

Abstract
Background
In psoriasis, inflammation and epidermal hyperplasia are thought to be controlled by T cell-derived cytokines. Evidence suggests that the Th17 cell cytokine interleukin-17 (IL-17) may play a role in disease pathogenesis.
Objective
To understand the impact that neutralization of IL-17 has on the clinical features of psoriasis and to understand the role that IL-17 has in inflammatory pathways underlying psoriasis in human subjects.
Methods
We examined skin lesions obtained from 40 subjects participating in a phase 1, randomized, double-blind, placebo-controlled trial of an anti-IL-17 monoclonal antibody, ixekizumab (previously LY2439821), in which subjects received subcutaneous 5mg, 15mg, 50mg or 150mg ixekizumab or placebo at weeks 0, 2, and 4.
Results
There were significant, dose-dependent reductions from baseline in keratinocyte proliferation, hyperplasia, epidermal thickness, infiltration into the dermis and epidermis by T cells and dendritic cells and keratinocyte expression of innate defense peptides at 2 weeks. By week 6, the skin appeared normal. Quantitative reverse transcriptase polymerase chain reaction and microarrays revealed an ablation of the disease-defining mRNA expression profile by 2 weeks after the first dose of study drug. The effect of IL-17 blockade on expression of genes synergistically regulated by IL-17 and Tumor necrosis factor (TNF) was of higher magnitude at 2 weeks than in prior studies with TNF antagonism.
Conclusion
Our data suggest that IL-17 is a key “driver” cytokine in psoriasis that activates pathogenic inflammation. Neutralizing IL-17 with ixekizumab may be a successful therapeutic strategy.
Keywords: Psoriasis, interleukin -17, Th17 cells, TNF
Introduction
Psoriasis is an incurable, inflammatory autoimmune skin disease with an estimated prevalence in Western populations of approximately 3% (1). Histologically, the hallmark of psoriasis is the presence of a greatly thickened, nucleated keratinocyte layer, with exaggeration of the rete pegs, caused by hyperproliferation of keratinocytes and dermal infiltration by activated T cells, neutrophils and dendritic cells(2). Examination of infiltrates in psoriatic lesions has revealed significant increases in CD4+ T cell subsets, Th1, Th17, and Th22 and CD8+ Tc17 and Tc22 cells (3-7).
These T cell subsets induce elements of the inflammatory response underlying psoriasis pathogenesis by expressing a set of inflammatory cytokines. Interleukin-17 (IL-17A), highly expressed by Th17 cells, has a direct effect on the regulation of genes expressed by keratinocytes which are involved in innate immune defense, including defensins (8;9), S100 family proteins, lipocalin and LL37/cathelicidin, as well as a range of CXCL chemokines that regulate neutrophil trafficking (10;11). IL-22, expressed by Th22 and Th17 cells, and related IL-20 family members promote keratinocyte hyperproliferation and abnormal differentiation (12-15). The Th1 cell derived interferon-γ (IFN-γ) may be a key inducer of several hundred IFN-regulated gene products, including STAT1, that are strongly up-regulated in the psoriasis transcriptome.
The role of these individual T cell subsets in human disease is not yet certain. However, there is now evidence that IL-23 plays an important role in psoriasis pathogenesis, and IL-23 is known to promote a Th17 response (16). IL-23 is a cytokine produced by antigen presenting cells and is required for activation and survival of Th17 cells. IL-23 has been shown to be significantly increased in psoriatic lesions (17;18). Recently, certain variants of the genes that encode p19 and p40 subunits of IL-23 and the IL-23 receptor (19;20) have been linked to genetic susceptibility of psoriasis. In addition, IL-23 receptor variants associated with psoriasis strongly affect the generation of Th17 T cells from naïve precursors in vitro (21).
Th17 T cells are defined by their ability to secrete IL-17A (IL-17) and IL-17F. In mice, Th17 cells also secrete IL-22, while in humans most IL-22 is secreted from a separate Th22 population. Both IL-17 and IL-22 are highly expressed in skin affected by psoriasis. The relative contributions of these two cytokines to disease pathogenesis is currently debated. Studies in mouse model systems indicate that IL-22 is required for the psoriasis phenotype (22;23), and in human in vitro models, IL-22 can induce expression of psoriasis associated genes and acanthosis (13;24). IL-17, however, may have a role distinct from IL-22, as signaling pathways are distinct and each cytokine has distinguishable effects on gene transcription in human keratinocytes (25). One clinical trial evaluating the efficacy of an antibody neutralizing IL- 17A, AIN457, showed improvement of psoriasis in a subset of subjects (54% improvement in Psoriasis Area and Severity Index [PASI] over placebo) but demonstrated incomplete resolution of the clinical and pathological phenotype with a single dose of antibody (26). A phase 2 clinical trial of another monoclonal antibody neutralizing IL-17A, ixekizumab, showed higher levels of efficacy over 12 weeks of treatment(27). Accordingly, the pathogenic functions of cytokines contributed by Th1, Th17, and Th22 T-cell subsets in psoriasis vulgaris are still incompletely defined at present as is therapeutic mechanism of IL17 blockade in psoriasis
Here we show that neutralizing IL-17 with multiple doses of a subcutaneous ixekizumab, a humanized IgG4 monoclonal antibody that selectively binds and neutralizes IL-17A, in subjects with psoriasis results in consistent, rapid and significant improvements in clinical measures of disease consistent with the phase 2 trial data, as well as a rapid reversal of regenerative epidermal hyperplasia, keratinocyte proliferation and dermal infiltration of leukocytes. Furthermore, our analysis of gene expression using reverse transcriptase polymerase chain reaction (RT-PCR) and microarrays suggests that IL-17 neutralization may suppress signaling through numerous inflammatory circuits by inhibiting expression of cytokines from multiple T cell subsets, as well as chemokines, and antimicrobial proteins from keratinocytes. Expression of many disease-related genes was strongly suppressed by ixekizumab.The largest effects observed were on genes regulated by IL-17 or coregulated by IL-17 and tumor necrosis factor α (TNF) and, importantly, these effects were larger than those seen with TNF neutralization with etanercept(28). Overall, these data suggest psoriasis vulgaris is largely a disease mediated by IL-17 and/or Th17 T cells.
Methods
Study Design and Subjects
A total of 46 subjects with chronic moderate to severe plaque psoriasis participated in a phase 1, randomized, subject- and investigator-blinded, placebo-controlled, dose-escalation study of ixekizumab, an anti-IL-17 monoclonal antibody. Subjects were randomized to groups receiving subcutaneous injections of 5 mg (n=8), 15 mg (n=8), 50 mg (n=8), or 150 mg (n=8) of ixekizumab, or placebo (n=8), or to receive intravenous infusions of 15 mg (n=5) ixekizumab or placebo (n=1) (intravenous treatment results not reported here). Subjects received 3 doses of study drug, at weeks 0, 2, and 4. Subjects were evaluated for safety throughout the duration of the trial (through week 20) and for efficacy at weeks 2, 4, 6, 12, 16 and 20. Forty subjects completed all three subcutaneous injections of study drug. The clinical trial was conducted according to the principles expressed in the Declaration of Helsinki and informed consent for their information to be stored in the hospital database and used for research was obtained from all subjects in written form. This study protocol was approved by ethical review boards at sites conducting this study.
Skin biopsies
From each subject, punch skin biopsies (2 side-by-side 4-mm biopsies) were obtained from a selected psoriatic lesion. Repeat biopsies were taken from the same lesional area at baseline, week 2, and week 6. One of the specimens was placed in a cryotube and the other was placed in cryopreservation embedding medium and flash frozen in liquid nitrogen. Both were stored at approximately -70°C.
Histology
Tissue sections were stained with hematoxylin (Fisher) and eosin (Shandon) (H&E) and with mouse anti-human monocolonal antibodies to: CD3, clone SK7 (BD); CD11c, clone B-Ly6 (BD); K16, clone Ks8.12 (Sigma); Ki67, clone Mib-1 (Santa-Cruz); LL37/cathelicidin, clone OSX12 (Abcam); S100A7, clone 47C1068 (Abcam); S100A8, clone 29396a (Lifespan Biosciences); BD2-Beta defensin, goat antibody (Peprotech); and DC-LAMP, clone 1404.G4 (BD). A secondary biotin-labeled horse anti-mouse Ab (Vector Laboratories) was amplified with the avidin-biotin complex (Vector Laboratories). 3-Amino-9-ethylcarbazole (Sigma-Aldrich) was the chromogen used. Images were obtained with a Nikon Eclipse 50i with Nikon DS-Fi1 camera using a Plan Apo lens (magnification 10× /0.45 WD 4.0) at room temperature. Image analysis for cell counts was done with NIH Image J1.43u. Composite images were created in Adobe Photoshop.
RT-PCR
RNA was extracted using the RNeasy Mini Kit (Qiagen) and on-column DNAse digestion (RNAse-free DNAse Set, Qiagen) and used for either RT-PCR or gene array. RT-PCR was performed using EZ PCR core reagents, primers, and probes (Applied Biosystems) as previously published (29). Quantitative results were compared using a Wilcoxon signed rank test.
Gene array
RNA was amplified using the NuGen Ovation® RNA Amplification System V2 and Ovation Whole Blood Kit. cDNA was then purified using Qiagen QIAquick. Purified cDNA was then fragmented using NuGen Biotin Module. The fragmented cDNA was hybridized to an Affymetrix array (Affymetrix HGU133a 2.0 chip, Affymetrix). The microarray data is deposited in GEO (accession number: GSE31652).
Statistical Analyses
Microarray data were analyzed using the R language (www.R-project.org) and Bioconductor packages. The Harshlight package (30) was used to scan Affymetrix chips for spatial artifacts. Expression values were obtained using the GCRMA (GC Robust MultiArray Averaging) procedure. Quality Control was carried out using Bioconductor package (arrayQualityControl).
In order to identify genes in which expression is significantly altered by ixekizumab and placebo treatments, expression values were modeled using mixed-effect models using the framework of Bioconductor's limma package. Variables for ‘week’ and ‘group’ were included as fixed effects with a random intercept for each subject. Comparisons were assessed using a moderated Student's paired t-test followed by multiple correcting using the Benjamini-Hochberg procedure, which controls the false discovery rate. Hierarchical clustering was used to create the heatmap presented in Figure 3, using euclidean distance and the Mcquitty agglomeration algorithm.
Figure 3. Identification of genes modulated >6 fold by IL-17 neutralization.
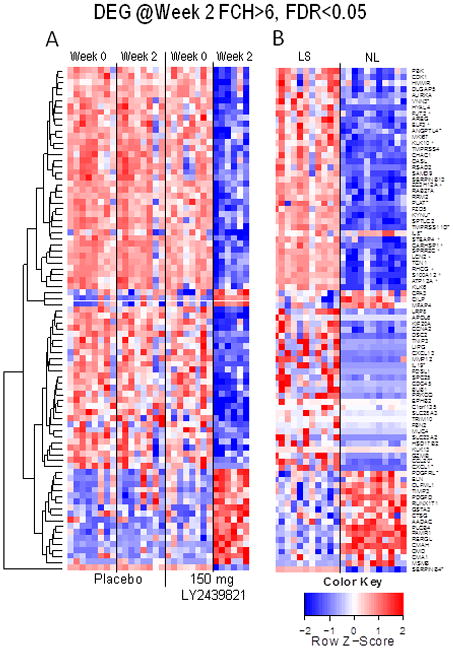
A. Differentially expressed genes that had a >6 fold change compared to baseline in the 150 mg group were identified and clustered. mRNA expression of these genes is presented for the placebo group for comparison. B. Gene expression data for untreated lesional skin biopsies (LS) and nonlesional skin biopsies (NS) from a previous experiment (28). The genes are ordered according to the clustering results in A. Asterisks represent those genes that are synergistically regulated by TNF and IL-17. Plus signs indicate those that are additively coregulated by TNF and IL-17.
To compare the treatment effect of ixekizumab 150 mg and etanercept 50 mg on the expression of genes associated with psoriasis, we used gene expression data from a previous study (28). This data is available in the Geo Omnibus repository under accession number (GSE11903). The objective was to compare the changes induced by both agents after 2 weeks of treatment for those genes that define the psoriasis phenotype from the genomic standpoint (the psoriatic transcriptome), and to compare the treatment effects to untreated lesional biopsies and nonlesional biopsies. There are several ‘psoriatic transcriptomes’ that have been previously reported (31-34). Here we use the transcriptome described in Suarez-Farinas et al. (32) because the samples in that study and the one reported here were hybridized in the same facility and because both studies use the same hgu133a2 chip.
In order to eliminate any batch effect, we took several steps to normalize the data. In order to be consistent with the published values of the psoriasis transcriptome and the etanercept-induced changes in terms of number of differentially expressed genes (DEGs) and fold change (FCH) measured in lesional and nonlesional biopsies at week 2 and week 0, respectively, we used the gcrma-derived expression values of the etanercept experiment as a target density and normalized the gcrma-derived expression values of the present study to this target density. This ensured that data derived from all chips would have the same overall density as the published study. Figure S5 shows that after improvement, a strong Batch effect is observed in the expression values and to a lesser degree in the FCH between week 2 and baseline, which is the measurement of interest. After adjusting for Batch effect using the program Combat (35), the Batch effect was eliminated from the expression values.
To define the Residual Disease Genomic Profile (RDGP), we calculated the improvement score for each gene as described in (32). This score is a number indicating the percentage of the initial pathology that is resolved by treatment. 100% corresponds to complete resolution and negative values imply that the treatment increases the aberrant expression of the gene. Genes with an improvement score smaller than 75% are part of the RDGP.
We calculated the improvement at week 2 for each gene, then generated an improvement score for each gene-set. Gene-sets were defined as those genes that were members of the psoriasis transcriptome (described in (36)) and were also members of previously described pathways: additive and synergistic (36); IFN-γ (25); TNF(28); and IFN-α (33).
For each gene-set, the proportion of genes with an improvement score >75 was calculated and the proportions were compared between treatments using the McNemar test for paired proportions. The average improvement score for each gene-set was analyzed
Finally, using methods previously described (28;37;38), we calculated a measure of epidermal response (a combined score of epidermal thickness, number of Ki67+ keratinocytes, and K16 mRNA levels) using multivariate μ-Scores (39) and correlated the μ-Score with measures of infiltrating leukocytes, associated cytokines, and defined cytokine response genes in keratinocytes for each subject at week 2 and week 6.
Informed Consent
The clinical trial was conducted according to the principles expressed in the Declaration of Helsinki and informed consent for their information to be stored in the hospital database and used for research was obtained from all subjects in written form. This study protocol was approved by ethical review boards at sites conducting this study.
Results
Ixekizumab was administered to 32 subjects with plaque psoriasis by subcutaneous injection across a range of doses from 5 mg to 150 mg at weeks 0, 2, and 4. Eight subjects received placebo injections at the same time points. Skin biopsies for IL-17 pathway and other disease-related biomarkers and histopathology were taken before treatment (baseline) and again at weeks 2 and 6 after starting study drug.
IL-17 pathway protein and gene products are rapidly attenuated by ixekizumab
A pathogenic model for IL-17 in psoriasis, based on cell culture experiments, suggests that this cytokine could directly activate a set of 40-50 genes in epidermal keratinocytes, which might be expanded to a larger set of inflammatory products by additive or synergistic effects on gene transcription through combined signaling of IL-17 and TNF (36) . Accordingly, the first assessment of ixekizumab's effect in psoriasis lesions was its ability to modulate mRNA or protein expression of IL-17regulated products in epidermal keratinocytes. Figure 1 shows marked reductions in expression of LL37 (cathelicidin), beta-defensin 2, S100A7, and S100A8 proteins in keratinocytes within 2 weeks of antibody administration, and near normalization of expression by 6 weeks of treatment. This effect was not seen in subjects receiving placebo treatment (supplementary Figure 1). A rapid, dose-dependent reduction in lipocalin 2 mRNA was also seen within 2 weeks of IL-17 blockade (Figure 2) and the higher dose groups of 50 mg and 150 mg showed the most complete suppression.
Figure 1. IL-17 neutralization results in decreased keratinocyte proliferation and differentiation, leukocyte infiltration and keratinocyte release of inflammatory cytokines.

A representative example of serial skin biopsies from a subject treated with 150 mg ixekizumab is presented. Histological analysis included H&E staining. Immunohistochemical analysis included staining for Keratin 16 (K16), Ki67, CD3, CD11c, LL37/cathelicidin, S100A7, S100A8, BD 2 and DC-LAMP. Scale bar is 50 microns.
Figure 2. IL-17 neutralization results in decreased expression of cytokines from multiple T cell subsets.

At baseline and weeks 2 and 6, mRNA expression for Keratin 16 (k16) and LCN, a IL-17 target gene, were analyzed as well as mRNA expression levels for T-cell cytokines: IFN-γ, IL-17 A and F, IL-22, and DC cytokine IL-23. *p<0.05 vs baseline. Data are shown as mean ± SEM.
Improvements in cellular and molecular disease biomarkers parallel IL-17 target reductions
Since IL-17 is not a direct keratinocyte mitogen, it was not predicted that epidermal hyperplasia would be rapidly attenuated by ixekizumab. However, rapid reductions in proliferating (Ki67+) keratinocytes, keratin 16+ keratinocytes, epidermal thickness (Figure 1, Supplemental Figure 2), and keratin 16 mRNA (Figure 2) were seen by 2 weeks and largely normalized by 6 weeks of treatment. Surprisingly, there was also large-scale collapse of underlying tissue infiltration by T-cells, inflammatory (CD11c+) dendritic cells (p<0.05 for reductions in T-cells and dendritic cells), as well as decreased levels of cytokine transcripts that define activated Th1, Th17, and Th22 T cell subsets, i.e., IFN-γ, IL-17A/F, and IL-22 mRNAs, as measured by RT-PCR (Figures 1 and 2 and Supplemental Figure 2). Production of IL-23 (p19 and p40 mRNAs) was also rapidly reduced after IL-17 blockade. These changes were all dose-dependent, with the highest effect seen in the 50 and 150 mg treatment groups and no significant reductions in the placebo group. At six weeks of treatment, 8/8 subjects in the 150 mg group had absent keratin 16 in suprabasal keratinocytes, as well as elimination of parakeratosis, loss of psoriasiform patterning and gain of a granular layer, such that defining pathological characteristics of psoriasis were eliminated.
IL-17 blockade resulted in rapid clinical improvement in psoriasis
In addition to assessing the exposures of ixekizumab needed to reverse expression of IL-17 regulated gene products in psoriatic lesions, this study assessed clinical efficacy using the Psoriasis Area and Severity Index (PASI) and the safety and tolerability of this short term treatment intervention. At 6 weeks after the first dose, an improvement of at least 75% in the PASI (PASI 75) was observed in 0/8 and 0/8 subjects receiving placebo or 5 mg ixekizumab, respectively, while 2/8, 5/7 and 8/8 subjects receiving 15 mg, 50 mg or 150 mg ixekizumab, respectively, achieved this endpoint (Table S2). Hence, a dose-response for disease improvement was seen with ixekizumab, with all subjects in the 150 mg dosing group having a high-grade clinical improvement of a PASI 75. Five out of seven patients still had a PASI 75 response at Week 20, 16 weeks after the last dose (Table S2). Significant differences in the proportions of patients with a PASI75 between treatment groups were present It is important to note that this level of clinical response was unexpected at 6 weeks since, for many other treatments, a maximal clinical response is not achieved until 12 weeks or more after the start of therapy (40-42). This treatment was well tolerated and an overview of adverse events (AEs) is available in Table S3.
Investigation of the early response to IL-17 blockade through expression profiling
The rapid blockade of IL-17 target proteins and mRNAs, as well as parallel improvements in other disease-related biomarkers and the clinical disease score, raises the question of whether there was also a rapid effect on the expression of a broader set of disease-associated molecules or pathways after IL-17 blockade. Accordingly, gene expression was measured using Affymetrix arrays in baseline psoriasis lesions and lesions treated for 2 weeks with either placebo injections or 150 mg of ixekizumab, the dose at which each patient achieved a PASI 75 at 6 weeks. This analysis found that placebo treatment did not lead to significant changes in gene expression, whereas 916 probe-sets representing 765 genes were significantly modulated (>2 fold change, p<0.05) in week 2 biopsies, as listed in supplementary Table S4. As illustrated in Figure 3, genes that were strongly suppressed (change >6-fold) by ixekizumab included: IL-19, a potent inducer of epidermal hyperplasia; IL-8 and CXCL1, known IL-17 induced chemokines with potent ability to attract neutrophils; CCL20, a chemokine with ability to attract Th17 T cells and myeloid DCs into inflammatory sites; granzyme B, an effector molecule in cytotoxic T cells; and LCN, an innate defense peptide strongly induced by IL-17. Overall, the reduction in proliferating keratinocytes (Ki67+ cells) was highly correlated with a reduction in IL-19 mRNA expression (r= 0.93, p= 0.002; supplemental Figure 4).
The extent to which disease-associated transcripts were modulated at only 2 weeks of treatment was surprising, especially in relationship to a prior study we conducted with the TNF inhibitor etanercept (using the same Affymetrix Hug133a2 array and analysis criteria), where <200 genes were modulated by 2 weeks of cytokine antagonism (28). To better compare response kinetics and to compare effects on genes that are coregulated by IL-17 and TNF signaling in keratinocytes (genes regulated in an additive or synergistic fashion by both IL-17 and TNF), we did a formal genomic meta-analysis of transcriptional alterations in both studies by combining array data with adjustments for batch effects of the different experiments. Results obtained with this combined approach are shown in Figures 3 and 4. In Figure 3, the right columns (LS and NL) show differences in gene expression in untreated lesions vs. nonlesional skin using the normalized reference data obtained from our previous study (28). The two-week response to IL-17 blockade in the 150 mg ixekizumab shown in Figure 3 shows that most highly expressed transcripts in lesional skin approach nonlesional levels. Consistent with microarray gene expression data, reductions in mRNAs (as measured by RT-PCR) encoding keratin 16 (K16) and beta-defensin 4 (DEFB4) are larger with ixekizumab treatment, compared to etanercept, as are reductions in epidermal thickness and proliferating (Ki67+) keratinocytes (Figures 4 A, B). Using the differences in gene expression between LS and NL skin, we had previously determined that there are ∼1200 differentially expressed genes (DEGs) in psoriatic skin (32). At 2 weeks, 643 of these transcripts are normalized by IL-17A blockade, whereas only 104 were modulated by TNF blockade (Figure 4C), and the overall improvement strongly favored ixekizumab (Figure 4D). Among all these psoriasis related DEGs, there was approximately70% mean improvement with ixekizumab compared to 35% with etanercept; 40% of the psoriasis DEGs were improved by at least 75% vs. 6% with etanercept. Modulation of the expression of genes within separate cytokine-response pathways by ixekizumab and etanercept at 2 weeks are compared in Figures 4E and F (25;28). Compared to all psoriasis-related DEGs, an even higher fraction of IL-17 pathway genes are improved with ixekizumab, whereas TNF inhibition had only a small effect, as would be predicted. Previously, it has been shown in vitro that many genes that are highly expressed in psoriasis are coregulated by IL-17 and TNF in a synergistic or additive manner (36), however, this has not yet been established in vivo. In this analysis we see that these coregulated genes are strongly modulated by IL-17 blockade and weakly modulated by TNF blockade, with a highly significant difference (p<10-16, Figures 4 E and F). Overall, these data suggest that: 1) synergistic and additive effects of IL-17 and TNF on mRNA abundance for specific genes occurs in vivo in psoriasis lesions; and 2) IL-17 could be the main “driver” for high-level expression of these target genes.
Figure 4. Comparison of treatment effect at week 2 for ixekizumab, etanercept, and placebo.
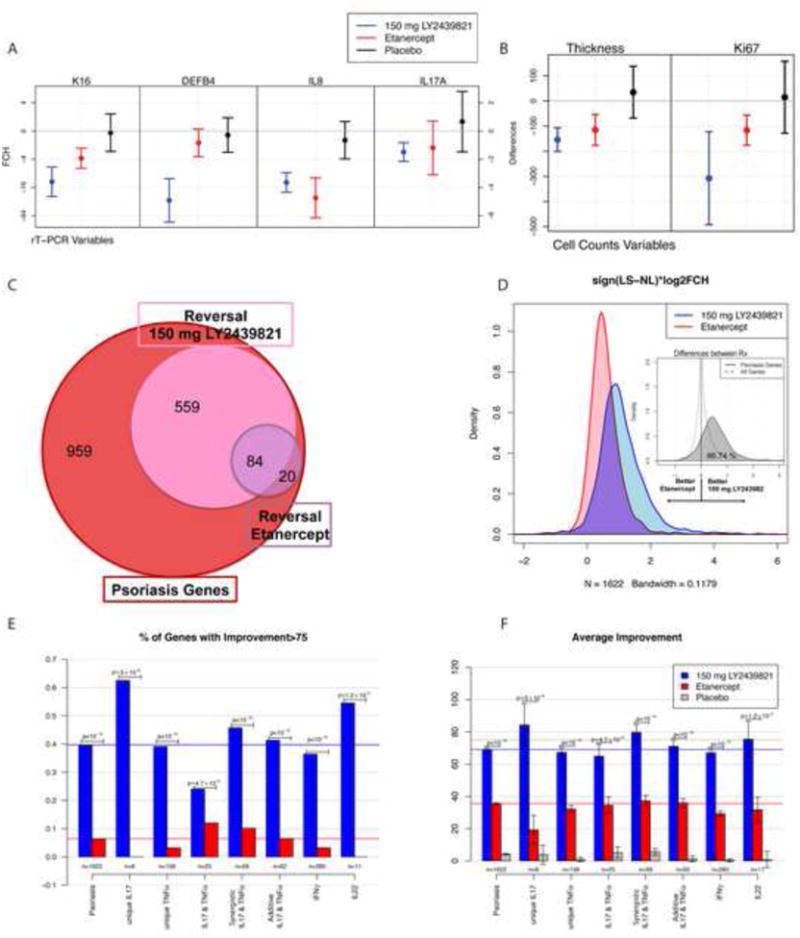
A-B. Mean (95% CI)I for fold change from baseline for each treatment for A) RT-PCR and B) histological variables. C. Venn-diagram summarizing the number of genes among those in the psoriasis transcriptome with improvement of at least 75%. D. Estimated density distribution of the treatment effect multiplied by the sign of the lesional (LS) vs. nonlesional (NL) direction to show only the magnitude of the change towards resolution. The insert graph has the density distribution of the difference in effect between ixekizumab and etanercept for all genes and the genes in the psoriasis transcriptome. For all genes, one can see a 0-centered distribution (equal effects) but for the psoriasis genes, there is a shift to the right, indicating larger changes with ixekizumab treatment among psoriasis genes. E. Proportion of psoriasis genes improved by more than 75% in each gene-set. F. Average improvement in each gene-set.
Another aspect of IL-17 antagonism in vivo in psoriasis, and one not predicted from pathway models, is that Th1, Th22, and Th17 defining cytokines, as well as downstream response pathways (Figures 4E, F) are all measurably suppressed by ixekizumab. One effect we measured that could be related to suppression of multiple T-cell subsets is a marked decrease in mature DC-LAMP+ dendritic cells in psoriasis lesions (Figure 1) and as a potential mechanism of this decrease, a reduction in LL37/cathelicidin expression in skin lesions (Figure 1). Complexes of self RNA with LL37/cathelicidin can generate DC-LAMP+ dendritic cells from immature myeloid (CD11c+) precursors and this has been a postulated disease mechanism for psoriasis (43).
We also sought to determine potential relationships between suppression of specific Th-subsets and eventual disease improvement, as defined by improvements in epidermal hyperplasia. For this analysis, we studied individual subject responses by comparing mu-scores for epidermal improvement and individual inflammatory molecules or cell subsets (Supplmental Figure 4). In this analysis, suppression of IL-17-regulated pathway products (DEFB4, LCN2, IL-8, and CXCL-1) at week 2, but not levels of IL-17A or IL-17F mRNAs, were all highly correlated with epidermal improvement (r values between 0.7 and 0.8, p<0.03). Week 2 reductions in Th1 and Th22 linked genes: IFN-γ, CXCL10, MX-1 and IL-22 were poorly and insignificantly associated with the epidermal response. Overall, these results support a therapeutic model where blockade of IL-17 effects on target cells, e.g. keratinocytes, is more related to early disease improvement than changes in T-cell activation/ production of cytokines. These data also suggest that inhibition of IL-17 pathway genes, which are highly expressed in lesional keratinocytes, is more related to epidermal improvement than inhibition of IFN-γ response genes (CXCL10, MX-1).
Discussion
While autoimmune inflammation was long considered to be driven by Th1 T cell activation and associated cytokines (i.e. IFN-γ), the discovery of Th17 T cells and the causality of Th17 T cells in inducing experimental autoimmune encephalitis greatly altered this concept (44-46).
In turn, it then raised the question whether Th17 T cells have an important pathogenic contribution in human immune- mediated inflammatory diseases. Psoriasis vulgaris is a strong candidate disease for pathogenic activity of Th17 cells based on several lines of evidence: genetic susceptibility controlled in part by IL-23 and IL-23 receptor gene variants; disease improvements associated with ustekinumab therapy (blocking the p40 subunit of this cytokine)(47-49); increased skin infiltration by Th17 T-cells (3-5); and over-expression of IL-17A/F transcripts in skin lesions, with strong up-regulation of many disease-related mRNAs that are induced by IL-17 signaling in skin cells (5;25;28;32;36).
Even so, Th17 T-cells are a relatively small percentage of overall infiltrates, while Th1 cells producing gamma-interferon, and Th22 T-cells selectively producing IL-22 are major parts of the disease phenotype (6). Pathogenic models have considered that combined actions of all these cytokines might be required to sustain pathogenic inflammation, and broad alterations in gene expression are best explained by activation of multiple cytokine receptors/cytokine pathways in this disease (32;50). To the extent that individual cytokines have been proposed as major drivers of the disease phenotype, IL-22 has emerged as a “most likely” cytokine for regulating epidermal hyperplasia and associated skin inflammation in mouse models (23). However, pathways of skin inflammation in humans may be significantly different from animal models, as most IL-22 produced by T-cells in humans are within a distinct subset termed “Th22,” whereas Th17 T-cells in mice co-produce IL-17 and IL-22.
The results of the study reported here, testing multiple doses of an IL-17 monoclonal antibody, ixekizumab, provide strong evidence linking IL-17 to a central role in disease pathogenesis in psoriasis. We observed significant clinical improvements in patients receiving ixekizumab with no unexpected safety signals, although the sample size was small and further clinical trials are needed to fully evaluate its safety and efficacy. In addition, histological and genetic analyses of lesional skin biopsies revealed the underlying collapse of inflammatory circuits with treatment that may explain the high magnitude of clinical efficacy and the long-lasting effect of treatment after the last dose. The translational science approach illustrated in this study demonstrate the power of detailed mechanistic studies in the drug development process for understanding disease pathogensis in psoriasis in particular and potentially for autoimmune diseases more broadly.
It seems likely that Th17 T-cells are the source of IL-17 in psoriatic skin, since antagonism of the p40 subunit or p19 subunit of IL-23 can produce marked disease clearing and suppression of the IL-17 pathway in diseased skin (49;51;52). Clinical responses are not highly correlated with expression of IL-22 or interferon-response pathway genes, suggesting that IL-17 is the major “driver” cytokine for this complex disease phenotype. Accordingly, the psoriasis disease phenotype defined by dysregulation of over 1000 gene products, tissue infiltration by multiple leukocyte subsets, and associated keratinocyte hyperplasia/tissue patterning and the clinical disease appearance, was significantly reversed by treatment with ixekizumab, a monoclonal antibody to IL-17A. Given that IL-17A and IL-17F bind independently to the same receptor, it may well be that the bioactive form of this cytokine is an IL-17A/F heterodimer, as potentially suggested by a recent case of IL-17 deficiency caused by a mis-folded IL-17F variant (53).
In vitro studies in which human keratinocytes are treated with T cell derived cytokines have defined a relatively narrow range of products induced only by IL-17, but a much larger set of genes is induced by the combination of IL-17 with TNF (36). Thus, both TNF and IL-17 are likely co-inducers of a broad set of genes that are highly up-regulated in the psoriasis transcriptome, but the in vivo contribution of each cytokine to this response should be considered. The set of genes suppressed at week 2 in this study is much larger and of greater magnitude than TNF/IL-17 genes suppressed with 2 weeks of treatment with a TNF antagonist, etanercept (28;36;54) suggesting that IL-17 may be the dominant cytokine inducing synergistic and additive genes for the combined TNF/IL-17 response.
Our results also begin to cement the idea that different inflammatory skin diseases such as atopic dermatitis and psoriasis may be reaction patterns to different underlying cytokines produced by distinct T-cell subsets (25). The data in this manuscript argue strongly for psoriasis as an IL-17/Th17 disease, whereas there is very little expression of the IL-17 axis in atopic dermatitis (10). In contrast to psoriasis, expression of Th2 and Th22 cytokines are very high in atopic dermatitis and these might thus be the key drivers of this distinct skin disease (6;10), but still one that has many parallel features to psoriasis (55;56). Eventually, there will need to be cytokine antagonism studies in atopic dermatitis and other inflammatory skin diseases to establish the pathogenic role of specific cytokines or combinations. We hope this study might serve as a model of how to do early proof-of-concept clinical studies with incorporation of in-depth biomarker research at an early stage of hypothesis testing. In fact, the data from this study has informed the design of a phase 2 study of ixekizumab, which was just recently published. This study showed significant clinical efficacy at 12 weeks with this antibody in patients with chronic moderate-to-severe psoriasis, as noted earlier (27). Furthermore, the extent and duration of the collapse of the inflammatory milieu observed in this study provides a mechanistic explanation for the high degree of clinical efficacy and durability of the clinical response observed in the phase 2 study(27).
In summary, neutralization of IL-17 with the monoclonal antibody, ixekizumab, decreased expression of genes associated with numerous circuits of inflammation resulting in a rapid reversal of the psoriatic phenotype. The clinical and genetic effects were dose dependent. Neutralization of IL -17 modulates genes upstream and downstream of known IL-17 effects and has a stronger effect on the expression of genes that are known to be coregulated by IL-17 and TNF than does TNF inhibition alone. Combined, our data suggest that IL-17 and/or Th17 T cells have essential functions in the pathogenesis of psoriasis.
Supplementary Material
Key Messages.
ixekizumab, a monoclonal antibody against IL-17(IL-17A), resolved the clinical and pathological features of psoriasis in patients receiving the highest dose with no unexpected safety signals
Analysis of gene expression in lesional skin biopsies revealed neutralization of IL-17 led to significant down-modulation of multiple inflammatory pathways to a greater extent than TNF neutralization
These data suggest IL-17 is the central cytokine driving psoriasis pathogenesis
Acknowledgments
This study was funded by Eli Lilly and Company.
The phase 1 study of ixekizumab was funded by Eli Lilly and Co. We thank all of the investigators who participated in the RHAG trial. We thank David Shrom, Julie Sherman, Robert Hauk and Hassan Jamal for their help in preparing this manuscript. Dr. Krueger has received payment for an advisory board and has received a research grant from Eli Lilly and Co. Dr. Cameron, Dr. Hoffman, Dr. McColm, Ms. Phipps, Dr. Banerjee and Dr. Hoffman are employees and stockholders of Eli Lilly and Co. Dr. Patrick Haslett was an employee and stockholder of Eli Lilly and Co. during the acquisition of this data and is currently an employee and stockholder of Shire Human Genetic Therapies.
Abbreviations
- IL
interleukin
- RT-PCR
reverse transcriptase polymerase chain reactions
- TNF
tumor necrosis factor alpha
- PASI
Psoriasis Area and Severity Index
- PASI 75
at least a 75% improvement in PASI
- BD4
beta-Defensin 4
- DEG
differentially expressed Genes
- FCH
fold change
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Greaves MW, Weinstein GD. Treatment of psoriasis. N Engl J Med. 1995 Mar 2;332(9):581–8. doi: 10.1056/NEJM199503023320907. [DOI] [PubMed] [Google Scholar]
- 2.Lowes MA, Bowcock AM, Krueger JG. Pathogenesis and therapy of psoriasis. Nature. 2007 Feb 22;445(7130):866–73. doi: 10.1038/nature05663. [DOI] [PubMed] [Google Scholar]
- 3.Kagami S, Rizzo HL, Lee JJ, Koguchi Y, Blauvelt A. Circulating Th17, Th22, and Th1 cells are increased in psoriasis. J Invest Dermatol. 2010 May;130(5):1373–83. doi: 10.1038/jid.2009.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kryczek I, Bruce AT, Gudjonsson JE, Johnston A, Aphale A, Vatan L, et al. Induction of IL-17+ T cell trafficking and development by IFN-gamma: mechanism and pathological relevance in psoriasis. J Immunol. 2008 Oct 1;181(7):4733–41. doi: 10.4049/jimmunol.181.7.4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lowes MA, Kikuchi T, Fuentes-Duculan J, Cardinale I, Zaba LC, Haider AS, et al. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol. 2008 May;128(5):1207–11. doi: 10.1038/sj.jid.5701213. [DOI] [PubMed] [Google Scholar]
- 6.Nograles KE, Zaba LC, Shemer A, Fuentes-Duculan J, Cardinale I, Kikuchi T, et al. IL-22-producing “T22” T cells account for upregulated IL-22 in atopic dermatitis despite reduced IL-17-producing TH17 T cells. J Allergy Clin Immunol. 2009 Jun;123(6):1244–52. doi: 10.1016/j.jaci.2009.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zaba LC, Fuentes-Duculan J, Eungdamrong NJ, Abello MV, Novitskaya I, Pierson KC, et al. Psoriasis is characterized by accumulation of immunostimulatory and Th1/Th17 cell-polarizing myeloid dendritic cells. J Invest Dermatol. 2009 Jan;129(1):79–88. doi: 10.1038/jid.2008.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kao CY, Chen Y, Thai P, Wachi S, Huang F, Kim C, et al. IL-17 markedly up-regulates beta-defensin-2 expression in human airway epithelium via JAK and NF-kappaB signaling pathways. J Immunol. 2004 Sep 1;173(5):3482–91. doi: 10.4049/jimmunol.173.5.3482. [DOI] [PubMed] [Google Scholar]
- 9.Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006 Oct 2;203(10):2271–9. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guttman-Yassky E, Lowes MA, Fuentes-Duculan J, Zaba LC, Cardinale I, Nograles KE, et al. Low expression of the IL-23/Th17 pathway in atopic dermatitis compared to psoriasis. J Immunol. 2008 Nov 15;181(10):7420–7. doi: 10.4049/jimmunol.181.10.7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity. 2005 Mar;22(3):285–94. doi: 10.1016/j.immuni.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 12.Blumberg H, Conklin D, Xu WF, Grossmann A, Brender T, Carollo S, et al. Interleukin 20: discovery, receptor identification, and role in epidermal function. Cell. 2001 Jan 12;104(1):9–19. doi: 10.1016/s0092-8674(01)00187-8. [DOI] [PubMed] [Google Scholar]
- 13.Boniface K, Bernard FX, Garcia M, Gurney AL, Lecron JC, Morel F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. 2005 Mar 15;174(6):3695–702. doi: 10.4049/jimmunol.174.6.3695. [DOI] [PubMed] [Google Scholar]
- 14.Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity. 2004 Aug;21(2):241–54. doi: 10.1016/j.immuni.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 15.Wolk K, Witte E, Wallace E, Docke WD, Kunz S, Asadullah K, et al. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol. 2006 May;36(5):1309–23. doi: 10.1002/eji.200535503. [DOI] [PubMed] [Google Scholar]
- 16.Di Cesare A, Di Meglio P, Nestle FO. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J Invest Dermatol. 2009 Jun;129(6):1339–50. doi: 10.1038/jid.2009.59. [DOI] [PubMed] [Google Scholar]
- 17.Lee E, Trepicchio WL, Oestreicher JL, Pittman D, Wang F, Chamian F, et al. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med. 2004 Jan 5;199(1):125–30. doi: 10.1084/jem.20030451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Piskin G, Sylva-Steenland RM, Bos JD, Teunissen MB. In vitro and in situ expression of IL-23 by keratinocytes in healthy skin and psoriasis lesions: enhanced expression in psoriatic skin. J Immunol. 2006 Feb 1;176(3):1908–15. doi: 10.4049/jimmunol.176.3.1908. [DOI] [PubMed] [Google Scholar]
- 19.Capon F, Di MP, Szaub J, Prescott NJ, Dunster C, Baumber L, et al. Sequence variants in the genes for the interleukin-23 receptor (IL23R) and its ligand (IL12B) confer protection against psoriasis. Hum Genet. 2007 Sep;122(2):201–6. doi: 10.1007/s00439-007-0397-0. [DOI] [PubMed] [Google Scholar]
- 20.Cargill M, Schrodi SJ, Chang M, Garcia VE, Brandon R, Callis KP, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. 2007 Feb;80(2):273–90. doi: 10.1086/511051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di Meglio P, Di Cesare A, Laggner U, Chu CC, Napolitano L, Villanova F, et al. The IL23R R381Q gene variant protects against immune-mediated diseases by impairing IL-23-induced Th17 effector response in humans. PLoS One. 2011;6(2):e17160. doi: 10.1371/journal.pone.0017160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma HL, Liang S, Li J, Napierata L, Brown T, Benoit S, et al. IL-22 is required for Th17 cell-mediated pathology in a mouse model of psoriasis-like skin inflammation. J Clin Invest. 2008 Feb;118(2):597–607. doi: 10.1172/JCI33263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007 Feb 8;445(7128):648–51. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- 24.Sa SM, Valdez PA, Wu J, Jung K, Zhong F, Hall L, et al. The effects of IL-20 subfamily cytokines on reconstituted human epidermis suggest potential roles in cutaneous innate defense and pathogenic adaptive immunity in psoriasis. J Immunol. 2007 Feb 15;178(4):2229–40. doi: 10.4049/jimmunol.178.4.2229. [DOI] [PubMed] [Google Scholar]
- 25.Nograles KE, Zaba LC, Guttman-Yassky E, Fuentes-Duculan J, Suarez-Farinas M, Cardinale I, et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J Dermatol. 2008 Nov;159(5):1092–102. doi: 10.1111/j.1365-2133.2008.08769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hueber W, Patel DD, Dryja T, Wright AM, Koroleva I, Bruin G, et al. Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med. 2010 Oct 6;2(52):52ra–72. doi: 10.1126/scitranslmed.3001107. [DOI] [PubMed] [Google Scholar]
- 27.Leonardi C, Matheson R, Zachariae C, Cameron G, Li L, Edson-Heredia E, et al. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N Engl J Med. 2012 Mar 29;366(13):1190–9. doi: 10.1056/NEJMoa1109997. [DOI] [PubMed] [Google Scholar]
- 28.Zaba LC, Suarez-Farinas M, Fuentes-Duculan J, Nograles KE, Guttman-Yassky E, Cardinale I, et al. Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J Allergy Clin Immunol. 2009 Nov;124(5):1022–10. doi: 10.1016/j.jaci.2009.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chamian F, Lowes MA, Lin SL, Lee E, Kikuchi T, Gilleaudeau P, et al. Alefacept reduces infiltrating T cells, activated dendritic cells, and inflammatory genes in psoriasis vulgaris. Proc Natl Acad Sci U S A. 2005 Feb 8;102(6):2075–80. doi: 10.1073/pnas.0409569102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suarez-Farinas M, Haider A, Wittkowski KM. “Harshlighting” small blemishes on microarrays. BMC Bioinformatics. 2005;6:65. doi: 10.1186/1471-2105-6-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gudjonsson JE, Ding J, Johnston A, Tejasvi T, Guzman AM, Nair RP, et al. Assessment of the psoriatic transcriptome in a large sample: additional regulated genes and comparisons with in vitro models. J Invest Dermatol. 2010 Jul;130(7):1829–40. doi: 10.1038/jid.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suarez-Farinas M, Lowes MA, Zaba LC, Krueger JG. Evaluation of the psoriasis transcriptome across different studies by gene set enrichment analysis (GSEA) PLoS One. 2010;5(4):e10247. doi: 10.1371/journal.pone.0010247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yao Y, Richman L, Morehouse C, de los RM, Higgs BW, Boutrin A, et al. Type I interferon: potential therapeutic target for psoriasis? PLoS One. 2008;3(7):e2737. doi: 10.1371/journal.pone.0002737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou X, Krueger JG, Kao MC, Lee E, Du F, Menter A, et al. Novel mechanisms of T-cell and dendritic cell activation revealed by profiling of psoriasis on the 63,100-element oligonucleotide array. Physiol Genomics. 2003 Mar 18;13(1):69–78. doi: 10.1152/physiolgenomics.00157.2002. [DOI] [PubMed] [Google Scholar]
- 35.Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007 Jan;8(1):118–27. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 36.Chiricozzi A, Guttman-Yassky E, Suarez-Farinas M, Nograles KE, Tian S, Cardinale I, et al. Integrative responses to IL-17 and TNF-alpha in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Invest Dermatol. 2011 Mar;131(3):677–87. doi: 10.1038/jid.2010.340. [DOI] [PubMed] [Google Scholar]
- 37.Haider AS, Lowes MA, Suarez-Farinas M, Zaba LC, Cardinale I, Khatcherian A, et al. Identification of cellular pathways of “type 1,” Th17 T cells, and TNF- and inducible nitric oxide synthase-producing dendritic cells in autoimmune inflammation through pharmacogenomic study of cyclosporine A in psoriasis. J Immunol. 2008 Feb 1;180(3):1913–20. doi: 10.4049/jimmunol.180.3.1913. [DOI] [PubMed] [Google Scholar]
- 38.Johnson-Huang LM, Suarez-Farinas M, Sullivan-Whalen M, Gilleaudeau P, Krueger JG, Lowes MA. Effective narrow-band UVB radiation therapy suppresses the IL-23/IL-17 axis in normalized psoriasis plaques. J Invest Dermatol. 2010 Nov;130(11):2654–63. doi: 10.1038/jid.2010.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wittkowski KM, Lee E, Nussbaum R, Chamian FN, Krueger JG. Combining several ordinal measures in clinical studies. Stat Med. 2004 May 30;23(10):1579–92. doi: 10.1002/sim.1778. [DOI] [PubMed] [Google Scholar]
- 40.Krueger GG, Langley RG, Leonardi C, Yeilding N, Guzzo C, Wang Y, et al. A human interleukin-12/23 monoclonal antibody for the treatment of psoriasis. N Engl J Med. 2007 Feb 8;356(6):580–92. doi: 10.1056/NEJMoa062382. [DOI] [PubMed] [Google Scholar]
- 41.Menter A, Tyring SK, Gordon K, Kimball AB, Leonardi CL, Langley RG, et al. Adalimumab therapy for moderate to severe psoriasis: A randomized, controlled phase III trial. J Am Acad Dermatol. 2008 Jan;58(1):106–15. doi: 10.1016/j.jaad.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 42.Papp KA, Tyring S, Lahfa M, Prinz J, Griffiths CE, Nakanishi AM, et al. A global phase III randomized controlled trial of etanercept in psoriasis: safety, efficacy, and effect of dose reduction. Br J Dermatol. 2005 Jun;152(6):1304–12. doi: 10.1111/j.1365-2133.2005.06688.x. [DOI] [PubMed] [Google Scholar]
- 43.Ganguly D, Chamilos G, Lande R, Gregorio J, Meller S, Facchinetti V, et al. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med. 2009 Aug 31;206(9):1983–94. doi: 10.1084/jem.20090480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aranami T, Yamamura T. Th17 Cells and autoimmune encephalomyelitis (EAE/MS) Allergol Int. 2008 Jun;57(2):115–20. doi: 10.2332/allergolint.R-07-159. [DOI] [PubMed] [Google Scholar]
- 45.Diveu C, McGeachy MJ, Cua DJ. Cytokines that regulate autoimmunity. Curr Opin Immunol. 2008 Dec;20(6):663–8. doi: 10.1016/j.coi.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 46.McGeachy MJ, Cua DJ. The link between IL-23 and Th17 cell-mediated immune pathologies. Semin Immunol. 2007 Dec;19(6):372–6. doi: 10.1016/j.smim.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 47.Gandhi M, Alwawi E, Gordon KB. Anti-p40 antibodies ustekinumab and briakinumab: blockade of interleukin-12 and interleukin-23 in the treatment of psoriasis. Semin Cutan Med Surg. 2010 Mar;29(1):48–52. doi: 10.1016/j.sder.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 48.Leonardi CL, Kimball AB, Papp KA, Yeilding N, Guzzo C, Wang Y, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1) Lancet. 2008 May 17;371(9625):1665–74. doi: 10.1016/S0140-6736(08)60725-4. [DOI] [PubMed] [Google Scholar]
- 49.Papp KA, Langley RG, Lebwohl M, Krueger GG, Szapary P, Yeilding N, et al. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2) Lancet. 2008 May 17;371(9625):1675–84. doi: 10.1016/S0140-6736(08)60726-6. [DOI] [PubMed] [Google Scholar]
- 50.Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009 Jul 30;361(5):496–509. doi: 10.1056/NEJMra0804595. [DOI] [PubMed] [Google Scholar]
- 51.Sofen H, Smith S, Matheson R, Leonardi C, Calderon C, Bouman-Thio E, et al. Results of a single ascending dose study to assess the safety and tolerability of CNTO 1959 following intravenous or subcutaneous administration in healthy subjects and in subjects with moderate to severe psoriasis. Br J Dermatol. 2012;165(6):e10. [Google Scholar]
- 52.Strober BE, Crowley JJ, Yamauchi PS, Olds M, Williams DA. Efficacy and safety results from a phase III, randomized controlled trial comparing the safety and efficacy of briakinumab with etanercept and placebo in patients with moderate to severe chronic plaque psoriasis. Br J Dermatol. 2011 May 17; doi: 10.1111/j.1365-2133.2011.10419.x. [DOI] [PubMed] [Google Scholar]
- 53.Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. 2011 Apr 1;332(6025):65–8. doi: 10.1126/science.1200439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zaba LC, Cardinale I, Gilleaudeau P, Sullivan-Whalen M, Suarez-Farinas M, Fuentes-Duculan J, et al. Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses. J Exp Med. 2007 Dec 24;204(13):3183–94. doi: 10.1084/jem.20071094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guttman-Yassky E, Nograles KE, Krueger JG. Contrasting pathogenesis of atopic dermatitis and psoriasis--part II: immune cell subsets and therapeutic concepts. J Allergy Clin Immunol. 2011 Jun;127(6):1420–32. doi: 10.1016/j.jaci.2011.01.054. [DOI] [PubMed] [Google Scholar]
- 56.Guttman-Yassky E, Nograles KE, Krueger JG. Contrasting pathogenesis of atopic dermatitis and psoriasis--part I: clinical and pathologic concepts. J Allergy Clin Immunol. 2011 May;127(5):1110–8. doi: 10.1016/j.jaci.2011.01.053. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.