

Abstract

A new and facile synthetic pathway to metabolically stable 5′-methylene-bis(pivaloyloxymethyl)(POM)phosphonate furanonucleoside prodrugs is reported. The key step involves a Horner-Wadsworth-Emmons reaction of a tetra(pivaloyloxymethyl) bisphosphonate salt with appropriately protected 5′-aldehydic nucleosides. This efficient approach was applied for the synthesis HCV related 2′-deoxy-2′-α-fluoro-2′-β-C-methyl nucleosides.
Over the past 40 years, nucleoside analogs have become essential agents for the treatment of various infectious diseases such as human immunodeficiency virus (HIV), herpes simplex virus (HSV), hepatitis C virus (HCV) and hepatitis B virus (HBV). Their mechanism of action generally involves the direct or delayed chain termination of viral DNA or RNA elongation by incorporation of nucleoside triphosphate analogs. The biotransformation of nucleosides into their corresponding triphosphates involves three successive phosphorylation steps catalyzed by viral and/or cellular kinases and in many cases, the limiting step in this process is the conversion to the corresponding 5′-monophosphate.1 Within the nucleoside analogs family, nucleoside phosphonates have emerged as an important class since the discovery of compounds such as tenofovir, adefovir and cidofovir. 2 Because a carbon-phosphorus bond replaces the 5′-oxygen-phosphorus bond, nucleoside phosphonates are more stable analogs of nucleoside monophosphates and can, in theory, bypass the first limiting phosphorylation step. However, as with their phosphate counterparts, the ionic character of these phosphonic acid derivatives is generally an obstacle for absorption and/or cellular penetration, which often translates into a lack of in vivo activity for these compounds. In order to mask the negative charges, phosphonate prodrugs bearing biolabile protecting groups, such as pivaloyloxymethyl (POM) and isopropyloxymethyl groups (POC), have been developed and incorporated into the FDA approved adefovir dipivoxil 2 and tenofovir diisoproxil fumarate 3 which are utilized clinically for the treatment of HBV and HIV infections, respectively (Figure 1).2 The apparently simple replacement of the oxygen atom by a carbon atom, to form 5′-methylene nucleoside phosphonates prodrugs, is actually a low yielding multistep sequence. Synthesis of such compounds involves 1) introduction of a simple phosphonate moiety 2) a trimethylsilyl bromide driven cleavage of the phosphonate esters; 3) purification of highly polar phosphonic acids; 4) and variable yields when coupling phosphonic acids with the prodrug portion. A recently published exception to this sequence is the approach developed by Agrofoglio in which butenyl acylic nucleoside phosphonate prodrug are prepared using an olefin cross metathesis reaction with POM/POC protected allylphosphonates.3
Figure 1.
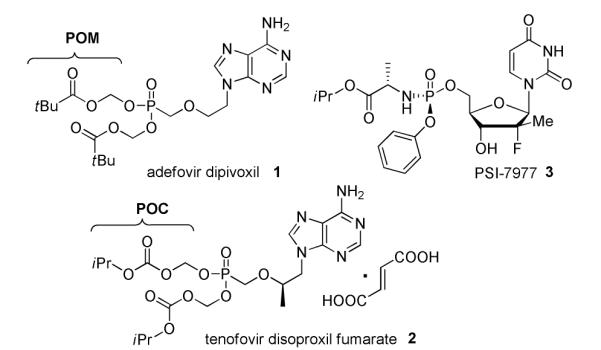
Biologically active phosphonate and phosphoramidate nucleosides
Simple 5′-methylene-alkylphosphonates are most often prepared via an initial Wittig reaction between 5′-aldehydic nucleosides and dialkyl ((triphenylphosphoranylidene)methyl)phosphonate reagents. 4 , 5 Alternative synthetic pathways include: 1) Arbuzov condensation on a 5′-deoxy-6′-halo-sugar, subsequent glycosylation and deprotection; 6 2) 3′,4′-β-oxetane ring opening with alkylphosphonate anions followed by 3′-hydroxy inversion;7 and 3) 4′-C-radical generation by photolysis of the 4′-(2-thiopyridone) ester (Barton reaction) via protection and 5′-oxidation to the carboxylate. 8 An attractive but surprisingly underexploited synthetic pathway for the synthesis of 5′-methylene-nucleoside phosphonates reported by Montgomery, involves a modified Horner-Wadsworth-Emmons (HWE) olefination by addition of dialkyl bisphosphonate salt on a 5′-aldehydic sugar and its subsequent glycosylation. 9 A similar approach was used later by Blackburn and Rachid for the synthesis of 3-phospho-D-glyceric acid analogs starting from a β-D-ribopentodialdehydo-l,4-furanoside10 and by Calenberg for the synthesis of 6-substituted uridine phosphonic acid analogs.11
We sought to extend the approach of Montgomery and Hewson9 by preparing a dialkyl bisphosphonate where the alkyl portion was the desired final prodrug substituent. Utilization of such a reagent in a modified HWE reaction followed by reduction would eliminate the traditional4, 12 and often problematic phosphate ester cleavage and also make the overall synthesis more convergent by shifting the coupling of the phosphonic acid with the prodrug portion to the HWE reagent synthesis.
Herein, we report a general method to prepare nucleoside 5′-methylene bis(POM)-phosphonate prodrugs that allows: (1) direct introduction of the phosphonate moiety bearing the biolabile POM leaving group; (2) apparent compatibility with most 5′-aldehydic nucleosides, independent of the nucleobase by condensation of tetra(POM)-bisphosphonate 3 via a modified HWE olefination (Figure 2). As part of our HCV research program, we applied this strategy to the 2′-deoxy-2′-α-fluoro-2′-β-C-methyl sugar derivatives whose most advanced member, PSI-7977 (GS-7977), 13, 14 is currently in phase III clinical trials as a safe and effective anti-HCV agent (Figure 1).15 The required tetra(POM)-bisphosphonate (TPBP, 5), was prepared by a reported method from tetramethyl bisphosphonate 4 and chloromethyl pivalate in presence of sodium iodide (Scheme 1).16 Previous studies involving 5 have been limited to its use as a methylenebisphosphonate prodrug for bone resorption disorders16 and the closely related mimics of farnesyl diphosphate. 17 The use of tetra substituted bisphosphonates in modified HWE reactions have, to date, been limited to CH3-, CH3CH2-, CF3CH2-, i-Bu-, Ph-, and Bn-substitutions.
Figure 2.
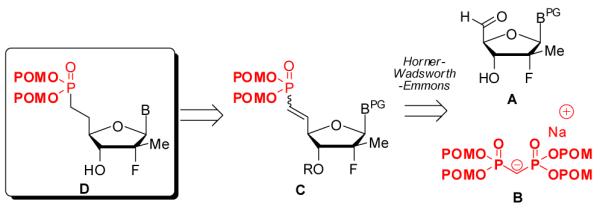
Retrosynthetic analysis
Scheme 1.
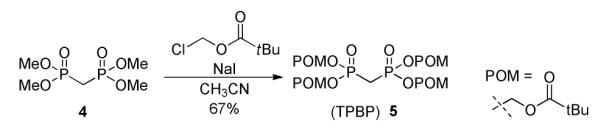
Synthesis of tetra(POM)-bisphosphonate (TPBP) 5
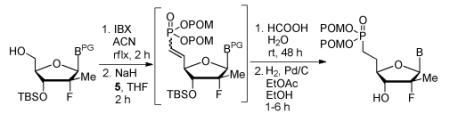
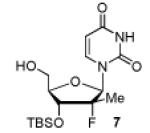
In consideration of the chemical stability of POM groups, the selection of appropriate protective groups for our strategy was crucial and required investigation. We sought a protecting group that: (1) can be selectively introduced on the 3′-position of the sugar in high yield; (2) is stable under oxidative and bisphosphonate salt addition conditions; (3) can easily be removed without affecting the POM groups. t-Butyldimethylsilyl (TBS) groups appeared to fit these criteria. Therefore, 3′-OTBDMS-2′-Me-2′-F-uridine derivative 7 was prepared as outlined in Scheme 2, by persilylation of 618 with TBSCl followed by selective 5′-desilylation in presence of aqueous trichloroacetic acid (TCA).19
Scheme 2.
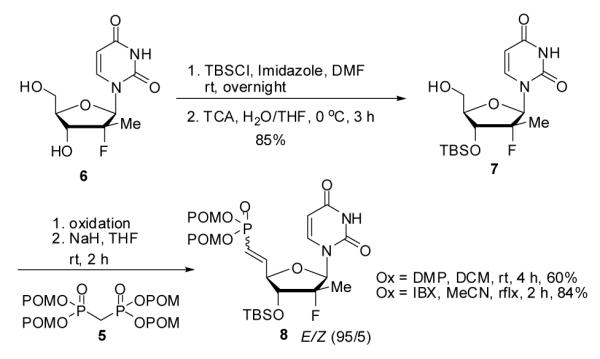
Introduction of 3′-TBS group and oxidation suitability
Oxidation of the 3′-OTBS derivative 7 was conducted with both Dess-Martin periodinane (DMP) at room temperature and 2-iodoxybenzoic acid (IBX)20 at 80 °C (Scheme 2). The IBX oxidation of 7 afforded clean aldehyde in almost quantitative yield after simple filtration, while aldehyde resulting from DMP oxidation was mixed with several DMP byproducts. Finally, reaction between the crude aldehyde and 2.2 equivalents of TPBP 5, lead to the formation of desired bis(POM)-vinylphosphonate 8. Interestingly, the use of a cleaner crude intermediate aldehyde obtained from IBX oxidation had an impact on the efficiency of the overall reaction sequence (60% compared to 84%). As expected with an HWE reaction mechanism, trans-bis(POM)-vinylphosphonate 8 was obtained as the major isomer (E/Z ratio of 95:5 as determined by 1H NMR).
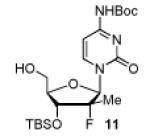
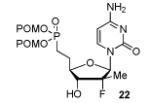
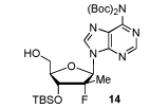
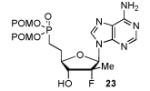
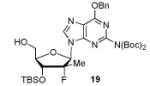
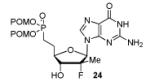
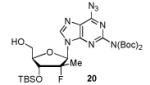
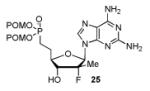
In order to generalize our strategy, this route was applied to the preparation of synthetically more challenging cytosine (C), adenine (A), guanine (G), and 2,6-diamino purine 2′-F-2′-C-Me nucleoside bis(POM)-phosphonate analogs 22-25. For each nucleoside base, selective protections were utilized to be compatible with the key oxidation/HWE reaction sequence. Thus, cytosine derivative 9 was first treated with TBSCl then with 2 equivalents of Boc2O to give the fully protected compound 10. Finally, selective deprotection using trichloroacetic acid (TCA) afforded the 3′-OTBS-N4-Boc nucleoside 11 in 81% overall yield with no observed Boc cleavage. A similar sequence was applied to prepare 3′-OTBS-N6-diBoc adenine nucleoside 14. The exocyclic amine was bis-Boc protected in high yield by treatment with excess Boc2O in presence of N,N-dimethylaminopyridine (DMAP). 21 The TCA mediated deprotection gave the corresponding 5′-hydroxyl product 14 in an overall 74% yield with both Boc groups intact. In order to prepare guanosine and 2,6-diaminopurine phosphonate derivatives 19 and 20, we started from the 6-chloro precursor 16 which was synthesized from 1522 in 89% yield by persilylation of the sugar hydroxy groups using 5 equivalent of TBSCl in presence of imidazole. Substitution of the 6-chloro of nucleoside analog 16 with either sodium benzyloxide or sodium azide followed by 2-amino protection with Boc2O led to compounds 17 and 18. Selective 5′-deprotection with TCA afforded the desired protected intermediates 19 and 20 readily available for conversion to bis(POM)-phosphonate prodrugs.
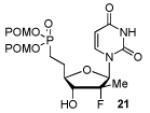
The IBX oxidation/TPBP sodium salt addition sequence was successfully applied to the protected 5′-OH nucleosides 11, 14, 19 and 20 (Table 1). In the oxidation of the purine analogs 14, 19 and 20, the N,N-bis(Boc) protection provides enough steric and/or electron withdrawing effects to avoid potential decomposition.23 It is noteworthy that after the modified HWE reaction, we were unable to separate the bisPOM-vinylphosphonate nucleosides from excess tetra(POM)-bisphosphonate 5 by flash silica gel column chromatography (except in the case of the U analog; entry 1) and thus had to achieve subsequent deprotection with an 80% HCOOH/water solution to facilitate isolation. All attempts to use more traditional desilyation conditions (TBAF, TBAF/HOAc,. HF Et3N or 1M HCl in dioxane) led to significant or complete degradation of the substrates. Final hydrogenation using H2 and Pd/C24 provided the desired 5′-methylene bis(POM)-phosphonate nucleosides 21-25 in yields that ranged from 60 to 71% over 4 steps and 44 to 60% from the starting nucleosides. All five final phosphonate prodrugs were devoid of anti-HCV,25 anti-HIV, or cytotoxic activity.26,27
Table 1.
Conversion of protected 5′-hydroxyl nucleosides to 5′- methylene-bis(POM)-phosphonate prodrugs 21-25.
| entry | starting material | deprotection product | yielda (%) |
|---|---|---|---|
| 1 |
|
|
71 |
| 2 |
|
|
60 |
| 3 |
|
|
64 |
| 4 |
|
|
67 |
| 5 |
|
|
60 |
Isolated yield over 4 steps.
In conclusion, we report herein the first highly efficient synthesis of 5′-methylene bis(POM)-phosphonate prodrugs through a modified HWE reaction. The key HWE reaction of the sequence involves the addition of a tetra(POM) bisphosphonate sodium salt to Boc/TBDMS protected 5′-aldehydic nucleosides. The 2′-deoxy-2′-α-fluoro-2′-β-C-methyl-5′-methylene bis(POM)-phosphonate nucleoside prodrugs 21-25 were obtained with excellent overall yields that ranged from 60 to 71% over 4 steps from the corresponding protected starting materials. Through this work we have extended the scope of tetra substituted bisphosphonates that take part in a modified HWE reaction. The versatility of this approach could be utilized for the rapid synthesis of new and diverse phosphonate prodrugs with superior biological activity.
Supplementary Material
Scheme 3.

Synthesis of protected nucleosides 11, 14, 19 and 20.
Acknowledgment
This work was supported in part by NIH grant 5P30-AI-50409 (CFAR), and by the Department of Veterans Affairs. We thank Tami McBrayer and Mervi Detorio for the biological testing.
Footnotes
Supporting Information Available: Experimental procedures and full spectroscopic data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1(a).De Clercq E. Nat. Rev. Microbiol. 2004;2:704. doi: 10.1038/nrmicro975. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cihlar T, Ray AS. Antivir. Res. 2010;85:39. doi: 10.1016/j.antiviral.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 2.De Clercq E, Holý A. Nature Rev. Drug Discovery. 2005;4:928. doi: 10.1038/nrd1877. [DOI] [PubMed] [Google Scholar]
- 3(a).Topalis D, Pradere U, Roy V, Caillat C, Azzouzi A, Broggi J, Snoeck R, Andrei G, Lin J, Ericksson S, Alexandre JAC, El-Amri C, Deville-Bonne D, Meyer P, Balzarini J, Agrofoglio LA. J. Med. Chem. 2011;54:222. doi: 10.1021/jm1011462. [DOI] [PubMed] [Google Scholar]; (b) Pradere U, Clavier H, Roy V, Nolan SP, Agrofoglio LA. Eur. J. Org. Chem. 2011;36:1712. [Google Scholar]
- 4(a).For recent reports using this procedure see : Gallier F, Alexandre JAC, El Amri C, Deville-Bonne D, Peyrottes S, Perigaud C. Chem. Med. Chem. 2011;6:1094. doi: 10.1002/cmdc.201100068. Kim B-S, Kim B-T, Hwang K-J. Bull. Korean Chem. Soc. 2010;31:1643. Cosyn L, Van Calenbergh S, Joshi BV, Ko H, Carter RL, Harden TK, Jacobson KA. Bioorg. Med. Chem. Lett. 2009;19:3002. doi: 10.1016/j.bmcl.2009.04.027. Suk D-H, Bonnac L, Dykstra CC, Pankiewicz KW, Patterson SE. Bioorg. Med. Chem. Lett. 2007;17:2064. doi: 10.1016/j.bmcl.2007.01.014. Koh Y-H, Shim JH, Wu JZ, Zhong W, Hong Z, Girardet J-L. J. Med. Chem. 2005;48:2867. doi: 10.1021/jm049029u.
- 5(a).Jones GH, Moffatt JG. J. Am. Chem. Soc. 1968;90:5337. doi: 10.1021/ja01021a086. [DOI] [PubMed] [Google Scholar]; (b) Jones GH, Hamamura EK, Moffatt JG. Tetrahedron Lett. 1968;9:5731. [Google Scholar]
- 6(a).Padyukova NS, Karpeisky MY, Kolobushkina LI, Mikhailov SN. Tetrahedron Lett. 1987;28:3623. [PubMed] [Google Scholar]; (b) Mikhailov SN, Paoyukova NS, Karpeiskii MY, Kolobushkina LI, Beigelman LN. Collect. Czech. Chem. Comm. 1989;54:1055. [Google Scholar]; (c) Ioannidis P, Classon B, Samuelsson B, Kvarnstrom I. Acta Chem. Scand. 1991;45:746. doi: 10.3891/acta.chem.scand.45-0746. [DOI] [PubMed] [Google Scholar]
- 7(a).Tanaka H, Fukui M, Haraguchi K, Masaki M, Miyasaka T. Tetrahedron Lett. 1989;30:2567. [Google Scholar]; (b) Hutter D, Blaettler MO, Benner SA. Helvetica Chimica Acta. 2002;85:2777. [Google Scholar]; (c) Barral K, Priet S, De Michelis C, Sire J, Neyts J, Balzarini J, Canard B, Alvarez K. Eur. J. Med. Chem. 2010;45:849. doi: 10.1016/j.ejmech.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 8.Barton DHR, Gero SD, Quiclet-Sire B, Samadi M. J. Chem. Soc. Chem. Commun. 1989:1000. [Google Scholar]; (b) Barton DHR, Gero SD, Quiclet-Sire B, Samadi M. Tetrahedron Lett. 1989;30:4969. [Google Scholar]; (c) Barton DHR, Gero SD, Quiclet-Sire B, Samadi M. Tetrahedron. 1992;48:1627. [Google Scholar]
- 9.Montgomery JA, Hewson K. Chem. Commun. 1969:15. [Google Scholar]
- 10.Blackburn GM, Rashid A. J. Chem. Soc. Chem. Commun. 1989:40. [Google Scholar]
- 11.Nencka R, Sinnaeve D, Karalic I, Martins JC, Van Calenbergh S. Org. Biomol. Chem. 2010;8:5234. doi: 10.1039/c0ob00061b. [DOI] [PubMed] [Google Scholar]
- 12.Mackman RL, Ray AS, Hui HC, Zhang L, Birkus G, Boojamra CG, Desai MC, Douglas JL, Gao Y, Grant D, Laflamme G, Lin K-Y, Markevitch DY, Mishra R, McDermott M, Pakdaman R, Petrakovsky OV, Vela JE, Cihlar T. Bioorg. Med. Chem. 2010;18:3606. doi: 10.1016/j.bmc.2010.03.041. [DOI] [PubMed] [Google Scholar]
- 13.Sofia MJ, Bao D, Chang W, Du J, Nagarathnam D, Rachakonda S, Reddy PG, Ross BS, Wang P, Zhang H-R, Bansal S, Espiritu C, Keilman M, Lam AM, Micolochick Steuer HM, Niu C, Otto MJ, Furman PA. J. Med. Chem. 2010;53:7202. doi: 10.1021/jm100863x. [DOI] [PubMed] [Google Scholar]
- 14.This compound can now be found under the name GS-7977 as Gilead Sciences, Inc. acquired Pharmasset, Inc. in January 2012.
- 15.For a review of NS5B anti-HCV agents, see: Sofia MJ, Chang W, Furman PA, Mosley RT, Ross BS. J. Med. Chem. 2012;55:2481. doi: 10.1021/jm201384j.
- 16.Vepsäläinen JJ. Tetrahedron Lett. 1999;40:8491. [Google Scholar]
- 17.Troutman JM, Chehade KAH, Kiegiel K, Andres DA, Spielmann HP. Bioorg. Med. Chem. Lett. 2004;14:4979. doi: 10.1016/j.bmcl.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 18.Clark JL, Hollecker L, Mason JC, Stuyver LJ, Tharnish PM, Lostia S, McBrayer TR, Schinazi R, Watanabe KA, Otto MJ, Furman PA, Stec WJ, Patterson SE, Pankiewicz KW. J. Med. Chem. 2005;48:5504. doi: 10.1021/jm0502788. [DOI] [PubMed] [Google Scholar]
- 19.Zhu X-F, Williams HJ, Scott AI. Synthetic Comm. 2003;33:2011. [Google Scholar]
- 20.More JD, Finney NS. Org. Lett. 2002;4:3001. doi: 10.1021/ol026427n. [DOI] [PubMed] [Google Scholar]
- 21.Ikeuchi H, Meyer M;E, Ding Y, Hiratake J, Richards N;G. J. Bioorg. Med. Chem. 2009;17:6641. doi: 10.1016/j.bmc.2009.07.071. [DOI] [PubMed] [Google Scholar]
- 22(a).Du J, Nagarathnam D, Pamulapati GR, Ross BS, Sofia MJ. WO2009152095. [Google Scholar]; (b) Pamulapati GR, Chun B-K, Zhang H-R, Rachakonda S, Ross BS, Sofia MJ. J. Org. Chem. 2011;76:3782. doi: 10.1021/jo200060f. [DOI] [PubMed] [Google Scholar]; (c) Clark JL, Mason JC, Hollecker L, Stuyver LJ, Tharnish PM, McBrayer TR, Otto MJ, Furman PA, Schinazi RF, Watanabe KA. Bioorg. Med. Chem. Lett. 2006;16:1712. doi: 10.1016/j.bmcl.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 23.A reviewer has pointed out to us that generation of the 5′-aldehyde with purine nucleobases is problematic owing to cyclonucleoside formation with the N-3 amino group, which leads presumably to deglycosylation and other degradation products.
- 24.While 1 h completely reduced the vinylphosphonate, several hours were required for one pot removal of the benzyl group and the reduction of the azido group.
- 25.Stuyver LJ, Whitaker T, McBrayer TR, Hernandez-Santiago BI, Lostia S, Tharnish PM, Ramesh M, Chu CK, Jordan R, Shi J, Rachakonda S, Watanabe KA, Otto MJ, Schinazi RF. Antimicrob. Agents Chemother. 2003;47:244. doi: 10.1128/AAC.47.1.244-254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26(a).Schinazi RF, Sommadossi JP, Saalmann V, Cannon DL, Xie M-W, Hart GC, Smith GA, Hahn EF. Antimicrob. Agents Chemother. 1990;34:1061. doi: 10.1128/aac.34.6.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Stuyver LJ, Lostia S, Adams M, Mathew J, Pai BS, Grier J, Tharnish P, Choi Y, Chong Y, Choo H, Chu CK, Otto MJ, Schinazi RF. Antimicrob. Agents Chemother. 2002;46:3854. doi: 10.1128/AAC.46.12.3854-3860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.No significant antiviral activities were found for compounds 21-25 when tested up to 100 LM against HIV in peripheral blood mononuclear cells and up to 10 LM against HCV in a subgenomic RNA replicon system. No cytotoxicity was observed in Huh7 cells up to 10 LM or in PBM, human lymphoblastoid CEM, or African Green monkey Vero cells up to 100 LM.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.