

Abstract
Acromegaly resulting from the ectopic secretion of growth hormone-releasing hormone (GHRH) is rare. We present a case of acromegaly secondary to proven GHRH-secretion by a bronchial carcinoid tumor in a type 1 diabetic subject and document the clinical course pre- and post-resection of the tumor and of subsequent octreotide therapy. A 54-year-old Caucasian man was referred for evaluation of acromegalic symptoms and significantly increased insulin requirements. He had a history of left lung surgery 20 years prior for hemoptysis. Initial laboratory results indicated acromegaly. Fasting serum growth hormone (GH): 26.1 ng/mL (0–5 ng/mL), insulin-like growth factor 1 (IGF-1): 635 ng/mL (87–283 ng/mL), GH at 60 min post-ingestion of 75 grams of oral glucose during a glucose tolerance test: 8.3 ng/mL (normal<1 ng/mL). Pituitary magnetic resonance imaging (MRI) revealed diffuse pituitary enlargement without adenoma. A 4.4 cm left hilar mass was noted on chest computed tomography (CT) scan. Further evaluation for a suspected GHRH-secreting neuroendocrine tumor was pursued. Plasma GHRH level was elevated: 198 pg/mL (<50 pg/mL). Octreoscan showed radiolabelled-octreotide uptake in the left lung mass and pituitary gland. Surgical resection of the lung mass was performed. Immunohisto-chemical study of the tumor tissue indicated a neuroendocrine tumor secreting GHRH. Postoperatively, serum GHRH, GH and IGF-1 levels fell precipitously. At 10 months, IGF-1 levels were mildly elevated and 7 months of 10 mg long-acting octreotide therapy (Sandostatin® LAR®) was trialed. At 20 months, off octreotide, serum IGF-1 levels had normalized, acromegalic features were receding, and the patient's daily insulin requirements had decreased by 57%.
Keywords: Acromegaly, Ectopic acromegaly, Growth hormone-releasing hormone, Neuroendocrine tumor, Carcinoid, Somatostatin analogue, Octreotide
Introduction
Ectopic acromegaly, caused by growth hormone-releasing hormone (GHRH) secretion from neoplastic tissue acting on pituitary somatotrophs with consequent inappropriate growth hormone (GH) release, is a rarely encountered diagnosis, with estimations that it accounts for 0.5–5% of all cases of acromegaly [1, 2]. The first well-documented association of an extrapituitary neuroendocrine tumor and acromegaly was published in 1959 [3]. It was subsequently shown that extracts from such tumors had in vitro GH-releasing activity [4]. GHRH was characterized in 1982 following the isolation and sequencing of the peptide from two patients with GHRH-secreting pancreatic neuroendocrine tumors [5, 6], investigators had previously been hindered by the inability to isolate sufficient amounts of this “growth hormone-releasing factor” from normal brain tissue. The investigational quests that led to the elucidation of GHRH are fascinating stories in their own right [7].
We describe a patient with type 1 diabetes who presented with signs and symptoms of acromegaly, an enlarged pituitary gland without a discrete adenoma on magnetic resonance imaging (MRI), and a distant history of lung surgery for a benign tumor, who was subsequently diagnosed with ectopic GHRH secretion by a bronchial carcinoid tumor. Surgical resection of the bronchial carcinoid tumor resulted in biochemical and clinical remission of acromegaly.
Case history
Clinical presentation
A 54-year-old Caucasian man was referred to the National Institutes of Health for evaluation of acromegaly. While following the patient for type 1 diabetes mellitus, his referring physician had documented the development of acromegalic features including hand and foot enlargement, coarsening of facial features, deepening of the voice and increasing insulin requirements. On detailed questioning the patient described gradually enlarging extremities over more than 20 years with an increase in shoe size from 9.5 to 12.5 and the need to increase his wedding ring size. Additional symptoms included fatigue, worsening snoring, decreased exercise tolerance, joint aches and weight gain of 20 lb over the prior 12 months. Medications were insulin glargine 120 units daily, regular insulin 42 units twice daily, ramipril 2.5 mg daily and aspirin 81 mg daily.
Past medical history was significant for stage 1 hypertension and a 45 year history of type 1 diabetes mellitus. Past surgical history was notable for a left lung upper lobectomy for hemoptysis in 1982; the patient had been told that a benign tumor was resected but had not had any further follow-up; documentation of this procedure and surrounding medical evaluation was unavailable. The family and social history were unremarkable.
Physical examination revealed a Caucasian man of 160 cm height and 114 kg weight with a blood pressure of 144/85; a striking acromegaloid facial appearance with frontal bossing, prominence of the supraorbital ridges, large fleshy nose and prognathism. Ear nose and throat exam revealed marked interdental spacing, macroglossia and fleshy narrowing of the external auditory canal. Chest exam revealed a well-healed left thoracotomy scar. Both hands and feet were enlarged with doughy soft-tissue swelling of the digits. Multiple skin tags were present, most prominent on the upper torso. The remainder of the exam was unremarkable.
Biochemical examinations
Biochemical evaluation revealed a fasting glucose of 118 mg/dL (70–115 mg/dL), hematocrit: 43.1% (36.7–48.3%), blood urea nitrogen: 16 mg/dL (8–22 mg/dL): serum creatinine: 0.9 mg/dL (0.9–1.4 mg/dL), fasting serum GH: 26.1 ng/mL (0–5 ng/mL), insulin-like growth factor 1 (IGF-1): 635 ng/mL (87–283 ng/mL), GH at 60 min post-ingestion of 75 grams of oral glucose during a glucose tolerance test: 8.3 ng/mL (normal <1 ng/mL), prolactin: 11 mcg/L (1–25 mcg/L), follicle stimulating hormone: 8 U/L (2–8 U/L), luteinizing hormone: 2 U/L (2–8 U/L), total testosterone: 274 ng/dL (240–950 ng/dL), thyroid stimulating hormone: 3.36 mcIU/mL (0.40–4.00 mcIU/mL), free thyroxine: 1.2 ng/dL (0.8–1.9 ng/dL), chromogranin A: 470 ng/mL (0–225 ng/mL), whole blood serotonin: 552 ng/mL (<330 ng/mL), urinary 5-hydroxyindoleacetic acid (5-HIAA): 10 mg/24 h (1–6 mg/24 h) and hemoglobin A1C: 7.1% (4.8–6.4%).
Imaging studies
Pituitary magnetic resonance imaging (MRI) scan revealed a diffusely enlarged gland without focal lesion. High resolution computed tomography (CT) of the chest with virtual bronchoscopy revealed surgical changes at the left hilum with an adjacent 4.4 cm contrast-enhancing mass without mediastinal lymphadenopathy. There was hyperinflation of the remaining left lung and areas of pleural and parenchymal scarring. CT and MRI imaging of the abdomen and pelvis were unremarkable. Whole-body radiolabelled-octreotide scan showed intense radiolabelled-octreotide uptake in the left hilar region and in the pituitary (Fig. 1).
Fig. 1.
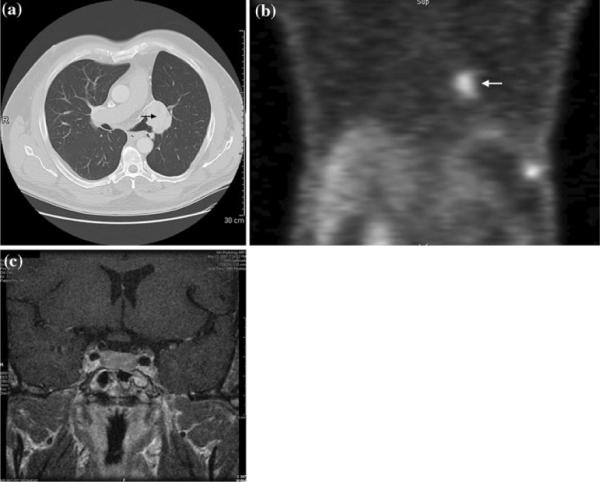
Imaging findings: a computed tomography scan of the chest revealing left hilar mass. b Octreotide scan demonstrating intense radiolabelled-octreotide uptake in the left lung near the mediastinal margin (marked by white arrow). The area of uptake in the left subdiaphragmatic region represents uptake by an accessory spleen (unmarked). c Coronal pituitary MRI demonstrating homogenously enlarged pituitary gland, without adenoma
Serum GHRH level
Because the above presentation suggested ectopic GHRH secretion from a presumably recurrent bronchial neuroendocrine tumor, the serum GHRH concentration was determined and was found to be markedly elevated at 198 pg/mL (<50 pg/mL).
Surgical resection of lung mass
Following bronchoscopic evaluation, surgical resection of the tumor was performed. In order to achieve complete tumor resection, a left pneumonectomy was performed via a posterolateral thoracotomy. Extensive adhesions were noted from prior surgery. Intraoperative frozen section examination of a left hilar mass revealed neuroendocrine tumor. Post-operative recovery was uneventful.
Histopathological studies
Histological analysis confirmed a 3.3 cm bronchial neuroendocrine tumor with clear surgical margins, staining positive for synaptophysin and chromogranin. Immunohistochemical staining with anti-GHRH antibody was strongly positive for GHRH (Fig. 2) and negative for GH.
Fig. 2.
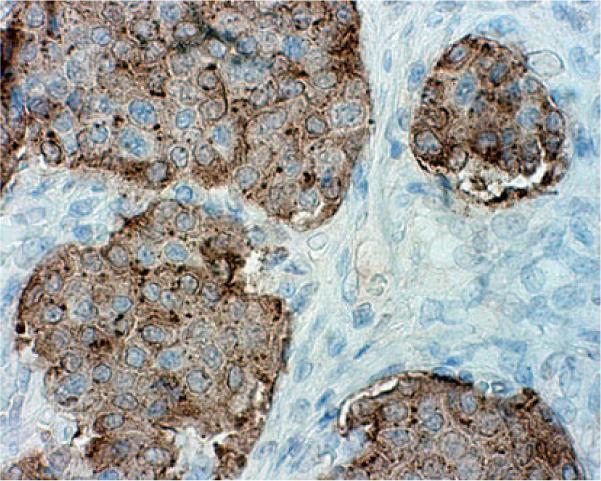
Histological findings: immunohistochemical stain for GHRH on surgically resected bronchial tumor (×400) demonstrating strong positivity for anti-GHRH antibody
Clinical response and follow-up
Post-operatively, serum levels of IGF-1, GH and GHRH fell rapidly. Twelve months later, on re-evaluation, the patient had marked improvement of soft tissue swelling and a decrease in daily insulin requirement by 57%. Fasting GH was measured at 4.9 ng/mL (0–5 ng/mL), IGF-1 at 334 ng/mL (87–283 ng/mL), and GHRH at 11 pg/mL (<50 pg/mL). Chromogranin A level was in the normal range at 190 ng/mL (<225 ng/mL). Repeat chest CT excluded local or metastatic tumor recurrence. Whole body octreotide scanning was negative for uptake in the thorax but did document mild increased uptake in the region of the pituitary.
Because serum IGF-1 and GH levels remained mildly elevated at 10 months, long-acting octreotide (Sandostatin® LAR®) therapy was started at a conservative dose of 10 mg intramuscularly monthly. Following 7 months of treatment, IGF-1 levels were within the normal range and octreotide was discontinued as a trial off-therapy. Four months following the discontinuation of octreotide, serum IGF-1, GH and GHRH levels were within the normal range and the decision was made to observe the patient without further octreotide treatment.
The patient continues to make excellent progress and has resumed his previously active life and full employment.
Methods
Biochemical measurements
IGF-1 and GH levels were determined at the NIH Department of Laboratory Medicine, Bethesda, MD by Chemiluminescence immunoassay. Non-NIH Samples for IGF-1 levels were processed at Mayo Medical Laboratories, Rochester by chemiluminescence immunoassay and for GH at Quest Diagnostics® by chemiluminescence immunoassay.
Serum GHRH levels on NIH samples were determined at Inter Science Institute, Inglewood, CA by direct radioimmunoassay. Non-NIH samples for GHRH were processed at Quest Diagnostics® Nichols Institute, San Juan Capistrano, CA by extraction radioimmunoassay.
Histopathologic analysis
Paraffin tissue blocks were cut into 4 mm-thick sections. The sections were then deparaffinized in xylene and rehydrated in graded alcohol concentrations. Endogenous peroxidase was quenched with 0.03% H2O2 (EnVision™ System, Peroxidase, DAKO Co, CA) for 5 min. Antibodies against GH were provided by the National Institute of Diabetes and Digestive and Kidney Diseases and against GHRH by Dr. Lawrence A. Frohman from the University of Illinois at Chicago.
After incubation with the primary antibody, sections were incubated with a secondary antibody conjugated with HRP-labeled polymer (EnVision™ System, Peroxidase, DAKO Co, CA) for 30 min at room temperature, followed by development with diaminobenzidine solution. The slides were washed in phosphate buffered saline (3 times for 5 min each) between each incubation step. After counterstaining with hematoxylin, the slides were mounted with coverslips in xylene-based mounting medium (Cyto-seal, Stephens Scientific, Kalamazoo, MI). Negative controls, in which incubation with the primary antibody was omitted, were processed along with all the samples.
Discussion
The majority of acromegaly cases are caused by over-secretion of GH by a pituitary adenoma. Extra-pituitary acromegaly results from pathologic GHRH secretion by either an extra-hypothalamic neuroendocrine tumor or, less frequently, a hypothalamic gangliocytoma (termed eutopic GHRH secretion) [8]. Bronchial and gastrointestinal carcinoids are the most commonly associated tumor, followed by pancreatic neuroendocrine tumors (including patients with Multiple Endocrine Neoplasia type 1 [9]) and rarely foregut neuroendocrine tumors [10] and pheochromocytomas [11].
The signs and symptoms produced by ectopic acromegaly have been found to be the same as in classic acromegaly [1, 2]; in ectopic acromegaly patients may also report symptoms resulting from the primary tumor (e.g. flushing and/or hemoptysis in bronchial carcinoid [12]) or an associated syndrome (e.g. kidney stones in MEN 1 [13]).
IGF-1 levels in ectopic acromegaly, when reported, have been elevated [14]. Following a confirmatory oral glucose tolerance test, the next step in the diagnostic evaluation of acromegaly is a pituitary MRI. It is at this point that the diagnosis of ectopic acromegaly is often first suspected. The absence of a discrete pituitary adenoma is a strong indication to pursue further evaluation for ectopic GHRH syndrome [9]. Published case series have shown, however, that as in our patient, the pituitary is often abnormal on imaging. In one series, the pituitary was normal or homogenously enlarged in 58% of the cases but imaging was suggestive of a pituitary adenoma in 29% [14]. This has frequently led to mistaken pituitary surgery.
The most common pituitary histological finding in cases of ectopic acromegaly, in which pituitary tissue has been excised, is somatotroph hyperplasia resulting from prolonged GHRH stimulation [15]. In some cases pituitary tissue is more characteristic of true neoplastic transformation however [16]. The finding of somatotroph hyperplasia under chronic GHRH stimulation has been documented in murine models [17].
The key differentiator in ectopic acromegaly is a serum GHRH level, which is elevated in all reported cases of proven ectopic acromegaly [1, 2, 14]. In classic acromegaly, GHRH levels are normal or suppressed by GH negative feedback at the hypothalamic level [18].
Once ectopic acromegaly is suspected, chest and abdominal imaging will frequently reveal the causative lesion or lesions. Octreotide scan, using radiolabelled somatostatin scintigraphy, confirmed the presence of neuroendocrine tumor in our patient and has been shown to be positive in other recent cases [12].
Definitive diagnosis and proof of causality is achieved following surgical resection of the GHRH-secreting tumor. GHRH and GH levels should fall rapidly post-operatively. Histology is most often that of a non-specific neuroendocrine tumor, tumor tissue will stain for GHRH but usually not for GH [9] (Fig. 2).
The treatment of acromegaly due to ectopic GHRH production has two major components: therapy directed at the underlying neuroendocrine tumor and/or therapy directed at the effects of excess IGF-1 production.
Optimal treatment of ectopic acromegaly is complete surgical resection of the GHRH-secreting neuroendocrine tumor. In our patient, left pneumonectomy, with histological evidence of complete carcinoid resection, brought about a dramatic and sustained decrease in GHRH, GH and IGF-1 levels. Further, interval anatomical and functional imaging has shown the patient to be free of neuroendocrine tumor. As evidenced by the regression of soft-tissue swelling and a dramatic decrease in total daily insulin requirement, the secondary effects of hypersomatotropinemia are also improving. Historically, however, either by the time acromegaly is diagnosed, or recognized in a patient with known neuroendocrine tumors, the neuroendocrine tumor is frequently found to be metastatic and unresectable. In one series, 8 of 31 patients with ectopic acromegaly were found to have disseminated disease at diagnosis [14]. In this situation, medical therapy with somatostatin analogues is the mainstay of treatment.
There is well-established experience in the ectopic acromegaly literature in the use of somatostatin analogues, both in cases in which the causative tumor is non-resectable or disseminated and in persistent IGF-1 elevation following surgery [10, 19].
Somatostatin has an inhibitory effect on the secretion of GHRH by neuroendocrine tumors through the activation of somatostatin receptors in tumor tissue [20]. The presence of tumor somatostatin receptors is evidenced by strong radiolabelled somatostain uptake both in our patient and in several reported cases [12, 21]. Exogenous somatostatin has been shown to reduce the secretion of both pituitary GH and tumor GHRH in vivo and in cultures of carcinoid tissue in a patient with ectopic acromegaly and pituitary tissue in patients with GH secreting pituitary adenomas [12, 22, 23]. Some cases of ectopic acromegaly respond better to somatostatin analogues than others and increasingly, research is focusing on the subtypes of somatostatin receptors expressed in target tissue to explain the degree of response and to optimize treatment with agents more specific to a certain receptor (e.g. SOM230 in tumors with high expression of somatostatin receptor subtypes 1,3 and 5 [12]).
Conclusions
This case provides an excellent illustration of the unusual and intriguing condition of ectopic acromegaly.
Complete resection of the underlying bronchial carcinoid tumor restored serum GHRH, GH and IGF-1 levels to normal and the clinical effects of acromegaly are receding. The borderline elevated levels of GH and IGF-1 seen during follow-up are possibly due to the effects of pituitary somatotroph hyperplasia evidenced by our MRI findings of homogenous pituitary enlargement at presentation and radiolabelled octreotide pituitary-uptake at presentation and 12 months post-surgery.
In cases of acromegaly with pituitary imaging equivocal for adenoma, a serum GHRH level should be determined to exclude ectopic acromegaly, and to spare the patient unnecessary pituitary surgery.
Acknowledgments
The authors wish to acknowledge the generous assistance of Lawrence A. Frohman, M.D., University of Illinois at Chicago, in providing the anti-GHRH antibodies used in immunohistochemical analysis. This work was funded by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases and the Eunice Kennedy Shriver National Institutes of Child Health and Human Development at the National Institutes of Health.
References
- 1.Melmed S. Extrapituitary acromegaly. Endocrinol Metab Clin North Am. 1991;20:507–518. [PubMed] [Google Scholar]
- 2.Faglia G, Arosio M, Bazzoni N. Ectopic acromegaly. Endocrinol Metab Clin North Am. 1992;21:575–595. [PubMed] [Google Scholar]
- 3.Altmann HW, Schuetz W. On a bronchus carcinoid containing bone (morphological and clinical observations in an acromegalic patient) Beitr Pathol Anat. 1959;120:455–473. [PubMed] [Google Scholar]
- 4.Shalet SM, Beardwell CG, MacFarlane IA, Ellison ML, Norman CM, Rees LH, Hughes M. Acromegaly due to production of a growth hormone releasing factor by a bronchial carcinoid tumor. Clin Endocrinol (Oxf) 1979;10:61–67. doi: 10.1111/j.1365-2265.1979.tb03034.x. [DOI] [PubMed] [Google Scholar]
- 5.Guillemin R, Brazeau P, Bohlen P, Esch F, Ling N, Wehrenberg WB. Growth-hormone releasing-factor from a human pancreatic tumor that caused acromegaly. Science. 1982;218:585–587. doi: 10.1126/science.6812220. [DOI] [PubMed] [Google Scholar]
- 6.Rivier J, Spiess J, Thorner M, Vale W. Characterization of a growth hormone-releasing factor from a human pancreatic islet tumour. Nature. 1982;300:276–278. doi: 10.1038/300276a0. [DOI] [PubMed] [Google Scholar]
- 7.Thorner MO. The discovery of growth hormone-releasing hormone. J Clin Endocrinol Metab. 1999;84:4671–4676. doi: 10.1210/jcem.84.12.6210. [DOI] [PubMed] [Google Scholar]
- 8.Asa SL, Scheithauer BW, Bilbao JM, Horvath E, Ryan N, Kovacs K, Randall RV, Laws ER, Jr, Singer W, Linfoot JA, et al. A case for hypothalamic acromegaly: a clinicopatho-logical study of six patients with hypothalamic gangliocytomas producing growth hormone-releasing factor. J Clin Endocrinol Metab. 1984;58:796–803. doi: 10.1210/jcem-58-5-796. [DOI] [PubMed] [Google Scholar]
- 9.Losa M, von Werder K. Pathophysiology and clinical aspects of the ectopic GH-releasing hormone syndrome. Clin Endocrinol (Oxf) 1997;47:123–135. doi: 10.1046/j.1365-2265.1997.2311048.x. [DOI] [PubMed] [Google Scholar]
- 10.Vonwerder K, Losa M, Muller OA, Schweiberer L, Fahlbusch R, Delpozo E. Treatment of metastasizing Grf-producing tumor with a long-acting somatostatin analog. Lancet. 1984;2:282–283. doi: 10.1016/s0140-6736(84)90320-9. [DOI] [PubMed] [Google Scholar]
- 11.Neto LV, Taboada GF, Correa LL, Polo J, Nascimento AF, Chimelli L, Rumilla K, Gadelha MR. Acromegaly secondary to growth hormone-releasing hormone secreted by an incidentally discovered pheochromocytoma. Endocr Pathol. 2007;18:46–52. doi: 10.1007/s12022-007-0006-8. doi:10.1007/s12022-007-0006-8. ISSN1046-3976. [DOI] [PubMed] [Google Scholar]
- 12.van Hoek M, Hofland LJ, de Rijke YB, van Nederveen FH, de Krijger RR, van Koetsveld PM, Lamberts SW, van der Lely AJ, de Herder WW, Feelders RA. Effects of somatostatin analogs on a growth hormone-releasing hormone secreting bronchial carcinoid, in vivo and in vitro studies. J Clin Endocrinol Metab. 2009;94:428–433. doi: 10.1210/jc.2008-1712. [DOI] [PubMed] [Google Scholar]
- 13.Suga K, Yamashita N, Chiba K, Ito T, Kaziwara Y, Yokoyama N. Multiple endocrine neoplasia type 1 producing growth hormone-releasing factor in an endocrine pancreatic tumor. Acta Med Nagasaki. 2002;47:61–65. [Google Scholar]
- 14.Losa M, Schopohl J, von Werder K. Ectopic secretion of growth hormone-releasing hormone in man. J Endocrinol Invest. 1993;16:69–81. doi: 10.1007/BF03345835. [DOI] [PubMed] [Google Scholar]
- 15.Ezzat S, Asa SL, Stefaneanu L, Whittom R, Smyth HS, Horvath E, Kovacs K, Frohman LA. Somatotroph hyperplasia without pituitary adenoma associated with a long standing growth hormone-releasing hormone-producing bronchial carcinoid. J Clin Endocrinol Metab. 1994;78:555–560. doi: 10.1210/jcem.78.3.8126126. [DOI] [PubMed] [Google Scholar]
- 16.Nasr C, Mason A, Mayberg M, Staugaitis SM, Asa SL. Acromegaly and somatotroph hyperplasia with adenomatous transformation due to pituitary metastasis of a growth hormone-releasing hormone-secreting pulmonary endocrine carcinoma. J Clin Endocrinol Metab. 2006;91:4776–4780. doi: 10.1210/jc.2006-0610. [DOI] [PubMed] [Google Scholar]
- 17.Kineman RD, Teixeira LT, Amargo GV, Coschigano KT, Kop-chick JJ, Frohman LA. The effect of GHRH on somatotrope hyperplasia and tumor formation in the presence and absence of GH signaling. Endocrinology. 2001;142:3764–3773. doi: 10.1210/endo.142.9.8382. [DOI] [PubMed] [Google Scholar]
- 18.Mayo KE. Molecular-cloning and expression of a pituitary-specific receptor for growth hormone-releasing hormone. Mol Endocrinol. 1992;6:1734–1744. doi: 10.1210/mend.6.10.1333056. [DOI] [PubMed] [Google Scholar]
- 19.Barkan AL, Shenker Y, Grekin RJ, Vale WW. Acromegaly from ectopic growth hormone-releasing hormone secretion by a malignant carcinoid tumor. Successful treatment with long-acting somatostatin analogue SMS 201–995. Cancer. 1988;61:221–226. doi: 10.1002/1097-0142(19880115)61:2<221::aid-cncr2820610203>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 20.Bertherat J, Turpin G, Rauch C, Li JY, Epelbaum J, Sassolas G, Schaison G. Presence of somatostatin receptors negatively coupled to adenylate cyclase in ectopic growth hormone-releasing hormone- and alpha-subunit-secreting tumors from acromegalic patients responsive to octreotide. J Clin Endocrinol Metab. 1994;79:1457–1464. doi: 10.1210/jcem.79.5.7962343. [DOI] [PubMed] [Google Scholar]
- 21.Morel O, Giraud P, Bernier MO, Le Jeune JJ, Rohmer V, Jallet P. Ectopic acromegaly: localization of the pituitary growth hormone-releasing hormone producing tumor by In-111 pentetreotide scintigraphy and report of two cases. Clin Nucl Med. 2004;29:841–843. doi: 10.1097/00003072-200412000-00025. [DOI] [PubMed] [Google Scholar]
- 22.Zatelli MC, Maffei P, Piccin D, Martini C, Rea F, Rubello D, Margutti A, Culler MD, Sicolo N, Degli Uberti EC. Somatostatin analogs in vitro effects in a growth hormone-releasing hormone-secreting bronchial carcinoid. J Clin Endocrinol Metab. 2005;90:2104–2109. doi: 10.1210/jc.2004-2156. [DOI] [PubMed] [Google Scholar]
- 23.Melmed S, Ziel FH, Braunstein GD, Downs T, Frohman LA. Medical management of acromegaly due to ectopic production of growth hormone-releasing hormone by a carcinoid tumor. J Clin Endocrinol Metab. 1988;67:395–399. doi: 10.1210/jcem-67-2-395. [DOI] [PubMed] [Google Scholar]