

Abstract
We present a strategy to survey proteolytic processes in human cancer microenvironments. By combining in situ microdialysis during cancer surgery and mass spectrometry we were able to identify proteolytic enzymes, protease inhibitors and cleavage products in the interstitial fluid surrounding tumors and anatomically matched normal sites. Protease activity-based 18O-profiling revealed peptides processed by co-collected proteases ex vivo. This approach provides unique views of proteolytic networks in human cancers that could aid biomarker discovery efforts.
INTRODUCTION
Accurately characterizing proteolytic events in the tumor microenvironment provides crucial information on the communication between neoplastic cells, nonmalignant stromal cells and immune cells. Cancer-associated proteases participate in virtually all aspects of carcinogenesis, including malignant conversion, tissue invasion, and metastasis[1-3]. However, in vivo sampling of proteolytic events in cancer tissues has been previously only feasible and ethically acceptable in animal models[4]. In vivo sampling methods mitigate artifacts introduced by in vitro sampling approaches and provide access to proximal fluid enriched in biologically relevant molecules that likely constitute novel therapeutic targets and biomarkers[5] and give new insights into other protease-mediated processes such as cancer pain[6].
We introduce a novel strategy for sampling the secretome of human cancer in vivo and profiling its proteolytic activities. We captured, in real time, by microdialysis the interstitial fluid of tumors in patients undergoing surgical removal of oral squamous cell carcinomas. Following collection, we applied mass spectrometry-based proteomics approaches including our previously described PALeO method (proteinase activity labeling employing 18O-enriched water)[7] to simultaneously define the repertoire of proteins and peptides released by cancer cells and associated stromal cells and determine which members of the cancer secretome undergo active proteolytic processing (Fig. 1 and Methods and Materials). PALeO differs from other 18O-labeling proteomics strategies in that it exclusively relies on the enzymatic activities of endogenous proteases, while other approaches utilize exogenous proteases such as trypsin and focus primarily on quantitative comparisons[8].
Figure 1. Capturing proteolytic processes by intra-operative microdialysis and activity-based mass spectrometry.
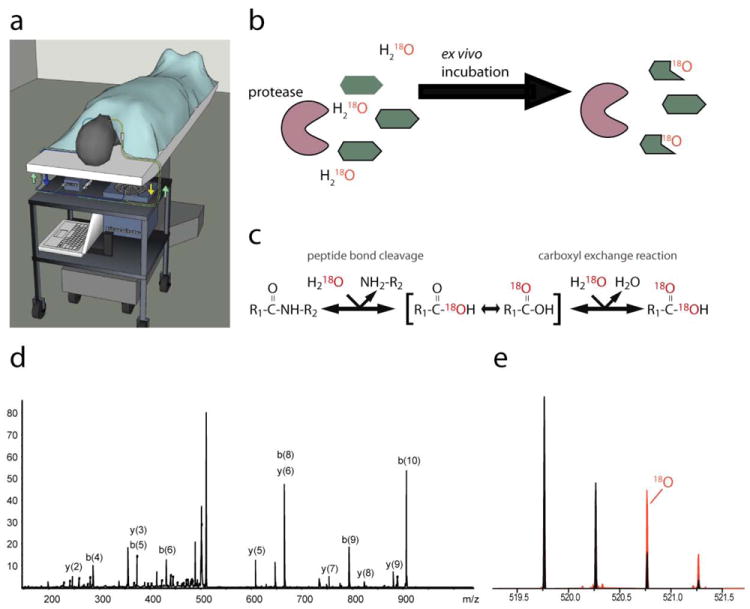
(a) Schematic representation of the microdialysis system (computer controlled pump and refrigerated microcollector) secured on a custom cart below the operating table. Once the patient is asleep and properly positioned, the microdialysis probes are atraumatically inserted into the oral cancer and normal (unaffected) site through the mouth, secured in place, and interstitial fluid typically collected for 4-7 hours. (b) Our “second-tier” experimental scheme of the mass spectrometric characterization of ongoing proteolytic activities in the samples using the PALeO-strategy. (c) Mechanism of the enzymatic incorporation of 18O-atoms in peptide cleavage products on which the PALeO-method is based. (d) Peptide sequence tag identifying the histone peptide TGASGSFKLN by MS/MS. (e) Peptide TGASGSFKLN underwent proteolytic cleavage ex vivo, as detected by the relative contribution of the 18O-labeled component to the isotopic envelope.
This general strategy should be applicable to other types of cancers and other biological processes involving proteolytic events and be particularly useful in the discovery of biomarkers for diseases.
Oral squamous cell carcinoma (SCC) provides an excellent model system to demonstrate proof-of-principle for this technique, because (i) oral SCC is accessible to placement of the microdialysis probe; (ii) the surgical treatment of oral SCC patients typically requires a neck dissection prior to removal, which allows microdialysate collection from the cancer as the neck dissection proceeds; and (iii) microdialysis probes can be placed in a contralateral, anatomically matched site that can serve as perfectly matched controls.
MATERIALS AND METHODS
The protocol for collecting interstitial fluid from the microenvironment of oral cancers in humans was approved by the University of California San Francisco Committee on Human Research, and written informed consent was obtained from all donors.
Collection of interstitial fluid perfusing oral cancers by microdialysis
We designed an intra-operative approach for the in situ collection of interstitial fluid from the cancer microenvironment without disrupting the cancer cells and surrounding tissue of the patient. The system consisted of a microdialysis pump (CMA 102; CMA Microdialysis AB) and a computer controlled refrigerated fraction collector (CMA 170; CMA Microdialysis AB) that were fitted together onto a cart underneath the operating room table (Fig. 1a). Once the patient was under general anesthesia and prior to the surgical procedure we inserted one microdialysis probe into the cancerous lesion and another probe into a normal, matched site. The microdialysis probe (CMA 71; CMA Microdialysis AB) consisted of a 10 mm polyethersulfone membrane with a pore dimension of 100 kDa. The probe had an inlet and outlet that allowed for a mobile phase to be pumped through it. As the physiological salt solution was slowly pumped through the probe, the solution equilibrated with the extracellular fluid. Once equilibrated, the fluid (mobile phase) contained a representative proportion of the molecules within the extracellular fluid. Extracellular proteins and peptides that were either secreted or shed by cells in the microenvironment were driven into the probe by the concentration differential. The probe was inserted perpendicular to the surface of the lesion for the length of the probe (10 mm) by an introducer (CMA SI-2, CMA Microdialysis AB) that minimized tissue trauma. Secondary probes were placed into contralateral, anatomically matched normal sites. The mobile phase perfusion fluid had a pH of 6.0, an osmolarity of 290 mosm/kg, and consisted of Na+ 147 mM, Ca+2 2.3 mM, K+ 4 mM, and Cl- 156 mM. The entire system was flushed with mobile phase. The mobile phase was then pumped at a fixed infusion rate. In the current study the flow rate was set at 0.5 μL per minute. Separate fractions were collected from the cancer and matched normal tissue over one-hour increments by the refrigerated fraction collector (4°C) throughout the neck dissection, which generally lasted 4-8 hours. Approximately one hour was required for probe equilibration. These fractions were excluded from the analysis. Following completion of the surgical procedure, samples were immediately frozen at -80°C.
Ex vivo profiling of proteolytic activities
For each sample, we combined the individual time fractions and incubated the samples for 16h at room temperature after adding an equal volume of 18O-enriched water (97%, Sigma Isotec). In the presence of H218O, the enzymatic hydrolysis of peptide bonds resulted in the incorporation of 18O-atoms in the C-termini of newly formed peptides. The PALeO strategy (protease activity labeling employing 18O-enriched water) that we employed to detect and characterize endogenous proteolytic reactions has been described previously[7]. After incubation, peptides were partitioned from proteins by ultrafiltration (12 000 × for 10 min; Vivaspin, nominal molecular weight limit 10 000). Peptide fractions were concentrated for mass spectrometry analysis by lyophilization and rehydration in 20 μL 5% acetonitrile/0.2% formic acid.
Protein sample preparation for mass spectrometry
During the ultrafiltration step, higher molecular weight proteins including proteases were retained above the membrane. Spin filters were incubated with 50μL of 8M Urea/2% SDS/150mM NH4HCO3/10mM DTT/1X LDS pH8.5 for 60 minutes at 37°C to recover these proteins. Next, samples were removed from the filters, cooled and alkylated with iodoacetamide and quenched with excess DTT. Samples were then centrifuged to pellet insoluble protein, and run 1/3 of the way into a one-dimensional 10% Bis Tris NuPAGE MOPS gel (Invitrogen). Gels were then fixed in destain (50% methanol and 7.5% acetic acid), rehydrated, stained with Simply Blue Safestain (Invitrogen), cut horizontally into two slices each, and destained until transparent. Gel samples were rinsed with three alternating washes of 50mM ammonium bicarbonate and acetonitrile. Samples were cooled to 4°C and subsequently each gel slice was resuspended in trypsin (5.5 μg/mL in 50mM ammonium bicarbonate/10% acetonitrile) and incubated at 37°C for 24 hours for digestion of proteins. Tryptic peptides were extracted with one rinse of 50mM ammonium bicarbonate/10% acetonitrile followed by one rinse of 50% acetonitrile/0.1% formic acid. Samples were prepared for mass spectrometry by lyophilization and rehydration in 20 μL 5% acetonitrile/0.2% formic acid.
LC-MS/MS analysis
Samples were loaded into 96-well plates for mass spectrometry analysis on an LTQ-Orbitrap XL (Thermo Fisher Scientific) instrument. For each run, we injected 10 μL of each re-constituted sample using a Thermo Scientific MicroAutosampler. Reverse phase chromatographic separation was performed using Hypersil GOLD™ C18™ 3μm media packed into a fused silica 75 μm inner diameter, 20 cm long column running at 250 nL/min from a Surveyor MS pump with a flow splitter. A gradient was produced between 5-40% acetonitrile, 0.2% formic acid over 150 minutes. The LTQ-Orbitrap was run in a top 8 configuration at 60K resolution for a full scan, with monoisotopic precursor selection enabled, and +1, and unassigned charge state rejected. The analysis on the LTQ-Orbitrap instrument was carried out with collision-induced dissociation fragmentation. In additional experiments, higher energy collisional dissociation fragmentation of ions was achieved at 15,000 resolving power in the LTQ-Orbitrap using an isolation window of 4.5, collision energy of 45, default charge state of 4 and activation time of 30 ms.
Mass spectrometry data analysis
Mass spectrometry data were analyzed using a laboratory information system created in-house that utilized MASCOT Distiller Software (Matrix Science) for spectral processing and peak detection. Peptides were identified by using the MASCOT algorithm (Matrix Science; version 2.2) to search against human proteins in the SwissProt database (Version 57.1; released April 2009) via tryptic digestion specifications in case of protein identifications and nonspecific digestion (i.e., “no enzyme” search) in the case of endogenous peptide identifications. Identities of peptides were confirmed by manual interpretation of the MS/MS data. 18O-labeling was calculated by using the Quantitation Toolbox of Mascot Distiller. Protein identifications across all experiments were ranked according to the summed score of all peptide identifications that were statistically significant (P < 0.05).
Immunohistochemistry
Immunohistochemistry for MMP8, MMP9, neurotrypsin, and trypsin-1 was performed on 8 μm sections of formalin-fixed, paraffin-embedded tissue specimens that were deparaffinized by standard immunohistochemical techniques. Sections were taken from the tumors of two patients whose dialysate was positive for these proteins. Microwave antigen unmasking was performed using Dako Antigen Retrieval Solution (Dako). Sections were then incubated with the primary MMP8 rabbit (ab53017), MMP9 rabbit (ab38898), neurotrypsin rabbit (ab5945, Abcam Inc.) or trypsin-1 goat (S-15, Santa Cruz Biotechnology) polyclonal anti-human antibody (1:200) at room temperature for 2 h. Immunoreactions were visualized with diaminobenzidine chromogen (Vector Laboratories), and sections were counterstained with Mayer’s hematoxylin. Tissue immunoreactivity was visualized on a Nikon Eclipse E600 microscope. The immunoreactivity of MMP8, MMP9, neurotrypsin, and trypsin-1 in normal oral mucosa (control) was compared to that in oral cancer tissue. The normal oral mucosa tissues were obtained from 3 healthy volunteers undergoing dental procedures such as wisdom tooth extraction or dental implant placement. Controls for MMP8, MMP9, neurotrypsin, and trypsin-1 included the omission of these primary antibodies.
RESULTS
This study was designed to develop a system to capture and study cancer proteolytic processes directly in oral cancer patients. We constructed an intra-operative microdialysis system that fits on a cart underneath the operating room table (Fig. 1a). Prior to the surgical procedure, we inserted one microdialysis probe into the cancerous lesion and another probe into a normal, anatomically matched site (e.g., SCC within the tongue and a contralateral, unaffected tongue site). Proteins below the molecular weight cut-off of the dialysis membrane (100 kDa) were captured by diffusion. We determined that flow rates of 0.5 μl/min after 60 minutes of probe equilibration yielded optimal protein recovery. After collection, microdialysates were subjected to two levels of proteomic analysis to profile proteolytic processes in the cancer secretions. First, proteins including proteases and protease inhibitors were separated on an SDS-PAGE gel and identified by liquid chromatography and tandem mass spectrometry (LC-MS/MS). Secondly, the PALeO-assay was used to identify peptides that were actively produced by collected proteases ex vivo (Fig. 1b-e).
We successfully completed intra-operative microdialysis on eight patients undergoing surgical resection of oral SCC lasting 4-7 hours. No intra-operative or post-operative complications related to probe placement or the microdialysis process were observed. Dialysates of two patients were selected for proteomics analysis. In total, we identified 217 proteins (≥ 2 peptide hits with P < .05) and an additional 290 proteins based on single peptide matches (Supplemental Table 1). To confirm the overrepresentation of components in proteolytic pathways[9], we submitted UniProt accession codes of identified proteins (≥ 2 peptide hits) to the DAVID knowledgebase[10] to reveal the enrichment of particular biological annotations. As expected from the literature, proteolytic enzymes as well as protease inhibitors were significantly enriched in the tumor site (Supplemental Table 2). Among the identified proteins were proteases with well-described roles in cancer biology such as members of the matrix metalloprotease family (MMP-8, MMP-9)[11], plasmin[12] and members of the complement cascade[13] (Fig. 2a). We confirmed the presence of the proteases identified by mass spectrometry by immunohistochemistry in oral biopsies collected from two patients who underwent intraoperative microdialysis and controls (Fig. 2b). Interestingly, two proteases with marginal MS-evidence (trypsin-1 and neurotrypsin) gave elevated immunopositivity levels, which suggested to us that additional low abundant proteases might be present in the samples that fell below the detection limit of our MS-approach.
Figure 2. Proteolytic enzymes detected in matched microdialysis and tumor samples of two cancer patients.
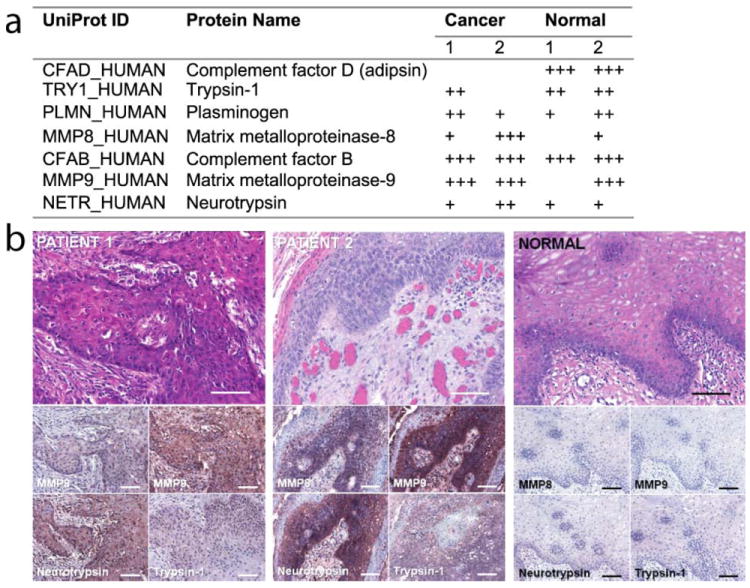
(a) Mass spectrometry-based evidence for selected proteases across the collected microdialysates (+++, > 2 peptide identifications with P < .05; ++, 1 hit with P < .05; +, multiple hits with P < .05). (b) Elevated immunopositivity levels of MMP8, MMP9, neurotrypsin, and trypsin-1 in cancer tissue compared to oral mucosa from a healthy patient (immunohistochemistry, bottom panels; H&E stains, top panels; horizontal scale bar = 100 μm).
Enzymatic activity does not necessarily correlate with enzyme abundance because enzymes, enzyme inhibitors, and substrates together form a dynamic enzymatic network that tightly regulates enzyme activity[14]. Likewise, proteases may exert their biological functions at abundances that are below the detection limits of current technologies. Activity-based profiling methods such as PALeO reveal protease activity by monitoring the generation of peptide products and depletion of substrates. We first queried the MEROPS peptidase database[15] for matches between identified proteases and endogenous inhibitors. Among the matches were pregnancy-zone protein, an endogenous inhibitor of MMP-9[16], and alpha-2-macroglobulin, an inhibitor of MMP-8 and MMP-9[17]. We also detected Serpin A1 a serine-proteinase inhibitor that targets plasmin and that itself can be inactivated by MMP-8 and MMP-9[18] (Supplemental Table 3). Thus, the co-existence of interacting proteases and endogenous inhibitors in the samples suggested an alteration of proteolytic activities that can only be assessed by the defining activity state of the proteases of interest. Accordingly, we scanned our list of identified proteins and peptides for predicted protease substrates and cleavage products according to the MEROPS database (Fig. 3). Among the substrate matches were auto-cleavage products for several proteases and 18 fibrinogen peptide species that matched to six cleavage sites recognized by plasmin (Fig. 3b). However, the majority of peptides did not correspond to any known cleavage sites recognized by the proteases of interest. Among these peptides was bradykinin, a mediator of inflammation, that can induce pain and previously has been reported as biomarker for breast cancer[19]. Bradykinin is generated by kallikreins through proteolytic processing of kininogen[20], a precursor protein that we also detected in the microdialysates. Other peptide precursor proteins detected in the microdialysates included angiotensinogen and hCAP-18, indicating that additional bioactive peptides (e.g., angiotensins, peptide LL-37) could also be present in the cancer microenvironment. The complete list of detected peptide precursor proteins and peptide identification are shown in Supplemental Tables 4 and 5, respectively.
Figure 3. Putative proteolytic processing in microdialysis samples.

(a) Observed protease-substrate matches according to the MEROPS knowledgebase of proteolytic reactions. (b) Observed fibrinogen peptides that corresponded to predicted cleavages sites for plasmin according to MEROPS.
Matching proteases to their physiological substrates is a major challenge in protease biology[21] and functional annotations of many proteolytic enzymes are sparse. The second step of our proteomic analysis was designed to fill this knowledge gap and provide unprecedented information of the in vivo functions of proteases in human cancer microenvironments. Specifically, we used the PALeO approach to identify the subset of peptides that was generated after sample collection by co-collected, catalytically active proteases. Our PALeO strategy exploits the fact that in the presence of isotopically enriched water enzymatic processing catalyzed by endogenous proteases introduces 18O-atoms into peptide products. Newly formed peptides are readily detected by their characteristic isotope ratios and identified by targeted MS/MS analyses (Fig. 1b-e). An overview of the proteolytic processes that were observed during the ex vivo incubation period is shown in Table 1. Surprisingly, among the peptides generated were histone fragments that could represent a new class of so-called alarmin molecules, which signal cell and tissue trauma[22, 23].
Table 1.
Examples of peptides that displayed 18O-incorporation in the PALeO-assay, which is characteristic of ex vivo formation by collected endogenous proteases.
| Precursors | Sequence | 16O | 18O1 | 18O2 | Sample |
|---|---|---|---|---|---|
| H12,H13, H14,H15 | GPPVSELITK | 0.87 | 0.29 | 0.02 | Cancer-2 |
|
| |||||
| H11,H12, H13,H14,H15 | GTLVQTKGTGASGSFKLN | 0.76 | 0.24 | 0.01 | Cancer-2 |
| GTLVQTKGTGASGSFKLN | 0.84 | 0.18 | 0.01 | Normal-2 | |
| VQTKGTGASGSF | 0.83 | 0.11 | 0.11 | Cancer-1 | |
| GTGASGSFKLN | 0.74 | 0.25 | 0.02 | Cancer-1 | |
| TGASGSFKLN | 0.80 | 0.21 | 0.00 | Cancer-1 | |
| SLVSKGTLVQT | 0.71 | 0.28 | 0.01 | Cancer-1 | |
|
| |||||
| FIBA_HUMAN | GKSSSYSKQFTSSTSYN | 0.70 | 0.19 | 0.12 | Cancer-1 |
| TVTKTVIGPDGHKEVT | 0.88 | 0.14 | 0.00 | Cancer-1 | |
| GEGDFLAEGGGVR | 0.81 | 0.34 | 0.00 | Cancer-2 | |
| KPVPDLVPGNF | 0.88 | 0.12 | 0.00 | Normal-2 | |
|
| |||||
| FIBB_HUMAN | RPAPPPISGGGY | 0.60 | 0.46 | 0.05 | Normal-1 |
| RPAPPPISGGGY | 0.60 | 0.59 | 0.35 | Normal-2 | |
|
| |||||
| PRPC_HUMAN | GRPQGPPQQGGHQQGPPPPPPGKPQ | 0.30 | 0.58 | 0.29 | Normal-2 |
| GRPQGPPQQGGHQQGPPPPPPGKPQGPPPQGGRPQ | 0.54 | 0.47 | 0.00 | Normal-2 | |
| GRPQGPPQQGGHQQGPPPPPPGKPQGPPPQGGRPQGPPQGQSPQ | 0.55 | 0.47 | 0.00 | Normal-2 | |
|
| |||||
| HBA_HUMAN | VLSPADKTNVK | 0.69 | 0.31 | 0.00 | Normal-2 |
|
| |||||
| HBB_HUMAN | DGLAHLDNLKG | 0.87 | 0.53 | 0.09 | Cancer-1 |
| DGLAHLDNLKGT | 0.81 | 0.19 | 0.05 | Normal-2 | |
| DGLAHLDNLKGT | 0.63 | 0.38 | 0.02 | Cancer-1 | |
| SDGLAHLDNLKG | 0.41 | 0.52 | 0.09 | Normal-2 | |
| AGVANALAHKYH | 0.74 | 0.17 | 0.13 | Normal-2 | |
DISCUSSION
We present here a new strategy to capture catalytically active proteases and identify novel, in vivo targets of proteolytic processing. We demonstrate for the first time the use of microdialysis in humans to collect interstitial fluid from cancer microenvironments for proteomic profiling. We validated this approach by accomplishing five lines of evidence: (i) by identifying established cancer-associated proteases in the microdialysate; (ii) by validating MS-based protease identifications from microdialysates with immunohistochemical staining of tissue biopsies; (iii) by identifying physiologically matching proteolytic enzymes, protease inhibitors, and substrates in the microdialysates; (4) by identifying established cancer-associated, bioactive peptides such as bradykinin; and (5) by demonstrating active proteolytic processing in the microdialysates.
The presented strategy provides unique insights into aspects of human cancer biology that are difficult to assay without disrupting the cancer or surrounding tissue. By targeting the interstitial fluid that bathes the cancerous lesion, we avoided the vast number of interfering proteins that would be encountered in peripheral fluids such as blood. Compared to a recent proteomics evaluation of the secretome of four head and neck cancer cell lines[24], the presented microdialysis technique was able to identify more proteins overall (217 vs. 140) and more proteases (9 vs. 4). Eight of the nine detected proteases, neurotrypsin being the notable exception, have been previously been associated with cancer. Additional cancer-associated proteases (e.g., cathepsins, ADAMs ADAMTS) may have eluded detection due the fact that the microdialysis approach only sampled secreted proteases, thereby excluding proteases that were membrane-bound, intracellular or that fell beyond the 100 kDa molecular weight limit of the microdialysis membrane. The heterogeneous composition of the tumors, interindividual variability and the general low abundance of proteases could also have contributed to the limited number of proteases detected.
We also identified a number of proteins in the dialysate that are not normally found in the extracellular space. Two such classes of proteins were the histones and blood-related proteins, which are likely present in the extracellular space secondary to inflammation and angiogenesis. Inflammation is a hallmark of cancer. Extracellular histones have been detected in hyperinflammatory states[25]. Angiogenesis occurs with carcinogenesis. The newly formed vessels are leaky and can lead to the extravasation of blood related proteins into the extracellular space.
The methodology presented here can be adapted broadly to other physiological conditions in which proteolytic mediators are involved (e.g., arthritic joints, inflamed muscle, other types of cancer) and where a comparison of normal and pathological tissue is sought. For example, protease-activated receptors on pain-sensing nerve endings are activated directly by proteolytic cleavage or indirectly via peptide products[6]. The downside of sampling by microdialysis was the inability to capture larger and membrane-bound proteases, however their presence may, in future iterations, be inferred by the presence of characteristic peptide cleavage products. Lastly, the strategy of combining microdialysis with activity-based proteomics can be expanded to incorporate other technologies such as the enrichment of proteolytic enzymes by chemical probes directed at their active sites[26] or the targeted surveillance of peptide cleavage products by multiple-reaction monitoring.
Supplementary Material
CLINICAL RELEVANCE.
The manuscript describes our new strategy to survey proteolytic processes that occur in human cancer microenvironments. We combined in situ microdialysis during oral cancer surgery and mass spectrometry-based proteomics to identify proteases and cleavage products in the interstitial fluid surrounding tumors and anatomically matched normal sites. By applying protease activity-based profiling we defined the set of peptides that are processed by co-collected proteases. The results demonstrate for the first time the use of microdialysis in humans to collect interstitial fluid from cancer microenvironments for proteomic profiling. Our findings show that this strategy can be used to obtain a novel view of in vivo targets of proteases without disrupting the cancer or surrounding tissue. The methodology can be broadly adapted to other physiological conditions in which proteolytic mediators are involved (e.g., arthritic joints, inflamed muscle, other types of cancer) and where a comparison of normal and pathological tissue is sought.
Acknowledgments
This work was supported by a UCSF Clinical and Translational Science Institute Investigator-Initiated Pilot Award and NIH/NIDCR Grant 1R01DE019796. We thank the BRIMS Center of Thermo Fisher Scientific and specifically Bryan Krastins for performing the MS-experiments.
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors declare no competing financial/commercial interests.
Contributor Information
Markus Hardt, Boston Biomedical Research Institute.
David K. Lam, Department of Oral and Maxillofacial Surgery, University of California San Francisco
John C. Dolan, Bluestone Center for Clinical Research, New York University
Brian L. Schmidt, Bluestone Center for Clinical Research, New York University.
References
- 1.Koblinski J, Ahram M, Sloane B. Unraveling the role of proteases in cancer. Clin Chim Acta. 2000;291:113–135. doi: 10.1016/s0009-8981(99)00224-7. [DOI] [PubMed] [Google Scholar]
- 2.DeClerck YA, Mercurio AM, Stack MS, Chapman HA, et al. Proteases, extracellular matrix, and cancer: a workshop of the path B study section. Am J Pathol. 2004;164:1131–1139. doi: 10.1016/S0002-9440(10)63200-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang C-M. In vivo secretome sampling technology for proteomics. Prot Clin Appl. 2007;1:953–962. doi: 10.1002/prca.200700031. [DOI] [PubMed] [Google Scholar]
- 5.Xue H, Lu B, Lai M. The cancer secretome: a reservoir of biomarkers. J Transl Med. 2008;6:52. doi: 10.1186/1479-5876-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmidt BL, Hamamoto DT, Simone DA, Wilcox GL. Mechanism of cancer pain. Mol Interv. 2010;10:164–178. doi: 10.1124/mi.10.3.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robinson S, Niles RK, Witkowska HE, Rittenbach KJ, et al. A mass spectrometry-based strategy for detecting and characterizing endogenous proteinase activities in complex biological samples. Proteomics. 2008;8:435–445. doi: 10.1002/pmic.200700680. [DOI] [PubMed] [Google Scholar]
- 8.Miyagi M, Rao KCS. Proteolytic 18O-labeling strategies for quantitative proteomics. Mass Spectrom Rev. 2007;26:121–136. doi: 10.1002/mas.20116. [DOI] [PubMed] [Google Scholar]
- 9.Mason SD, Joyce JA. Proteolytic networks in cancer. Trends Cell Biol. 2011;21:228–237. doi: 10.1016/j.tcb.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 11.Overall CM, Kleifeld O. Tumour microenvironment - opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat Rev Cancer. 2006;6:227–239. doi: 10.1038/nrc1821. [DOI] [PubMed] [Google Scholar]
- 12.McColl BK, Baldwin ME, Roufail S, Freeman C, et al. Plasmin activates the lymphangiogenic growth factors VEGF-C and VEGF-D. J Exp Med. 2003;198:863–868. doi: 10.1084/jem.20030361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rutkowski MJ, Sughrue ME, Kane AJ, Mills SA, Parsa AT. Cancer and the Complement Cascade. Molecular cancer research : MCR. 2010 doi: 10.1158/1541-7786.MCR-10-0225. [DOI] [PubMed] [Google Scholar]
- 14.auf dem Keller U, Doucet A, Overall CM. Protease research in the era of systems biology. Biol Chem. 2007;388:1159–1162. doi: 10.1515/BC.2007.146. [DOI] [PubMed] [Google Scholar]
- 15.Rawlings ND, Barrett AJ, Bateman A. MEROPS: the peptidase database. Nucleic Acids Res. 2010;38:D227–233. doi: 10.1093/nar/gkp971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arbeláez LF, Bergmann U, Tuuttila A, Shanbhag VP, Stigbrand T. Interaction of matrix metalloproteinases-2 and -9 with pregnancy zone protein and alpha2-macroglobulin. Arch Biochem Biophys. 1997;347:62–68. doi: 10.1006/abbi.1997.0309. [DOI] [PubMed] [Google Scholar]
- 17.Barrett AJ. Alpha 2-macroglobulin. Methods in Enzymology. 1981;80 Pt C:737–754. doi: 10.1016/s0076-6879(81)80056-0. [DOI] [PubMed] [Google Scholar]
- 18.Michaelis J, Vissers MC, Winterbourn CC. Human neutrophil collagenase cleaves alpha 1-antitrypsin. Biochem J. 1990;270:809–814. doi: 10.1042/bj2700809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Winden AWJ, van den Broek I, Gast M-CW, Engwegen JYMN, et al. Serum Degradome Markers for the Detection of Breast Cancer. J Proteome Res. 2010 doi: 10.1021/pr100395s. [DOI] [PubMed] [Google Scholar]
- 20.Campbell DJ. The renin-angiotensin and the kallikrein-kinin systems. Int J Biochem Cell Biol. 2003;35:784–791. doi: 10.1016/s1357-2725(02)00262-5. [DOI] [PubMed] [Google Scholar]
- 21.Marnett A, Craik CS. Papa’s got a brand new tag: advances in identification of proteases and their substrates. Trends Biotechnol. 2005;23:59–64. doi: 10.1016/j.tibtech.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 22.Coffelt SB, Scandurro AB. Tumors sound the alarmin(s) Cancer Res. 2008;68:6482–6485. doi: 10.1158/0008-5472.CAN-08-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gallucci S, Matzinger P. Danger signals: SOS to the immune system. Current Opinion in Immunology. 2001;13:114–119. doi: 10.1016/s0952-7915(00)00191-6. [DOI] [PubMed] [Google Scholar]
- 24.Ralhan R, Masui O, Desouza LV, Matta A, et al. Identification of proteins secreted by head and neck cancer cell lines using LC-MS/MS: Strategy for discovery of candidate serological biomarkers. Proteomics. 2011 doi: 10.1002/pmic.201000186. [DOI] [PubMed] [Google Scholar]
- 25.Xu J, Zhang X, Pelayo R, Monestier M, et al. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15:1318–1321. doi: 10.1038/nm.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cravatt BF, Wright A, Kozarich J. Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu Rev Biochem. 2008;77:383–414. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.