 10.1101/gad.1575207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.De La OJ, et al. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc Natl Acad Sci U S A. 2008;105(48):18907–12. doi: 10.1073/pnas.0810111105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Huh WJ, et al. XBP1 controls maturation of gastric zymogenic cells by induction of MIST1 and expansion of the rough endoplasmic reticulum. Gastroenterology. 2010;139(6):2038–49. doi: 10.1053/j.gastro.2010.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pin CL, Bonvissuto AC, Konieczny SF. Mist1 expression is a common link among serous exocrine cells exhibiting regulated exocytosis. Anat Rec. 2000;259(2):157–67. doi: 10.1002/(SICI)1097-0185(20000601)259:2<157::AID-AR6>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 104.Guo X, et al. Cloning, expression, and functional characterization of zebrafish Mist1. Biochem Biophys Res Commun. 2007;359(1):20–6. doi: 10.1016/j.bbrc.2007.05.055. [DOI] [PubMed] [Google Scholar]
- 105.Jia D, Sun Y, Konieczny SF. Mist1 regulates pancreatic acinar cell proliferation through p21 CIP1/WAF1. Gastroenterology. 2008;135(5):1687–97. doi: 10.1053/j.gastro.2008.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Luo X, et al. Aberrant localization of intracellular organelles, Ca2+ signaling, and exocytosis in Mist1 null mice. J Biol Chem. 2005;280(13):12668–75. doi: 10.1074/jbc.M411973200. [DOI] [PubMed] [Google Scholar]
- 107.Holmstrom SR, et al. LRH-1 and PTF1-L coregulate an exocrine pancreas-specific transcriptional network for digestive function. Genes Dev. 2011;25(16):1674–9. doi: 10.1101/gad.16860911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang J, et al. Prox1 activity controls pancreas morphogenesis and participates in the production of “secondary transition” pancreatic endocrine cells. Dev Biol. 2005;286:182–94. doi: 10.1016/j.ydbio.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 109.Westmoreland J, et al. Pancreas-Specific Deletion of Prox1 Affects Development and Disrupts Homeostasis of the Exocrine Pancreas. Gastroenterology. doi: 10.1053/j.gastro.2011.12.007. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang H, et al. Multiple, temporal-specific roles for HNF6 in pancreatic endocrine and ductal differentiation. Mech Dev. 2009;126:958–73. doi: 10.1016/j.mod.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Magenheim J, et al. Ngn3(+) endocrine progenitor cells control the fate and morphogenesis of pancreatic ductal epithelium. Dev Biol. 2011;359(1):26–36. doi: 10.1016/j.ydbio.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang S, et al. Neurog3 gene dosage regulates allocation of endocrine and exocrine cell fates in the developing mouse pancreas. Dev Biol. 2010;339(1):26–37. doi: 10.1016/j.ydbio.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]