

Summary
Genetic counselling may be offered to families with melanoma and to individuals with multiple melanomas to better understand the genetic susceptibility of the disease, the influence of environmental factors, the inheritance of the risk and behaviour that decreases the risk of dying from melanoma including specific dermatological follow-up such as total body photography and digital dermoscopy. Genetic testing may be offered to those individuals with more than a 10% chance of being a carrier of a mutation. This risk varies according to the incidence of melanoma in the country and sun behaviour. In countries with a low-medium incidence of melanoma, genetic testing should be offered to families with two cases of melanoma or an individual with two primary melanomas. In countries with a high incidence, families with three cases of melanoma, with two melanomas and one pancreatic adenocarcinoma, or patients with three primary melanomas may benefit from genetic testing.
Keywords: melanoma, predisposition, gene, genetic counselling, mutation
Genetic susceptibility to melanoma
The aetiology of melanoma is heterogeneous and complex. Genetic, phenotypical and environmental factors are involved in melanoma susceptibility. Ultraviolet (UV) radiation exposure has been identified as the principal environmental cause. UV radiation promotes DNA damage through the formation of dimeric photoproducts, gene mutations, oxidative stress, inflammation and immuno-suppression.
Physical traits, such as blonde or red hair, green or blue eyes, presence of freckles, multiple (100 or more) melanocytic naevi (moles), and five or more atypical naevi, are also risk factors (1). A prior diagnosis of melanoma is associated with an eight-fold increased risk of a second primary melanoma (2).
Many familial cancer syndromes are recognised by so-called high-risk features, including a cluster of relatives within the patient’s family who have the same or similar cancers (more than two family members with cancer over two or more generations), the development of cancer at a young age, the presentation of more than one tumour or more than one tumour within a specific group of tumours that are features of a syndrome (3)
In the case of melanoma, it is estimated that 5–10% of all cases occur in families (4). In these families, the pattern of susceptibility is consistent with an autosomal dominant inheritance with incomplete penetrance (5), thus relatives of these patients are at an increased risk of developing melanoma. In families, the research has been directed at identifying those genes implicated in melanoma susceptibility and to use this data to provide genetic counselling to families with melanoma (6).
Two high penetrance genes have been implicated in melanoma susceptibility: CDKN2A and CDK4. The tumour suppressor gene CDKN2A encodes two different proteins: the α transcript encodes the p16 protein, that regulates G1-phase exit by inhibiting the CDK4-mediated phosphorylation of the retinoblastoma protein (7, 8) The β transcript encodes the alternative product p14ARF which acts via the p53 pathway to induce cell cycle arrest or apoptosis (9, 10). Also, both P53 and p16INK4A have roles in responses to DNA damage and cellular senescence (11, 12). CDK4 which is an oncogene, encodes one of the binding partners of p16 (13). Only two mutations have been described to date and both are located in the same codon, affecting the protein domain of interaction with p16 which results in disabling the p16 protein from inhibiting the D1-cyclin-dependent kinase 4 mutant complex. The functions of CDKN2A and CDK4 in melanoma development, however, are currently incompletely understood. By definition these genes have a high penetrance and therefore there are usually multiple cases of cancer in the affected family.
To date, the number of CDK4 mutant melanoma families is reduced compared to the frequency of CDKN2A mutations in melanoma-prone families. Overall, 20% of melanoma prone families have germinal CDKN2A mutations but this varies from 5% to 72% depending on the selection criteria used and it is also highly variable across regions (14). The likelihood of detecting a mutation increases with the number of affected members detected, in some populations being 4% for two melanoma cases, 8% for three melanoma cases and 38% for three or more melanoma cases (14, 15). On the other hand, the risk of being a carrier for a melanoma patient with no family history of the disease is approximately of 1%.
The multiple case family studies indicate that the life-time risk of melanoma (penetrance) in CDKN2A mutation carriers is very high, ranging from 58% in Europe to 91% in Australia by age 80 years. But the penetrance of CDKN2A mutations in the general population may be much lower. It was calculated that the risk of melanoma in CDKN2A mutation carriers is 28% by age 80 in a population-based study (15).
In some families there appears to be a pre-disposition to a variety of different cancers. In a study by the International Consortium GENOMEL, the presence of pancreatic cancer in a family was strongly associated with the occurrence of a CDKN2A mutation (28% of families with a CDKN2A mutation had pancreatic cancer versus 6% of families with no mutation) (14). Nevertheless, in the majority of families with high penetrance melanoma susceptibility genes the predisposition appears to be to melanoma only.
A controversial issue is the effect of common CDKN2A variants in melanoma susceptibility. Multiples studies suggest that polymorphism p.A148T could be considered as a low penetrance melanoma susceptibility allele in some populations (16, 17) or in multiple primary melanoma patients (18). However, such findings have not been found in other studies (19, 20).
Two medium penetrance susceptibility genes have also been implicated in melanoma susceptibility: MC1R and MITF. The melanocortin-1 receptor (MC1R) encodes the melanocyte-stimulating hormone receptor and is involved in the pigmentation process (21). MC1R is highly polymorphic in the Caucasian population. These variants have varying effects on the receptor’s function, its ability to modify melanogenesis and thus, the eumelanin/pheomelanin ratio (22, 23). Variants in the MC1R gene are strongly associated with skin and hair pigmentation and also contribute to melanoma risk independent of these phenotypical features (24). There are large studies which confer the red-hair associated (RHC) subset of MC1R variants with increased melanoma risk. This risk varies from around two -fold per RHC allele in the populations, to three-fold in melanoma families. The risk is additive, carriers of two RHC alleles have a risk four-to six-fold higher than non-RHC carriers. Moreover, coinheritance of variants in MC1R with CDKN2A mutations increases the penetrance of melanoma in such mutation positive families (25, 26). Interestingly, the presence of two MC1R RHC variants in early melanomas was also associated with the presence of less dermoscopic structures and colours, thus being more difficult to diagnose. However, it is not clear how MC1R variants can be incorporated into clinical practice without taking into account their phenotypical effect.
Microphthalmia-associated transcription factor (MITF) is a master regulator gene of melanocyte development and differentiation and is also associated with melanoma development and progression (27). Recently it has been implicated in melanoma susceptibility. Mutation p.E318K (rs149617956) has been implicated both in familial and sporadic melanoma susceptibility (Odds Ratio -OR- 2.33), and in melanoma and/or renal cell carcinoma patients (28, 29). This variant was also associated with increased naevus count and non-blue eye colour (28).
The impact of the inheritance of low-medium penetrance genes in melanoma predisposition is weak, with maybe only one or occasionally two cases in a family. Co-inheritance of more than one low-medium penetrance gene may result in more marked clustering of melanoma in some families. As UV radiation is also implicated in melanoma susceptibility, families carrying these low-medium penetrance genes may have more melanoma cases if they live in areas of the world where the UV radiation is high.
Genetic counselling/assessment in melanoma
Genetic assessment in familial melanoma is a non directed process offered to families to better understand the meaning of the disease, the meaning of genetic susceptibility, the patterns of inheritance, the option of genetic testing, the understanding of all the possible results and the primary and secondary prevention of melanoma as well. The process includes melanoma risk assessment, genetic testing, informed consent, disclosure of test results and psychosocial assessment as in other cancer genetic counselling or assessment (30).
One of the most important benefits of genetic assessment in melanoma is the identification of individuals with increased risk of developing this cancer. It has to distinguish between those with a high risk (high penetrance melanoma genes/families), those with a moderately increased risk (multifactor aetiology or low penetrance alleles) and those with an average risk (30). This information can also be used to assess other at-risk individuals in the family. Patients with a personal and/or family history suggestive of an increased melanoma risk should be directed to a melanoma genetic assessment unit. Genetic counselling often implies genetic testing. Nevertheless, all patients can benefit from genetic counselling, even if they are not candidates for genetic testing. Genetic counselling in cancer has been accepted and is used in clinical practice for breast, ovarian, colorectal and medullary thyroid and diffuse gastric cancers. Its utility in patient screening and management is well documented (30).
Genetic testing for melanoma has been available since the CDKN2A gene (the major gene implicated in melanoma predisposition) was identified in 1994 (31, 32). Nevertheless its use in clinical practice has been controversial due to the variation in the estimates of CDKN2A mutation penetrance (28–91%) depending on how the study is designed, the ethnic background, UV exposure and co-inheritance of low-medium predisposing gene mutations (such as MC1R gene variants) (33). Moreover, when genetic testing is performed, it is also difficult to assess patients because individuals negative for the mutation segregating in the family are still at increased risk of melanoma. Thus, some people would consider family history of melanoma alone as good a method for assessment as genetic testing.
Genetic testing for melanoma predisposing mutations (in CDK4 and CDKN2A, including its alternative transcript) could be used in a clinical setting if attention is paid to the selection of patients, and a valid test interpretation and genetic counselling is given. People should be aware of the difficulty of interpreting test results and the potentially limited impact on clinical management. Only individuals with at least a 10% pre-test chance of carrying a mutation should be considered as candidates for genetic testing. These individuals should belong to melanoma families or melanoma/related cancer families (sarcoma, early onset breast cancer, brain tumours or pancreatic cancer), and/or have multiple primary melanomas. Patients with an early age at onset or with multiple or atypical naevi do not fulfil criteria for genetic testing, unless they also have a family history of melanoma (34, 35) (Table 1). Leachman et al (36) described a very useful rule to select patients for genetic testing in melanoma, the rule of two for low melanoma incidence countries, such as Southern Europe, the rule of three for medium-high melanoma incidence countries, such as the USA and Northern Europe (Table 2). Also a rule of four for very high melanoma incidence countries, such as Australia, may be suggested. They use different criteria according to the incidence of melanoma and prevalence of mutations in each region. In the United States (medium-high incidence of melanoma) we should consider for genetic assessment individuals with three or more primary invasive melanomas and families with at least one invasive melanoma and two more cases of melanoma and/or pancreatic cancer among first- or second-degree relatives on the same side of the family. In Southern Europe we should consider for genetic testing individuals with two (synchronous or metachronous) primary melanomas and/or families with at least one invasive melanoma and one or more other diagnoses of melanoma and/or pancreatic cancers among first- or second-degree relatives on the same side of the family
Table 1.
Features associated with genetic testing for melanoma predisposition
| Feature | |
|---|---|
| Family history of melanoma | |
| Depending on incidence of melanoma in specific population may be two or three cases of melanoma on the same side of the family | |
| Multiple Primary Melanoma patients | |
| The likelihood of mutation detection increases with the number of primaries in an individual | |
| Pancreatic cancer | |
| presence of pancreatic cancer in a melanoma family | |
| Age of MM <40 y. | |
| No genetic testing recommended as an isolate feature | |
Table 2.
Criteria for genetic testing according to melanoma incidence.
Low melanoma incidence area/population
|
Moderate to high melanoma incidence area/population
|
When genetic testing detects a melanoma predisposing mutation in a family, cascade screening of at risk individuals is recommended. Carriers of a mutation are at high risk of developing multiple melanomas and also, in some families, of pancreatic cancer in their lifetime (37, 38) (Figure 1). Increased skin-cancer screening and surveillance by physicians and self skin examination results in earlier detection of thinner melanomas (39, 40). As regards pancreatic cancer, patients should be aware of the current lack of effective screening guidelines (41).
Figure 1.
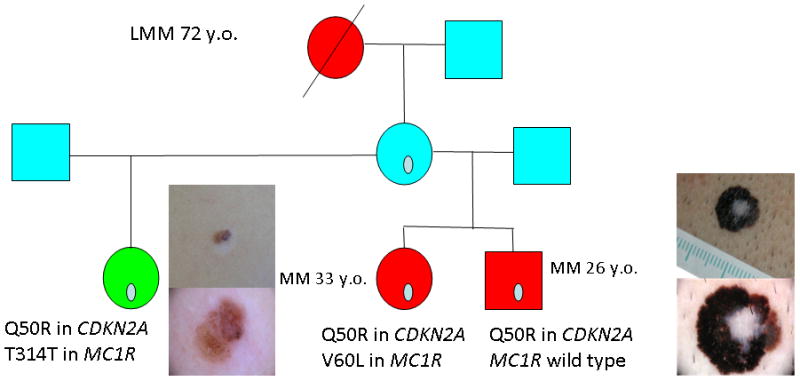
Pedigree of a CDKN2A mutation carrier family with 3 melanoma cases
In families where no CDKN2A mutation is identified, it should be stressed that the family is still at increased risk of melanoma on the basis of the family history. No further information on cancer risk is obtained, so these families should be managed according to family history.
Genetic counselling aims to help patients and families. Genetic counsellors should inform patients about the genetic risk factors for melanoma (Table 3) and other cancers, the indications for genetic testing, the chance of detecting a mutation, the possible reports (positive, negative or inconclusive), the possible implications for other family members, give psychological assessment, and also offer recommendations for cancer screening and UV protection and how these might change with testing. Although mutations can be detected in these patients, families and patients negative for a familial mutation still have up to a two-fold greater risk of developing melanoma than that of the general population, due to other melanoma susceptibility genes and environmental factors shared between families (42, 43). Identification of the missing attributable risk in those individuals with a familial aggregation to melanoma is essential to improve genetic counselling in these families. The high-throughput molecular methodologies such as SNP-array or more recently next generation sequencing constitute powerful tools to identify new high or medium susceptibility genes. Recently, a new approach to predisposition gene identification has identified new chromosomic regions linked to melanoma risk in pedigrees (44, 45).
Table 3.
Risk Factors for the Development of Melanoma
| Factor | Approximate Relative Risk |
|---|---|
| Member of melanoma-prone family* | Up to 35–70 |
| Previous primary melanoma | 8.5 |
| Family history of melanoma† | 2–3 |
| Skin type I | 1.4 |
| Freckling | 2–3 |
| Blue eyes | 1.6 |
| Red hair | 2.4–4 |
| History of blistering sunburn | 2–3 |
| Multiple moles and atypical moles | 2–12.7 |
Multiple affected relatives on the same side of the family.
One or more affected first-degree relatives.
Data from Kefford et al (1999)
Early detection of melanoma is crucial for survival. Thin melanomas have a good outcome while advanced melanomas have a poor prognosis. Genetic counselling and testing can identify mutation carriers in high-risk families. These individuals can benefit from the advantages of prevention and early detection practices (33, 40, 46). In 2011, Salerni et al (46) demonstrated again that the inclusion of patients who are at high risk for melanoma in follow-up programs allows the detection of melanomas at early stages, with good prognosis, even in the absence of clinical and dermoscopic features of melanoma. In a second study, reporting the experience of more than 10 years of follow up of high risk patients, Salerni et al (40) reported that digital follow up with specific dermatologic techniques including total body photography and digital dermoscopy permitted early detection of melanomas with a low rate of excisions. They also reported that long-term follow-up is required to allow the detection of slow-growing melanomas. Based on this 10-year experience, melanomas can be diagnosed at any time, suggesting that in a population at high risk for melanoma, follow up should be maintained over time.
Acknowledgments
Funding/Support
The research at the Melanoma Unit in Barcelona is partially funded by Grants 03/0019, 05/0302, 06/0265 and 09/1393 from Fondo de Investigaciones Sanitarias, Spain; by the CIBER de Enfermedades Raras of the Instituto de Salud Carlos III, Spain; by the AGAUR 2009 SGR 1337 of the Catalan Government, Spain; by the European Commission under the 6th Framework Programme, Contract no: LSHC-CT-2006-018702 (GenoMEL) and by the National Cancer Institute (NCI) of the US National Institute of Health (NIH) (CA83115).
Role of the Sponsors: The sponsors had no role in the design and conduct of the study; in the collection, analysis, and interpretation of data; or in the preparation, review, or approval of the manuscript
Contributor Information
Celia Badenas, Email: cbadenas@clinic.ub.es.
Paula Aguilera, Email: aguilisha@yahoo.com.
Joan A. Puig-Butillé, Email: jantonpuig@gmail.com.
Cristina Carrera, Email: criscarrer@yahoo.es.
Josep Malvehy, Email: jmalvehy@clinic.ub.es.
Susana Puig, Email: susipuig@gmail.com, spuig@clinic.ub.es.
References
- 1.Gandini S, Sera F, Cattaruzza MS, et al. Meta-analysis of risk factors for cutaneous melanoma: I. Common and atypical naevi. Eur J Cancer. 2005;41:28–44. doi: 10.1016/j.ejca.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 2.Bradford PT, Freedman DM, Goldstein AM, Tucker MA. Increased risk of second primary cancers after a diagnosis of melanoma. Arch Dermatol. 2010;146:265–272. doi: 10.1001/archdermatol.2010.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Winship IM, Dudding TE. Lessons from the skin--cutaneous features of familial cancer. Lancet Oncol. 2008;9:462–472. doi: 10.1016/S1470-2045(08)70126-8. [DOI] [PubMed] [Google Scholar]
- 4.Florell SR, Boucher KM, Garibotti G, et al. Population-based analysis of prognostic factors and survival in familial melanoma. J Clin Oncol. 2005;23:7168–7177. doi: 10.1200/JCO.2005.11.999. [DOI] [PubMed] [Google Scholar]
- 5.Anderson DE, Badzioch MD. Hereditary cutaneous malignant melanoma: a 20-year family update. Anticancer Res. 1991;11:433–437. [PubMed] [Google Scholar]
- 6.Kefford RF, Newton Bishop JA, Bergman W, Tucker MA. Counseling and DNA testing for individuals perceived to be genetically predisposed to melanoma: A consensus statement of the Melanoma Genetics Consortium. J Clin Oncol. 1999;17:3245–3251. doi: 10.1200/JCO.1999.17.10.3245. [DOI] [PubMed] [Google Scholar]
- 7.Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 8.Serrano M, Gomez-Lahoz E, DePinho RA, Beach D, Bar-Sagi D. Inhibition of ras-induced proliferation and cellular transformation by p16INK4. Science. 1995;267:249–252. doi: 10.1126/science.7809631. [DOI] [PubMed] [Google Scholar]
- 9.Pomerantz J, Schreiber-Agus N, Liegeois NJ, et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell. 1998;92:713–723. doi: 10.1016/s0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- 11.Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene. 2007;26:7773–7779. doi: 10.1038/sj.onc.1210881. [DOI] [PubMed] [Google Scholar]
- 12.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 13.Zuo L, Weger J, Yang Q, et al. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat Genet. 1996;12:97–99. doi: 10.1038/ng0196-97. [DOI] [PubMed] [Google Scholar]
- 14.Goldstein AM, Chan M, Harland M, et al. High-risk melanoma susceptibility genes and pancreatic cancer, neural system tumors, and uveal melanoma across GenoMEL. Cancer Res. 2006;66:9818–9828. doi: 10.1158/0008-5472.CAN-06-0494. [DOI] [PubMed] [Google Scholar]
- 15.Begg CB, Orlow I, Hummer AJ, et al. Lifetime risk of melanoma in CDKN2A mutation carriers in a population-based sample. J Natl Cancer Inst. 2005;97:1507–1515. doi: 10.1093/jnci/dji312. [DOI] [PubMed] [Google Scholar]
- 16.Bakos RM, Besch R, Zoratto GG, et al. The CDKN2A p. A148T variant is associated with cutaneous melanoma in Southern Brazil. Exp Dermatol. 2011;20:890–893. doi: 10.1111/j.1600-0625.2011.01332.x. [DOI] [PubMed] [Google Scholar]
- 17.Debniak T, Scott R, Masojc B, et al. MC1R common variants, CDKN2A and their association with melanoma and breast cancer risk. Int J Cancer. 2006;119:2597–2602. doi: 10.1002/ijc.22210. [DOI] [PubMed] [Google Scholar]
- 18.Puig S, Malvehy J, Badenas C, et al. Role of the CDKN2A locus in patients with multiple primary melanomas. J Clin Oncol. 2005;23:3043–3051. doi: 10.1200/JCO.2005.08.034. [DOI] [PubMed] [Google Scholar]
- 19.Spica T, Portela M, Gerard B, et al. The A148T variant of the CDKN2A gene is not associated with melanoma risk in the French and Italian populations. J Invest Dermatol. 2006;126:1657–1660. doi: 10.1038/sj.jid.5700293. [DOI] [PubMed] [Google Scholar]
- 20.Bertram CG, Gaut RM, Barrett JH, et al. An assessment of the CDKN2A variant Ala148Thr as a nevus/melanoma susceptibility allele. J Invest Dermatol. 2002;119:961–965. doi: 10.1046/j.1523-1747.2002.01825.x. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki I, Cone RD, Im S, Nordlund J, Abdel-Malek ZA. Binding of melanotropic hormones to the melanocortin receptor MC1R on human melanocytes stimulates proliferation and melanogenesis. Endocrinology. 1996;137:1627–1633. doi: 10.1210/endo.137.5.8612494. [DOI] [PubMed] [Google Scholar]
- 22.Frandberg PA, Doufexis M, Kapas S, Chhajlani V. Human pigmentation phenotype: a point mutation generates nonfunctional MSH receptor. Biochem Biophys Res Commun. 1998;245:490–492. doi: 10.1006/bbrc.1998.8459. [DOI] [PubMed] [Google Scholar]
- 23.Schioth HB, Phillips SR, Rudzish R, Birch-Machin MA, Wikberg JE, Rees JL. Loss of function mutations of the human melanocortin 1 receptor are common and are associated with red hair. Biochem Biophys Res Commun. 1999;260:488–491. doi: 10.1006/bbrc.1999.0935. [DOI] [PubMed] [Google Scholar]
- 24.Williams PF, Olsen CM, Hayward NK, Whiteman DC. Melanocortin 1 receptor and risk of cutaneous melanoma: a meta-analysis and estimates of population burden. Int J Cancer. 2011;129:1730–1740. doi: 10.1002/ijc.25804. [DOI] [PubMed] [Google Scholar]
- 25.Box NF, Duffy DL, Chen W, et al. MC1R genotype modifies risk of melanoma in families segregating CDKN2A mutations. Am J Hum Genet. 2001;69:765–773. doi: 10.1086/323412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fargnoli MC, Gandini S, Peris K, Maisonneuve P, Raimondi S. MC1R variants increase melanoma risk in families with CDKN2A mutations: a meta-analysis. Eur J Cancer. 2010;46:1413–1420. doi: 10.1016/j.ejca.2010.01.027. [DOI] [PubMed] [Google Scholar]
- 27.Levy C, Khaled M, Fisher DE. MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol Med. 2006;12:406–414. doi: 10.1016/j.molmed.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 28.Yokoyama S, Woods SL, Boyle GM, et al. A novel recurrent mutation in MITF predisposes to familial and sporadic melanoma. Nature. 2011;480:99–103. doi: 10.1038/nature10630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bertolotto C, Lesueur F, Giuliano S, et al. A SUMOylation -defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature. 2011;480:94–98. doi: 10.1038/nature10539. [DOI] [PubMed] [Google Scholar]
- 30.Riley BD, Culver JO, Skrzynia C, et al. Essential Elements of Genetic Cancer Risk Assessment, Counseling, and Testing: Updated Recommendations of the National Society of Genetic Counselors. J Genet Couns. 2011 Dec 2; doi: 10.1007/s10897-011-9462-x. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 31.Kamb A, Shattuck-Eidens D, Eeles R, et al. Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat Genet. 1994;8:23–26. doi: 10.1038/ng0994-22. [DOI] [PubMed] [Google Scholar]
- 32.Hussussian CJ, Struewing JP, Goldstein AM, et al. Germline p16 mutations in familial melanoma. Nat Genet. 1994;8:15–21. doi: 10.1038/ng0994-15. [DOI] [PubMed] [Google Scholar]
- 33.Aspinwall LG, Leaf SL, Dola ER, Kohlmann W, Leachman SA. CDKN2A/p16 genetic test reporting improves early detection intentions and practices in high-risk melanoma families. Cancer Epidemiol Biomarkers Prev. 2008;17:1510–1519. doi: 10.1158/1055-9965.EPI-08-0010. [DOI] [PubMed] [Google Scholar]
- 34.Hansen CB, Wadge LM, Lowstuter K, Boucher K, Leachman SA. Clinical germline genetic testing for melanoma. Lancet Oncol. 2004;5:314–319. doi: 10.1016/S1470-2045(04)01469-X. [DOI] [PubMed] [Google Scholar]
- 35.Tsao H, Zhang X, Kwitkiwski K, Finkelstein DM, Sober AJ, Haluska FG. Low prevalence of germline CDKN2A and CDK4 mutations in patients with early-onset melanoma. Arch Dermatol. 2000;136:1118–1122. doi: 10.1001/archderm.136.9.1118. [DOI] [PubMed] [Google Scholar]
- 36.Leachman SA, Carucci J, Kohlmann W, et al. Selection criteria for genetic assessment of patients with familial melanoma. J Am Acad Dermatol. 2009;61:677 e671–614. doi: 10.1016/j.jaad.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parker JF, Florell SR, Alexander A, DiSario JA, Shami PJ, Leachman SA. Pancreatic carcinoma surveillance in patients with familial melanoma. Arch Dermatol. 2003;139:1019–1025. doi: 10.1001/archderm.139.8.1019. [DOI] [PubMed] [Google Scholar]
- 38.Goldstein AM, Chan M, Harland M, et al. Features associated with germline CDKN2A mutations: a GenoMEL study of melanoma-prone families from three continents. J Med Genet. 2007;44:99–106. doi: 10.1136/jmg.2006.043802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carli P, De Giorgi V, Palli D, et al. Dermatologist detection and skin self-examination are associated with thinner melanomas: results from a survey of the Italian Multidisciplinary Group on Melanoma. Arch Dermatol. 2003;139:607–612. doi: 10.1001/archderm.139.5.607. [DOI] [PubMed] [Google Scholar]
- 40.Salerni G, Carrera C, Lovatto L, et al. Benefits of total body photography and digital dermatoscopy (“two-step method of digital follow-up”) in the early diagnosis of melanoma in patients at high risk for melanoma. J Am Acad Dermatol. 2011 Jun 15; doi: 10.1016/j.jaad.2011.04.008. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Snoo FA, Bishop DT, Bergman W, et al. Increased risk of cancer other than melanoma in CDKN2A founder mutation (p16-Leiden)-positive melanoma families. Clin Cancer Res. 2008;14:7151–7157. doi: 10.1158/1078-0432.CCR-08-0403. [DOI] [PubMed] [Google Scholar]
- 42.Bishop DT, Demenais F, Goldstein AM, et al. Geographical variation in the penetrance of CDKN2A mutations for melanoma. J Natl Cancer Inst. 2002;94:894–903. doi: 10.1093/jnci/94.12.894. [DOI] [PubMed] [Google Scholar]
- 43.Udayakumar D, Tsao H. Melanoma genetics: an update on risk-associated genes. Hematol Oncol Clin North Am. 2009;23:415–429. vii. doi: 10.1016/j.hoc.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 44.Teerlink C, Farnham J, Allen-Brady K, et al. A unique genome-wide association analysis in extended Utah high-risk pedigrees identifies a novel melanoma risk variant on chromosome arm 10q. Hum Genet. 2012;131:77–85. doi: 10.1007/s00439-011-1048-z. [DOI] [PubMed] [Google Scholar]
- 45.Hoiom V, Tuominen R, Hansson J. Genome-wide linkage analysis of Swedish families to identify putative susceptibility loci for cutaneous malignant melanoma. Genes Chromosomes Cancer. 2011;50:1076–1084. doi: 10.1002/gcc.20931. [DOI] [PubMed] [Google Scholar]
- 46.Salerni G, Lovatto L, Carrera C, Puig S, Malvehy J. Melanomas detected in a follow-up program compared with melanomas referred to a melanoma unit. Arch Dermatol. 2011;147:549–555. doi: 10.1001/archdermatol.2010.430. [DOI] [PubMed] [Google Scholar]