

Abstract
This study aimed to investigate the mechanisms that coordinate lymphangiogenesis. Using mouse models of lymphatic regeneration and inflammatory lymphangiogenesis, we explored the hypothesis that hypoxia inducible factor-α (HIF-1α) is a central regulator of lymphangiogenesis. We show that HIF-1α inhibition by small molecule inhibitors (YC-1 and 2-methyoxyestradiol) results in delayed lymphatic repair, decreased local vascular endothelial growth factor-C (VEGF-C) expression, reduced numbers of VEGF-C+ cells, and reductions in inflammatory lymphangiogenesis. Using transgenic HIF-1α/luciferase mice to image HIF-1α expression in real time in addition to Western blot analysis and pimonidazole staining for cellular hypoxia, we demonstrate that hypoxia stabilizes HIF-1α during initial stages of wound repair (1–2 wk); whereas inflammation secondary to gradients of lymphatic fluid stasis stabilizes HIF-1α thereafter (3–6 wk). In addition, we show that CD4+ cell-mediated inflammation is necessary for this response and regulates HIF-1α expression by macrophages, as CD4-deficient or CD4-depleted mice demonstrate 2-fold reductions in HIF-1α expression as compared to wild-types. In summary, we show that HIF-1α is a critical coordinator of lymphangiogenesis by regulating the expression of lymphangiogenic cytokines as part of an early response mechanism to hypoxia, inflammation, and lymphatic fluid stasis.—Zampell, J. C., Yan, A., Avraham, T., Daluvoy, S., Weitman, E. S., Mehrara, B. J. HIF-1α coordinates lymphangiogenesis during wound healing and in response to inflammation.
Keywords: hypoxia inducible factor-1α, CD4 cells, hypoxia
Lymphedema is a dreaded complication of cancer treatment that occurs in as many as 50% of patients who undergo lymph node dissection (1). Although the pathology of lymphedema remains poorly understood, it is clear that failed regeneration of disrupted lymphatics after injury is a contributing factor. Elucidation of mechanisms regulating lymphatic regeneration is, therefore, a critical step toward developing therapeutic interventions for lymphedema.
Vascular endothelial growth factor-C (VEGF-C) is a key regulator of lymphangiogenesis and has shown potential as a treatment for lymphedema by augmenting lymphatic repair after injury (2); however, the mechanisms that regulate and coordinate VEGF-C expression are not well defined. For example, although gradients of interstitial fluid flow have been shown to regulate VEGF-C expression during wound repair (3), the molecular mechanisms that translate this stimulus to VEGF-C expression remain unknown. Similarly, it is known that VEGF-C expression during wound repair is dependent on VEGF-A and can be modulated by inflammatory cytokines and macrophage migration; yet, it is not known how this complex process is coordinated or initiated (3–5). This gap in our understanding prevents clinical translation of VEGF-C-based therapies for lymphedema. Moreover, elucidation of the mechanisms that coordinate VEGF-C expression is a critical step in identifying pathological changes that result in lymphedema.
Hypoxia-inducible factor-1α (HIF-1α) is a transcription factor that plays a central role in coordinating angiogenesis (6). Angiogenic stimuli stabilize HIF-1α, enabling dimerization with HIF-1β, nuclear translocation, and activation of HIF-1α/β-responsive elements in target genes (7). In this manner, the cascade of target gene transcription by HIF-1α coordinates diverse physiological events, such as cellular migration, proliferation, and tubule formation, which ultimately results in angiogenesis (8).
Using mouse models of lymphatic regeneration, we show that HIF-1α plays a similar role in coordinating and initiating lymphangiogenesis by regulating the expression of lymphangiogenic cytokines. Further, we show that diverse lymphangiogenic stimuli, including hypoxia and gradients of lymphatic fluid stasis, regulate HIF-1α expression and thereby initiate lymphangiogenesis. HIF-1α accumulation in regions of lymphatic fluid stasis is driven in part through CD4+ cell-mediated chronic inflammation. Together, these data suggest that divergent stimuli, including hypoxia and inflammation, increase HIF-1α expression, and this response coordinates lymphangiogenesis.
MATERIALS AND METHODS
Mouse models
To determine the role of HIF-1α in lymphatic repair, a mouse tail model of lymphatic regeneration was performed in 8- to 10-wk-old female C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME, USA) as described previously (9, 10). Briefly, a surgical microscope (Leica, Wetzlar, Germany) was used to excise a 2-mm full-thickness circumference of skin to disrupt superficial lymphatics, followed by isolation and disruption of deep lymphatics. Wounds were filled with 0.3% type I rat collagen gel (BD Biosciences, Bedford, MA, USA), allowing for lymphatic repair across the collagen matrix (9, 11).
To determine the effects of lymphatic fluid stasis on HIF-1α expression, superficial and deep lymphatics were excised but not filled with collagen (9, 11), resulting in sustained edema. Control animals underwent circumferential tail skin incision without lymphatic ligation (12), and animals were sacrificed at designated time points for analysis (n=8/group). Axillary lymphadenectomy or sham operation (incision without lymph node removal) in C57BL/6 mice (n=7) was performed as described previously (12). All animal experiments were approved by the Institutional Animal Care and Use Committee at Memorial Sloan-Kettering Cancer Center.
HIF-1α blockade
Two small molecule inhibitors were used to antagonize HIF-1α via different mechanisms. Both compounds have been validated in studies demonstrating inhibitory activity against HIF-1α both in vivo and in vitro (13–16). 3-(5′-Hydroxymethyl-2′-furyl)-1-benzylindazole (YC-1; Cayman Chemical Co, Ann Arbor, MI, USA) inhibits hypoxia-dependent and mitogen-dependent HIF-1α accumulation (14) via partial suppression of phosphatidylinositol-3-OH kinase [PI(3)K]/Akt and nuclear factor-κB (NF-κB) pathways, as well as by functional inhibition of stabilized HIF-1α through blockade of coactivator binding (17). 2-Methyoxyestradiol (2ME; Santa Cruz Biotechnology, Santa Cruz, CA, USA) reduces HIF-1α synthesis at the post-transcriptional level and inhibits HIF-1α-induced activation of VEGF-A transcription (18).
Following lymphatic surgery, mice were randomized to an experimental group receiving YC-1 (30 μg/g in DMSO; Sigma-Aldrich, St. Louis, MO, USA) or control group (DMSO), injected intraperitoneally (i.p.; n=10/group). Additional mice (n=10/group) underwent tail surgery followed by randomization to 2ME [100 mg/kg in 10%DMSO/45% hydroxypropyl β-cyclodextran (HBC); Sigma-Aldrich] or vehicle control (DMSO/HBC). Animals in all groups were sacrificed at 21 d after surgery.
Lymphatic function
Tail volume measurements, dermal thickness, and microlymphangiography were performed according to our published methods (19) on d 21 postoperation (n=10/group). Briefly, tail volumes were calculated from tail diameter applied to a truncated cone formula, and dermal thickness was measured from the dermis to underlying fascia in histological sections. Microlymphangiography was performed by imaging lymphatic uptake and flow of intradermally injected fluorescein isothiocyanate (FITC)-conjugated, lysine-fixable dextran (2000 kDa, 2 mg/ml, Molecular Probes, Eugene, OR, USA) into the distal tail (20). Images were analyzed using a fluorescent microscope (Leica) and Volocity software (Perkin Elmer, Waltham, MA, USA).
Lymph node lymphangiogenesis model
C57BL/6 mice were treated with a 1:1 mixture of complete Freund's adjuvant (CFA) with 2% ovalbumin (OVA; Sigma-Aldrich) or phosphate-buffered saline (PBS; control) by subcutaneous footpad injection in the hind limb (12). Animals were treated with YC-1, 2ME, or respective controls for 5 d (n=8/group), after which popliteal lymph nodes were harvested.
Real-time analysis of HIF-1α expression
Transgenic mice that express a luciferase-HIF-1α fusion protein (ROSA26 ODD-Luc/+; Jackson Laboratory; ref. 21) underwent tail lymphatic disruption and wound coverage with collagen (n=10). HIF-1α expression was assessed in vivo using bioluminescence imaging performed preoperatively and then every 3 d for 9 wk following d-luciferin injection (150 mg/kg i.p.; Xenogen Corp., Alameda, CA, USA) with visualization using Live Image software (Xenogen).
Matrigel assay
Growth factor-reduced Matrigel (BD Biosciences, Chicago, IL, USA) containing deferoxamine (DFO; 500 μM; Sigma-Aldrich) or PBS were implanted subcutaneously in female mice (n=5; ref. 22). After 14 d, Matrigel plugs were sectioned, and the number of podoplanin-positive vessels per unit area was determined (Mirax software; Carl Zeiss, Oberkochen, Germany).
CD4+ cell depletion and CD4-knockout animals
C57BL/6 mice were depleted of CD4+ cells using anti-CD4 antibody (500 μg i.p., clone GK1.5; Bio-X-Cell, West Lebanon, NH, USA) administered 5 d prior to tail lymphatic ablation and continued every 3 d until d 7 postoperation; dosing then proceeded every 5 d until sacrifice (n=10). Controls were treated with isotype-matched antibody (500 μg i.p., IgG2b; Bio-X-Cell; n=10). Flow cytometry on isolated splenocytes (n=3/group) confirmed CD4 depletion using our methods (19). Identical operations were performed in CD4-knockout mice (B6.129S2-Cd4tm1Mak/J; Jackson Laboratory) or wild types (C57BL/6), and tissues were analyzed at 6 wk postoperation (n=8/group).
Histology
Cross-sectional (5 mm proximal or distal to the tail wound) or longitudinal (10 mm centered on the wound) tissue sections were harvested after surgery, fixed, paraffin embedded, and sectioned at 5 μm. Lymph nodes were embedded in optimal cutting temperature (OCT) compound (Tissue-Tek, Hatfield, PA, USA) and sectioned at 5–8 μm.
Primary antibodies for immunohistochemical staining were against VEGF-A, VEGF-C, podoplanin, von Willibrand factor (vWF), and HIF-1α (all from Abcam, Cambridge, MA, USA) and visualized using biotinylated secondary antibodies followed by an avidin-peroxidase complex (Vectastain; Vector Laboratories, Burlingame, CA, USA) and detection with 3,3′-diaminobenzidine tetrahydrochloride (DAB; Dako, Carpinteria, CA, USA) (19). Images were captured using a Mirax slide scanner (Carl Zeiss). Immunofluorescence staining was performed for lymphatic vessel endothelial receptor-1 (LYVE-1; R&D Systems, Minneapolis, MN, USA), visualized with fluorescent secondary antibodies (Invitrogen, Carlsbad, CA, USA), and analyzed using an Axioskope 40 fluorescent microscope (Carl Zeiss) with Metamorph software (Molecular Devices Corp., Sunnyvale, CA, USA). Coimmunohistochemistry was performed by immunohistochemical stains for LYVE-1 and VEGF-C followed by detection with DAB and NovaRed (Vector Laboratories); spectral component separation was performed using Nuance Multispectral Imaging System (CRI, Woburn, MA, USA).
For detection of cellular hypoxia, mice were administered pimonidazole hydrochloride (60 mg/kg i.p., Chemicon International, Temecula, CA, USA) immediately prior to sacrifice (23), and staining was performed using the Hypoxiaprobe-1 plus Kit (Chemicon). The number of positively-stained cells (HIF-1α, VEGF-C, VEGF-A, or pimonidazole), lymphatic vessels (podoplanin, vWF, or LYVE-1+) was assessed in 5 high-power fields/animal (×40; n=5–10/group) by observers in a blinded procedure. Cross-sectional areas of podoplanin-positive vessels were calculated using the formula πr1r2 (19). Lymphatic vessel density within lymph nodes was determined using Metamorph software and expressed relative to node area (vessels/mm2; n=6–8/group).
Protein analysis
Skin and subcutaneous tissues were harvested at standardized regions of the tail or axilla (n=5–10/group). Total cellular protein was extracted using tissue protein extraction reagent (ThermoFisher Scientific, Rockford, IL, USA) and quantified using the Bradford technique (Bio-Rad Protein Assay; Bio-Rad, Hercules, CA, USA).
Pooled protein (3–5 animals) was analyzed by Western blot performed in triplicate (12). Primary antibodies were used against HIF-1α (rabbit polyclonal antibodies and mouse monoclonal antibody, clone H1α67), VEGF-C (mouse monoclonal, Santa Cruz Biotechnology; and rabbit polyclonal, Novus, Littleton, CO, USA), carbonic anhydrase IX (CA-IX; Abcam), pan-Akt (Abcam), Prox-1 (AngioBio, Del Mar, CA, USA), LYVE-1 (Abcam), VEGF-A (Santa Cruz Biotechnology), and phosphorylated Akt (pAKT; Cell Signaling Technology, Danvers, MA, USA) followed by HRP-conjugated secondary antibodies (Santa Cruz Biotechnology) and detected with ECL Plus Western-Blotting Detection System (GE Healthcare, Little Chalfont, UK). Equal loading was confirmed using β-tubulin or β-actin (Abcam), and expression was normalized using ImageJ software (http://rsweb.nih.gov/ij). VEGF-C ELISA was performed using rat VEGF-C Platinum ELISA (eBiosciences, San Diego, CA, USA).
In vitro assays
Human dermal lymphatic endothelial cells (LECs; PromoCell, Heidelberg, Germany) were cultured in endothelial cell growth medium-MV2 (ECGM-MV2; PromoCell), and 1 × 106 cells were cultured in a hypoxia chamber (1% O2, 4% CO2, nitrogen balance; Billups-Rothenberg, Inc., Del Mar, CA, USA) or remained in normoxia and were harvested at multiple time points. Additional groups were cultured with YC-1 (100 μM) or DMSO control, cobalt (10 μM; Hampton Research, Aliso Viejo, CA, USA), actinomycin (5 μg/ml, Sigma-Aldrich), or cyclohexamide (10 μg/ml, Sigma-Aldrich). RNA and protein were isolated from cellular extracts using the Allprep DNA/RNA/protein kit (Qiagen Inc., Valencia, CA, USA).
PCR
Reverse transcription was performed using TaqMan Reverse Transcription reagents (Applied Biosystems, Foster City, CA, USA) followed by quantitative reverse transcriptase polymerase chain reaction (RT-PCR) using TaqMan Universal Mastermix (Applied Biosystems) and LightCycler thermocycler (Roche Diagnostics, Indianapolis, IN, USA). Expression of HIF-1α (primer Mm00468875_m1; Applied Biosystems) and VEGF-C (Mm01202432_m1; Applied Biosystems) was normalized to 18S. Experiments were performed in triplicate.
Statistical analysis
Multigroup comparison was performed using 1-way ANOVA with Tukey-Kramer post hoc test. Student's t test was used for analyzing differences between 2 groups. Data are presented as means ± sd unless otherwise noted; values of P < 0.05 were considered significant. All figures presented are representative of experiments performed in triplicate.
RESULTS
HIF-1α inhibition increases edema and lymphatic fluid stasis after wounding
YC-1 treatment reduced HIF-1α expression 2.2-fold in tissues surrounding the wound 3 wk after surgery (Fig. 1A). Similarly, carbonic anhydrase-IX (CA-IX), a surrogate marker for HIF-1α stabilization, was reduced 1.5-fold. YC-1-treated animals exhibited minor increases in Akt phosphorylation relative to total Akt. 2ME was less effective but also reduced HIF-1α (1.5-fold) and CA-IX (1.9-fold) expression and did not alter Akt phosphorylation.
Figure 1.

HIF-1α inhibition increases edema and lymphatic fluid stasis after wounding. A) Western blot analysis of protein harvested from tissue at the wound bed (boxed region) 3 wk postoperatively in animals treated with YC-1, 2ME, or respective vehicle control (fold-change values relative to controls are shown). B) Photographs of mouse tails following YC-1 (top), 2ME (bottom), or vehicle control treatment 3 wk postoperatively, demonstrating tail swelling following HIF-1α inhibition. C) Change in tail volume at 3 wk after surgery as compared to preoperative levels, demonstrating increased tail lymphedema following HIF-1α inhibition. D) Dermal thickness (μm) at 3 wk after surgery. E) H&E stains from transverse tail sections immediately distal to the wound bed. Arrows indicate dermal thickness. F, G) Podoplanin+ lymphatic vessel area (μm2; F) and representative photomicrographs (G) in YC-1, 2ME, and controls. Arrows indicate dilated lymphatics. Scale bars = 200 μm (E); 50 μm (G). n.s., not significant. **P < 0.01, ***P < 0.001.
Inhibition of HIF-1α markedly increased tail lymphedema. This response was obvious in YC-1-treated animals, in which tail volumes were increased 4-fold as compared with controls at 3 wk after surgery (P<0.001; Fig. 1B, C). Treatment with 2ME similarly increased tail edema; however, this response failed to reach significance (P=0.079). These findings correlated with increases in dermal thickness following treatment with either YC-1 (56.1% increase, P<0.001) or 2ME (26.1% increase, P<0.01, Fig. 1D, E). Lymphatic vessel area analysis revealed a 3.1-fold (P<0.01) and 4.4-fold (P<0.001) increase as compared with controls following YC-1 or 2ME treatment, respectively (Fig. 1F, G).
Inhibition of HIF-1α impairs lymphangiogenesis and delays lymphatic repair
To determine whether HIF-1α blockade alters interstitial fluid flow, we performed microlymphangiography at 3 wk postoperatively, following YC-1 or 2ME treatment. Reduced interstitial fluid flow following HIF-1α inhibition was evidenced by pooling of FITC-labeled lymphatic tracer distal to the tail wound (Fig. 2A). This finding was corroborated by immunofluorescence staining of lymphatic vessels demonstrating LYVE-1+ vessels crossing the wound in control but not experimental groups (Fig. 2B). Furthermore, HIF-1α inhibition resulted in reductions in podoplanin+ vessel density within 5 mm of the wound (21.0% reduction following YC-1, P<0.05, and 49.0% reduction following 2ME, P<0.01; Fig. 2C). Despite more significant reductions in vessel numbers, 2ME-treated groups demonstrated smaller reductions in edema than YC-1 groups; however, this may reflect slight differences in lymphatic function of existing lymphatics in animals of the 2ME experiment, suggested by smaller vessel diameter and less ectatic nature of these vessels (Fig. 1F, G). Given smaller reductions in vessel number following YC-1 treatment, we stained with LYVE-1, which revealed a 54.0% reduction in LYVE-1+ vessels (P<0.01; Fig. 2D). Western blot of these regions for lymphatic differentiation markers demonstrated decreased LYVE-1 expression (2.2-fold for YC-1; 1.4-fold for 2ME); however, Prox1 was unaltered (Fig. 2E). VWF immunohistochemistry revealed no differences in blood vessel numbers among groups (Fig. 2F), and it may be possible that staining occurred preferentially in established blood vessels rather than newly formed capillaries.
Figure 2.

Inhibition of HIF-1α impairs lymphangiogenesis and delays lymphatic repair. A) Representative microlymphangiography 3 wk following surgery (boxed); arrow indicates direction of lymphatic flow. Lymphatic tracer is visualized crossing the wound (white arrow) in controls. B) Immunofluorescent photomicrograph for LYVE-1 demonstrating lymphatic vessels traversing wound (arrow) in controls. C) Number of podoplanin+ vessels per high-power field (left panel) and representative photomicrographs (right panel) in YC-1, 2ME, and respective controls. Arrowheads indicate lymphatics; boxed region in gross photograph represents region used for analysis. D) Number of LYVE-1+ vessels per high-power field and representative photomicrographs (red, LYVE-1; blue, nuclear stain) demonstrating reduced numbers of lymphatic vessels following YC-1 treatment. E) Western blot of protein harvested from tissue centered at the wound (boxed) 3 wk after surgery in animals treated with YC-1, 2ME, or respective vehicle control (fold change relative to control). F) Number of VWF+ vessels per high-power field. G–I) Cross-sectional lymph node area (μm2; G) and lymphatic vessel density (H) in popliteal nodes following YC-1 (G–I), 2ME (G–I), or vehicle control treatment in PBS- or CFA-OVA-treated animals, demonstrating reduced lymph node size and lymphatic density following HIF-1α inhibition. I) Immunofluorescent images of LYVE-1 (red)- and DAPI (blue)-stained lymph nodes. J) Number of podoplanin+ lymphatic vessels in DFO- or PBS-supplemented Matrigel plugs. Representative low-power (×10; left) and high-power (×40; right; boxed) photomicrographs of podoplanin-stained Matrigel plugs are shown. K) Low- and high-power images of podoplanin stains showing specificity for lymphatic vessels (arrow) with lack of staining of blood vessel endothelium (arrowhead). Scale bars = 100 μm (B, I, J, K); 75 μm (C); 50 μm (D). n.s., not significant. *P < 0.05, **P < 0.01.
To determine the effects of HIF-1α on lymphangiogenesis independent of wound microenvironments, we utilized an inflammatory lymphangiogenesis lymph node model. Following YC-1 treatment, both lymph node enlargement and lymphatic vessel density in response to CFA-OVA were significantly reduced (2.4-fold reduction of node area, P<0.01; 2.2-fold reduction in lymphatic vessel density, P<0.05; Fig. 2G–I). Consistent with the decreased potential for 2ME to inhibit HIF-1α, 2ME-induced reductions in both mean node area and lymphatic vessel density failed to reach significance (P=0.057; Fig. 2G–I).
To determine the effects of HIF-1α stabilization independent of inflammation or wound healing, we modified a Matrigel lymphangiogenesis assay by subcutaneously implanting Matrigel containing deferoxamine (DFO; an iron chelator that stabilizes HIF-1α) or PBS. At 2 wk after implantation, lymphatic infiltration within DFO-supplemented Matrigel plugs was increased 1.9-fold compared to controls (P<0.01, Fig. 2J), suggesting that HIF-1α stabilization directly promotes lymphangiogenesis. Specificity of podoplanin is demonstrated by intense staining of lymphatic endothelium and lack of staining in surrounding blood vessel endothelium (Fig. 2K).
HIF-1α inhibition decreases expression of VEGF-A and VEGF-C
We next sought to determine the influence of HIF-1α on VEGF-A or VEGF-C during lymphangiogenesis. Inhibition of HIF-1α with YC-1 markedly decreased expression of both VEGF-A and VEGF-C mRNA (2.3- and 2.1-fold, respectively) in tissues immediately surrounding the wound (±5 mm) at 3 wk after surgery (P<0.01, Fig. 3A), resulting in a 31.6% reduction in VEGF-C expression (P<0.01; Fig. 3B). Western blot also demonstrated reductions in VEGF-A (3.1-fold following YC-1; 1.4-fold following 2ME) and VEGF-C expression (1.3-fold following YC-1; 1.9-fold following 2ME, Fig. 3C). Immunohistochemical localization of VEGF-A or VEGF-C demonstrated that YC-1, and to a lesser extent 2ME, led to reduced VEGF-A+ cells in tissues immediately surrounding the wound (51.9% decrease with YC-1, P<0.001; 19% decrease with 2ME, not significant, P=0.081; Fig. 3D). Both inhibitors led to significantly reduced VEGF-C+ cells (59.0 and 55.4% reduction following YC-1 and 2ME, respectively, P<0.001; Fig. 3E). To further study the role of hypoxia in regulating VEGF-C expression, we performed immunohistochemical colocalization and observed VEGF-C expression in LECs of control animals, and this expression was reduced following animals HIF-1α blockade (Fig. 3F, G).
Figure 3.

HIF-1α inhibition decreases expression of VEGF-A and VEGF-C. A) Relative expression of VEGF-A or VEGF-C mRNA in the 1-cm region centered at the wound (boxed area) after treatment with YC-1 or control 3 wk after surgery (fold change relative to control). B) ELISA for local VEGF-C expression following YC-1 treatment for 3 wk (pg/ml). C) Western blot for VEGF-A and VEGF-C following treatment with YC-1, 2ME, or respective controls at 3 wk after surgery (fold change relative to controls). D, E) Number of VEGF-A+ (D) and VEGF-C+ (E) cells per high-power field in the 5-mm region distal to the wound following HIF-1α inhibition or control treatment for 3 wk. High-power photomicrographs (×40) of immunohistochemical staining for VEGF-A (D) and VEGF-C (E) are shown. F) Numbers of LYVE-1/VEGF-C double-positive cells, determined by immunohistochemical colocalization, in the region 5-mm distal to the wound. G) High-power photomicrographs of LYVE-1 and VEGF-C colocalization. Red, LYVE-1; green, VEGF-C. H) VEGF-C expression (pg/ml) by ELISA in draining lymph nodes following YC-1 or vehicle control treatment in animals administered CFA-OVA. I, J) Left panels: number of VEGF-A+ (I) or VEGF-C+ (J) cells per high-power field in draining lymph nodes following treatment with YC-1, 2ME, or respective controls after CFA-OVA injection. Right panels: high-power photomicrographs (×40) for VEGF-A (I) and VEGF-C (J) within inflamed draining lymph nodes. Scale bars = 50 μm (D, E); 40 μm (G); 100 μm (I, J). *P < 0.05, **P < 0.01, ***P < 0.001.
Similar to our wound model, YC-1 treatment resulted in a 36.6% reduction of VEGF-C expression in popliteal nodes after CFA-OVA treatment (P<0.05; Fig. 3H). Decreased VEGF-C expression correlated with fewer VEGF-A and VEGF-C+ cells within inflamed nodes following HIF-1α inhibition (2.7- and 5.7-fold reduction in VEGF-A+ cells per high-power field following YC-1 and 2ME treatment, respectively, P<0.01; Fig. 3I; 6.9- and 8.5-fold reduction in VEGF-C+ cells per high-power field following YC-1 and 2ME treatment, respectively, P<0.001; Fig. 3J). Together, these findings suggest that one mechanism by which HIF-1α regulates wound healing and inflammatory lymphangiogenesis is through modulation of prolymphangiogenic growth factors.
Hypoxia stabilizes HIF-1α in early phases of wound healing
To determine mechanisms regulating HIF-1α expression in the wound, tail operations were performed in ROSA26 ODD-Luc/+ (ODD-Luc/+) mice in which the luciferase reporter is fused to a HIF-1α gene residue subject to oxygen-dependent degradation, thereby enabling HIF-1α imaging in vivo (21). Bioluminescence imaging revealed the greatest HIF-1α expression within and just proximal to the wound in the first 2 wk postoperation (Fig. 4A); by d 21 postoperation, HIF-1α expression appeared equal in regions immediately proximal and distal to the wound, with peak expression in the wound. Western blot revealed similar patterns of HIF-1α expression in the wound, particularly at 2 wk postoperation, in which HIF-1α expression was increased compared to that in control tissues and at 1 wk time points (Fig. 4B). Increases in HIF-1α expression correlated with increased VEGF-A (Fig. 4B) and modest increases in VEGF-C.
Figure 4.

Hypoxia stabilizes HIF-1α in the early phases of wound healing. A) Representative bioluminescent images of ROSA26 ODD-Luc/+ mice following luciferin injection before and after surgery, showing high levels of HIF-1α expression in the regions surrounding the wound (arrows). Color bar indicates photons per square centimeter per second per steradian. B) Western blot for HIF-1α, VEGF-A, VEGF-C, and β-actin at 1 and 2 wk after surgery in preoperative controls and the wound. C) High-power (×40; top panel) and low-power (×2; bottom panels) photomicrographs of HIF-1α immunohistochemical staining in the wound bed and surrounding areas (5 and 1 mm proximally and distally) at 3 wk postoperation. Note HIF-1α localization to inflammatory cells with the phenotypic appearance of macrophages. D) High-power photomicrograph (×20, left panel; ×40 view of boxed area, right panel) of HIF-1α immunohistochemical stains, demonstrating cellular staining in macrophages (arrows) surrounding the microvasculature. E) Number of pimonidazole+ cells located at various regions relative to the wound at 1, 2, or 3 wk following surgery. Note highest number of hypoxic cells in regions surrounding the wound at 2 wk. **P<0.01, ***P<0.001; †P < 0.05 vs. −1 mm; ‡P < 0.01 vs. +1 mm. F) Pimonidazole immunohistochemistry at 2 wk after surgery (×40, top panels; ×2, bottom panels). Scale bars = 50 μm (C, F); 100 μm (D).
Immunohistochemical localization of HIF-1α demonstrated the highest density of HIF-1α+ cells immediately adjacent to the wound and within perivascular regions (Fig. 4C, D). Cellular expression of HIF-1α was largely confined to infiltrating macrophages, and may represent migration of activated macrophages from adjacent blood vessels into these tissues (Fig. 4D).
To confirm that HIF-1α expression correlated with gradients of tissue hypoxia, animals were treated with pimonidazole prior to sacrifice to label hypoxic cells. Similar to our findings with ODD-Luc/+ mice, localization of hypoxic cells (pimonidazole+) demonstrated that the highest degree of hypoxia was within the wound and peaked at 2 wk after surgery (Fig. 4E, F). Although hypoxic cells were present in the wound and surrounding areas at 3 wk postoperation, the number of hypoxic cells was reduced compared to earlier time points (75% reduction, P<0.001), indicating resolving tissue hypoxia. It is important that HIF-1α expression in ODD-Luc/+ mice remains up-regulated at the 3-wk time point, and this is in contrast to the resolution of tissue hypoxia observed in pimonidazole stains. Together, these findings support that other factors contribute to HIF-1α expression at these later postoperative time points.
Hypoxia regulates HIF-1α and VEGF-C expression in isolated LECs
To further study the role of hypoxia in regulating HIF-1α and VEGF-C, we exposed LECs to hypoxic gradients or hypoxia-mimetic compounds. In support of our in vivo observations, hypoxia (1% O2) led to increased HIF-1α expression over 24 h (Fig. 5A). While HIF-1α protein increased over 24 h, absolute mRNA peaked at 6 h and returned to baseline by 24 h (data not shown, P<0.05). Under the same conditions, VEGF-C mRNA correspondingly peaked at 6 h (2.4-fold increase, P<0.01; Fig. 5B), and protein expression increased over time (7.3-fold increase by 24 h, P<0.001; Fig. 5C). LECs cultured with cobalt, a direct HIF-1α agonist, similarly had marked increases in VEGF-C mRNA after 6 h (2.5-fold increase, P<0.001; Fig. 5D). Treatment of hypoxic LECs with YC-1 reduced VEGF-C mRNA expression (35.8%; P<0.01); however, this reduction did not return VEGF-C expression to normoxic levels, suggesting that additional mechanisms contribute to VEGF-C expression under these circumstances (Fig. 5E). The observation that VEGF-C gene expression peaks early but returns to baseline prior to peak HIF-1α expression has been made previously for both VEGF-A and VEGF-C (24), and it is possible that additional mechanisms are activated to return cellular expression back to baseline despite continued HIF-1α expression. Together, these findings suggest that LECs are responsive to hypoxia and up-regulate VEGF-C expression, in part, by HIF-1α-dependent mechanisms.
Figure 5.

Hypoxia regulates HIF-1α and VEGF-C expression in isolated LECs. A) Western blot of protein extracts from human dermal LECs cultured in normoxia (21% O2 for 24 h) or hypoxia (1% O2) for 12 or 24 h demonstrating increased HIF-1α expression in response to hypoxia. B) Relative VEGF-C mRNA expression in LECs cultured in normoxia or hypoxia (1% O2) for 3, 6, 12, or 24 h (fold increase relative to normoxia). C) VEGF-C expression (pg/ml) by ELISA of protein extracts from LECs cultured in normoxia or hypoxia for 12 or 24 h. D) Relative VEGF-C mRNA expression in LECs cultured under normoxic conditions with or without cobalt (HIF-1α agonist) for 6 h (fold increase relative to normoxia). E) Relative VEGF-C mRNA expression in LECs cultured under normoxic or hypoxic conditions with or without YC-1 (100 μM) for 6 h (fold increase relative to normoxia). F) Relative VEGF-C mRNA expression in LECs cultured under normoxic or hypoxic conditions with or without cyclohexamide (10 μg/ml) for 6 h, followed by normoxia or hypoxia for 6 h (fold increase relative to normoxia). G) VEGF-C mRNA expression in LECs cultured under normoxic or hypoxic conditions for 6 h, followed by actinomycin (5 μg/ml) to block mRNA transcription for 0, 1, 2, or 3 h. No significant difference in slopes was noted by linear regression. n.s., not significant. *P < 0.05, **P < 0.01, ***P < 0.001.
No significant differences in VEGF-C mRNA expression occurred following inhibition of protein translation with cyclohexamide (Fig. 5F), suggesting that de novo protein synthesis is not required for up-regulation of VEGF-C mRNA in response to hypoxia. This finding is consistent with the fact that a primary mechanism of HIF-1α regulation occurs as a result of HIF-1α accumulation through escape of proteosomal degradation rather than new synthesis. Finally, VEGF-C mRNA degradation following blockade of transcription by actinomycin D (an inhibitor of RNA polymerase) under normoxic or hypoxic conditions occurred at similar rates, suggesting that VEGF-C mRNA stability in LECs is unaffected by hypoxia (Fig. 5G).
HIF-1α expression is regulated by lymphatic fluid stasis
When evaluating patterns of HIF-1α expression in ODD-Luc/+ mice for prolonged periods of time, HIF-1α expression remained elevated in regions distal to the zone of lymphatic injury even after resolution of tissue hypoxia (Fig. 6A). Elevations of HIF-1α expression persisted until tail lymphedema resolved 9 wk after surgery, suggesting that lymphatic fluid stasis can regulate HIF-1α independent of hypoxia.
Figure 6.

HIF-1α expression is regulated by lymphatic fluid stasis. A) Bioluminescent images of ODD-Luc/+ mice following luciferin injection at indicated preoperative and postoperative time points (d 28–63). Inset: wound (white arrow) and regions distal to wound. Note sustained HIF-1α expression in distal lymphedematous regions at d 28 and 35. B) Western blot of protein harvested from proximal (−15 mm) and distal (+15 mm) regions at 6 wk after tail skin and lymphatic excision. Fold-change values relative to proximal region are shown. C) Top panel: regions of analysis. Middle panels; low-power photomicrographs (×2.5) of HIF-1α immunohistochemical staining in cross-sections obtained from proximal and distal regions of the tail (±15 mm from the wound) at 6 wk postoperation. Bottom panels: high-power photomicrographs (×60) demonstrating HIF-1α immunolocalization to inflammatory cells, including perivascular macrophages in tissues exposed to lymphatic fluid stasis. Scale bars = 1000 μm (middle panels);100 μm (bottom panels). D) Photograph following tail skin incision without lymphatic ligation (incision; top panel) and skin excision with lymphatic ablation (excision; bottom panel) at 21 d after surgery. Note lack of swelling in distal tail in incision-treated animal. Percentage change in tail volume from preoperative baseline is displayed for 7 and 21 d postoperation. *P < 0.05. E) Western blot of pooled tail protein harvested from animals treated with incision or excision and lymphatic ligation at 3 wk postoperation (fold change relative to incision). F) Western blot for HIF-1α of protein harvested 3 wk after tail skin incision (INC) or tail skin and lymphatic excision (EXC) from regions of the tail as noted to determine effects of gradients of lymphatic stasis on HIF-1α expression. G) Western blot of protein from animals treated with axillary incision (sham) or axillary dissection (AXD) at 3 wk postoperation (fold change relative to sham). Note increased HIF-1α in areas exposed to severe lymphatic fluid stasis (upper arm) as compared to less severe stasis (lower arm).
To test this hypothesis, we compared HIF-1α expression in regions of lymphatic fluid stasis (+15 mm distal to the wound) to nonlymphedematous regions an equal distance proximal to the wound (−15 mm) at 6 wk postoperation (Fig. 6B). Consistent with our observations in ODD-Luc/+ mice, Western blot revealed a 1.6-fold increase in HIF-1α expression in lymphedematous regions. Increased HIF-1α correlated with increased expression of both VEGF-A (3.1-fold) and VEGF-C (1.4-fold), although pAkt was reduced in distal tail regions (0.3-fold), suggesting that HIF-1α stabilization occurs independent of PI(3)K/Akt activation in this setting. Consistent with the increased expression of VEGF-C, LYVE-1 expression was increased 1.5-fold in lymphedematous tissues, although Prox1 remained unchanged. Localization of HIF-1α by immunohistochemistry in tail tissues demonstrated markedly increased staining in lymphedematous tissues (Fig. 6C, D). Consistent with our previous findings, a chronic mononuclear inflammatory reaction was present in lymphedematous tissues (19). In addition, HIF-1α expression in lymphedematous tissues localized primarily to macrophages, suggesting that these cells are a primary source of HIF-1α in response to sustained lymphatic fluid stasis.
We used two additional models to test the hypothesis that lymphatic fluid stasis can regulate HIF-1α expression independent of wounding. First, we compared HIF-1α expression in distal tail segments of animals that underwent skin and lymphatic excision with those treated with skin incision only. Consistent with our previous reports, skin incision resulted in no increase in tail volume (Fig. 6D; ref. 12). In contrast, animals treated with tail skin and lymphatic excision demonstrated significant increases in tail volume (P<0.001). Western blot analysis demonstrated marked increases in HIF-1α in tissues from animals with sustained lymphatic stasis compared with those treated with skin incision alone at regions distal to the wound (Fig. 6E, F). These changes in HIF-1α expression correlated with markedly increased expression of CA-IX, VEGF-A, and VEGF-C.
To determine whether lymphatic fluid stasis regulates HIF-1α expression in a clinically relevant model, we compared HIF-1α expression in upper extremity tissues after axillary lymph node dissection (AXD) or axillary incision without lymph node removal (sham). We have shown that this procedure results in minor, though significant, increases in arm circumference persisting at least 3 wk postoperatively (12). Western blot of skin and subcutaneous tissues demonstrated that AXD resulted in significantly increased HIF-1α expression (3.4-fold increase) compared with sham treatment (Fig. 6G). HIF-1α expression followed gradients of lymph stasis, with increased expression in the upper arm compared with the lower arm, suggesting that even minor increases in lymphatic fluid stasis can regulate HIF-1α.
Chronic CD4+ cell inflammation modulates HIF-1α expression
We have shown that sustained lymphatic fluid stasis results in CD4+ cell-mediated inflammation (19). We therefore hypothesized that CD4+ cell-mediated inflammation may either directly regulate HIF-1α expression or indirectly modify the inflammatory response responsible for HIF-1α expression during chronic lymphedema. To test this hypothesis, we compared HIF-1α expression in lymphedematous tissues of animals depleted of CD4+ cells using neutralizing antibodies with isotype-control treated animals. Western blot analysis demonstrated markedly reduced (50–70% decrease) HIF-1α and CA-IX expression in tail tissues from CD4+-depleted animals at 6 wk postoperation (Fig. 7A), and pAkt expression was unchanged. To confirm these findings, we compared HIF-1α expression after tail surgery in CD4-knockout mice to wild-type littermates. Again, loss of CD4+ cell-mediated inflammation was associated with marked reductions (50%) in HIF-1α and CA-IX (60%) and little or no change in pAkt (Fig. 7B).
Figure 7.

Chronic CD4+ cell inflammation modulates HIF-1α expression in response to sustained lymphatic fluid stasis. A) Western blot from animals that underwent tail lymphatic ablation and treatment with isotype control or CD4-neutralizing antibodies at 6 wk after surgery (fold change relative to isotype). B) Western blot of protein harvested 15 mm distal to the wound from wild-type (WT) or CD4-knockout (CD4KO) animals 6 wk after tail skin excision and lymphatic ablation (fold change relative to WT). C) Number of HIF-1α+ cells per high-power field in lymphedematous (distal) portions of the tail following tail skin excision and lymphatic ligation at 6 wk after surgery. Animals depleted of CD4+ cells or CD4-knockout mice demonstrate fewer HIF-1α+ cells compared to controls. **P<0.01; ***P<0.001. D) Low-power photomicrographs (×2.5, top panels) of HIF-1α immunolocalization in cross-sections 15 mm distal to the wound (location shown in gross photograph) from animals that underwent tail lymphatic ablation and treatment with CD4 neutralizing or isotype-control antibodies for 6 wk. Note dilated lymphatics and edema in isotype-treated animals and reduced HIF-1α+ cells in CD4-depleted animals. High-power representative photomicrographs (×20, middle panels; ×60, bottom panels) of HIF-1α immunolocalization demonstrate fewer positive perivascular macrophages in CD4-neutralizing antibody-treated animals. E) Representative low-power photomicrographs (×2.5, top panels) of HIF-1α immunolocalization at 6 wk postoperation in cross sections 15 mm distal to the wound (shown in gross photograph) from CD4-knockout and WT littermates that underwent tail skin and lymphatic excision. High-power photomicrographs (×20, middle panels; ×60, bottom panels) of HIF-1α immunolocalization demonstrate fewer positive perivascular macrophages in CD4 knockouts. Scale bars = 1000 μm (D, E; middle panels); 100 μm (D, E; bottom panels).
Immunohistochemical localization of HIF-1α in distal tail tissues of CD4+-depleted or CD4-knockout animals confirmed Western blot findings and demonstrated reductions in the number of HIF-1α+ cells compared to controls (41.2 and 60.2% decrease in CD4-depleted and CD4-knockout animals, respectively; P<0.01; Fig. 7C–E). Similar to our findings in regions surrounding the wound, HIF-1α expression in lymphedematous tissues localized to macrophages. The severity of inflammation within lymphedematous regions was reduced following blockade of CD4-mediated responses, suggesting that CD4 cells may modify this chronic inflammatory response and indirectly modulate HIF-1α expression by macrophages. In summary, our findings suggest that HIF-1α is a central coordinating mechanism for lymphangiogenesis in response to a variety of physiological circumstances and can translate diverse environmental stimuli to lymphangiogenesis by regulating the expression of lymphangiogenic cytokines (Fig. 8).
Figure 8.
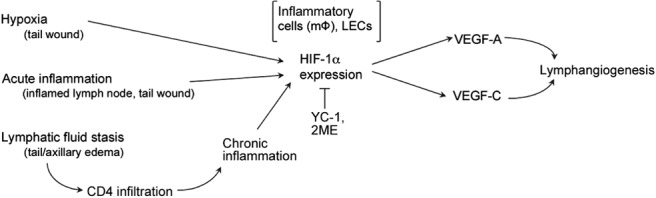
Hypoxia, inflammation, and lymphatic fluid stasis regulate HIF-1α expression to modulate lymphangiogenesis. HIF-1α expression is regulated by a variety of lymphangiogenic stimuli, including hypoxia, inflammation, and gradients of lymphatic (interstitial) fluid stasis. HIF-1α expression is required for lymphangiogenesis in these settings by regulating lymphangiogenic cytokine expression. Regulation of HIF-1α expression by persistent lymphatic fluid stasis is dependent on chronic CD4+ cell inflammation and interactions of these cells with infiltrating macrophages.
DISCUSSION
Clinical cancer studies have shown a correlation between tumor hypoxia, HIF-1α expression, and lymphatic metastases (25–27). Similarly, animal studies have shown that HIF-1α blockade reduces lymphatic metastasis, which suggests that HIF-1α regulates tumor-associated lymphangiogenesis (13, 15). The results of our study now show a direct role for HIF-1α in lymphangiogenesis during wound healing and inflammatory lymphangiogenesis, which demonstrates that HIF-1α is a key initiator and coordinator of lymphatic regeneration.
Our data suggest that HIF-1α-mediated effects on lymphangiogenesis occur via modulation of VEGF-A and VEGF-C expression. This observation is consistent with the fact that VEGF-C expression is necessary for lymphatic regeneration during wound repair and that its expression is regulated, at least in part, by lymphatic fluid flow (25, 28). Similarly, our findings are consistent with clinical studies correlating HIF-1α overexpression with VEGF-C production and tumor lymphangiogenesis/lymphatic invasion in a variety of malignancies (27, 29). Together, our findings suggest that HIF-1α-mediated VEGF-C expression may be a conserved mechanism regulating lymphangiogenesis in a variety of settings. These findings are important since they identify HIF-1α as a novel coordinating target for regulating lymphangiogenesis.
The regulation of VEGF-C expression by HIF-1α likely involves complex and overlapping mechanisms. In fact, relative contributions of VEGF-A to angiogenesis and VEGF-C to lymphangiogenesis are not entirely dichotomous. For example, VEGF-A is known to mediate macrophage recruitment and VEGF-C production in corneal lymphangiogenesis (4) and can induce VEGF-C production by human dermal LECs (30). Similarly, other studies have demonstrated that inflammatory lymphangiogenesis in draining lymph nodes is dependent almost entirely on VEGF-A rather than VEGF-C production (31). Our studies suggest that HIF-1α stabilization may represent a critical regulatory step in the initial expression of these growth factors during wound repair and inflammatory lymphangiogenesis.
Our findings with ODD-Luc/+ mice, pimonidazole stains, and isolated LEC cultures suggest that HIF-1α stabilization during wound healing is regulated, at least in part, by hypoxia. This finding is consistent with the well-characterized mechanisms of HIF-1α regulation in a number of physiological conditions (32). This conclusion is supported by the fact that isolated LECs responded directly to hypoxic conditions by up-regulating HIF-1α and VEGF-C and that inhibition of HIF-1α partially inhibited the effect of hypoxia on VEGF-C expression. Interestingly, previous studies have shown that lung fibroblasts do not increase VEGF-C expression in response to hypoxia; however, the exact degree of hypoxia was not reported (5). Therefore, it is possible that regulation of VEGF-C production by hypoxia is dependent on cell type or the relative degree of hypoxia.
Interestingly, we found that inflammation and sustained lymphatic fluid stasis also regulate HIF-1α expression. In fact, tissues exposed to even mild lymphatic fluid stasis in an axillary dissection model demonstrated increased HIF-1α expression. These findings are consistent with the fact that inflammatory cytokines and growth factor-dependent pathways, including the Ras/Raf/MAPK and PI(3)K/Akt (33) cascades, not only regulate HIF-1α expression in normoxia but also modulate the amplitude of HIF-1α expression during hypoxia (7). Interestingly, while lymphatic fluid stasis and resultant inflammatory changes increased HIF-1α expression independently from hypoxia, these circumstances did not markedly alter Akt phosphorylation. Furthermore, the small-molecule inhibitors used in our study significantly decreased HIF-1α expression but did not alter Akt phosphorylation. Together, these findings suggest that HIF-1α stabilization in the setting of chronic inflammation and lymphatic fluid stasis occurs independently from Akt phosphorylation.
While HIF-1α expression in chronic lymphedema appears to be dependent on inflammation, recent studies suggest that HIF-1α may likewise coordinate inflammatory responses. For example, Arbeit and colleagues (34) have shown that amplification of NF-κβ transcriptional activation by stromal components is dependent on epithelial HIF-1α stabilization, and Cramer and colleagues (35) have shown that HIF-1α overexpression is essential for myeloid cell-mediated inflammation. Thus, it is possible that HIF-1α accumulation in response to lymphatic fluid stasis further promotes inflammation, thereby behaving in an autocrine manner. This hypothesis requires further study, as the cellular mechanisms that regulate chronic inflammation in response to lymphatic fluid stasis remain unknown.
In summary, our results demonstrate that HIF-1α regulates lymphatic regeneration during wound repair and inflammatory lymphangiogenesis; this response is dependent, at least in part, on expression of lymphangiogenic cytokines, including VEGF-A and VEGF-C. In addition, HIF-1α expression is regulated by hypoxic gradients in the wound microenvironment and by inflammatory changes secondary to lymphatic fluid stasis. These findings are clinically relevant because they suggest a novel conserved coordinating mechanism for the regulation of lymphangiogenesis during wound healing and inflammation.
Acknowledgments
This work was supported by a U.S. National Institutes of Health (NIH) T32 grant (NIH CA 009501; J.Z.), a Plastic Surgery Education Foundation grant (J.Z., T.A.), a George Washington University surgical research grant (S.D.), a Society of Memorial Sloan-Kettering grant (B.J.M.), and the Division of Plastic Surgery, Memorial Sloan-Kettering. Technical services provided by the Small-Animal Imaging Core Facility, supported by NIH Small-Animal Imaging Research Program (SAIRP) grant R24 CA83084 and NIH Center grant P30 CA08748, are acknowledged. J.Z. performed experiments and writing. A.Y. performed T-cell depletion and CD4-knockout experiments. T.A. participated in study design and transgenic mouse experiments. S.D. performed experiments. E.W. performed experiments and edited manuscript. B.J.M. designed research and wrote the manuscript. The authors declare no conflicts of interest.
Footnotes
- 2ME
- 2-methyoxyestradiol
- CA-IX
- carbonic anhydrase IX
- DFO
- deferoxamine
- HIF-1α
- hypoxia-inducible factor-1α
- LEC
- lymphatic endothelial cell
- LYVE-1
- lymphatic vessel endothelial hyaluronan receptor-1
- MAPK
- mitogen-activated protein kinase
- NF-κB
- nuclear factor-κB
- p-Akt
- phosphorylated Akt
- PI(3)K
- phosphatidylinositol-3-OH kinase
- VEGF-A
- vascular endothelial growth factor-A
- VEGF-C
- vascular endothelial growth factor-C
- YC-1
- 3-(5′-hydroxymethyl-2′-furyl)-1-benzylindazole
REFERENCES
- 1. Petrek J. A., Heelan M. C. (1998) Incidence of breast carcinoma-related lymphedema. Cancer 83, 2776–2781 [DOI] [PubMed] [Google Scholar]
- 2. Saaristo A., Tammela T., Timonen J., Yla-Herttuala S., Tukiainen E., Asko-Seljavaara S., Alitalo K. (2004) Vascular endothelial growth factor-C gene therapy restores lymphatic flow across incision wounds. FASEB J. 18, 1707–1709 [DOI] [PubMed] [Google Scholar]
- 3. Goldman J., Conley K. A., Raehl A., Bondy D. M., Pytowski B., Swartz M. A., Rutkowski J. M., Jaroch D. B., Ongstad E. L. (2007) Regulation of lymphatic capillary regeneration by interstitial flow in skin. Am. J. Physiol. Heart Circ. Physiol. 292, H2176–H2183 [DOI] [PubMed] [Google Scholar]
- 4. Cursiefen C., Chen L., Borges L. P., Jackson D., Cao J., Radziejewski C., D'Amore P. A., Dana M. R., Wiegand S. J., Streilein J. W. (2004) VEGF-A stimulates lymphangiogenesis and hemangiogenesis in inflammatory neovascularization via macrophage recruitment. J. Clin. Invest. 113, 1040–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ristimaki A., Narko K., Enholm B., Joukov V., Alitalo K. (1998) Proinflammatory cytokines regulate expression of the lymphatic endothelial mitogen vascular endothelial growth factor-C. J. Biol. Chem. 273, 8413–8418 [DOI] [PubMed] [Google Scholar]
- 6. Liao D., Johnson R. S. (2007) Hypoxia: a key regulator of angiogenesis in cancer Cancer Metast. Rev. 26, 281–290 [DOI] [PubMed] [Google Scholar]
- 7. Bardos J. I., Ashcroft M. (2005) Negative and positive regulation of HIF-1: a complex network. Biochim. Biophys. Acta 1755, 107–120 [DOI] [PubMed] [Google Scholar]
- 8. Semenza G. L. (2004) Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology 19, 176–182 [DOI] [PubMed] [Google Scholar]
- 9. Avraham T., Clavin N. W., Daluvoy S. V., Fernandez J., Soares M. A., Cordeiro A. P., Mehrara B. J. (2009) Fibrosis is a key inhibitor of lymphatic regeneration. Plast. Reconstr. Surg. 124, 438–450 [DOI] [PubMed] [Google Scholar]
- 10. Boardman K. C., Swartz M. A. (2003) Interstitial flow as a guide for lymphangiogenesis. Circ. Res. 92, 801–808 [DOI] [PubMed] [Google Scholar]
- 11. Clavin N. W., Avraham T., Fernandez J., Daluvoy S. V., Soares M. A., Chaudhry A., Mehrara B. J. (2008) TGF-beta1 is a negative regulator of lymphatic regeneration during wound repair. Am. J. Physiol. Heart Circ. Physiol. 295, H2113–H2127 [DOI] [PubMed] [Google Scholar]
- 12. Zampell J. C., Yan A., Avraham T., Andrade V., Malliaris S., Aschen S. Z., Rockson S. G., Mehrara B. J. (2011) Temporal and spatial patterns of endogenous danger signal expression after wound healing and in response to lymphedema. Am. J. Physiol. Cell Physiol. 300, C1107–C1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shin D. H., Kim J. H., Jung Y. J., Kim K. E., Jeong J. M., Chun Y. S., Park J. W. (2007) Preclinical evaluation of YC-1, a HIF inhibitor, for the prevention of tumor spreading. Cancer Lett. 255, 107–116 [DOI] [PubMed] [Google Scholar]
- 14. Sun H. L., Liu Y. N., Huang Y. T., Pan S. L., Huang D. Y., Guh J. H., Lee F. Y., Kuo S. C., Teng C. M. (2007) YC-1 inhibits HIF-1 expression in prostate cancer cells: contribution of Akt/NF-kappaB signaling to HIF-1alpha accumulation during hypoxia. Oncogene 26, 3941–3951 [DOI] [PubMed] [Google Scholar]
- 15. Yeo E. J., Chun Y. S., Cho Y. S., Kim J., Lee J. C., Kim M. S., Park J. W. (2003) YC-1: a potential anticancer drug targeting hypoxia-inducible factor 1. J. Natl. Cancer Inst. 95, 516–525 [DOI] [PubMed] [Google Scholar]
- 16. Zhao Q., Du J., Gu H., Teng X., Zhang Q., Qin H., Liu N. (2007) Effects of YC-1 on hypoxia-inducible factor 1-driven transcription activity, cell proliferative vitality, and apoptosis in hypoxic human pancreatic cancer cells. Pancreas 34, 242–247 [DOI] [PubMed] [Google Scholar]
- 17. Li S. H., Shin D. H., Chun Y. S., Lee M. K., Kim M. S., Park J. W. (2008) A novel mode of action of YC-1 in HIF inhibition: stimulation of FIH-dependent p300 dissociation from HIF-1α. Mol. Cancer Ther. 7, 3729–3738 [DOI] [PubMed] [Google Scholar]
- 18. Mabjeesh N. J., Escuin D., LaVallee T. M., Pribluda V. S., Swartz G. M., Johnson M. S., Willard M. T., Zhong H., Simons J. W., Giannakakou P. (2003) 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell 3, 363–375 [DOI] [PubMed] [Google Scholar]
- 19. Avraham T., Daluvoy S., Zampell J., Yan A., Haviv Y. S., Rockson S. G., Mehrara B. J. (2010) Blockade of transforming growth factor-{beta}1 accelerates lymphatic regeneration during wound repair. Am. J. Pathol. 177, 3202–3214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Swartz M. A., Berk D. A., Jain R. K. (1996) Transport in lymphatic capillaries. I. Macroscopic measurements using residence time distribution theory. Am. J. Physiol. 270, H324–329 [DOI] [PubMed] [Google Scholar]
- 21. Safran M., Kim W. Y., O'Connell F., Flippin L., Gunzler V., Horner J. W., Depinho R. A., Kaelin W. G., Jr. (2006) Mouse model for noninvasive imaging of HIF prolyl hydroxylase activity: assessment of an oral agent that stimulates erythropoietin production. Proc. Natl. Acad. Sci. U. S. A. 103, 105–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Thangarajah H., Yao D., Chang E. I., Shi Y., Jazayeri L., Vial I. N., Galiano R. D., Du X. L., Grogan R., Galvez M. G., Januszyk M., Brownlee M., Gurtner G. C. (2009) The molecular basis for impaired hypoxia-induced VEGF expression in diabetic tissues. Proc. Natl. Acad. Sci. U. S. A. 106, 13505–13510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rosenberger C., Rosen S., Paliege A., Heyman S. N. (2009) Pimonidazole adduct immunohistochemistry in the rat kidney: detection of tissue hypoxia. Methods Mol. Biol. 466, 161–174 [DOI] [PubMed] [Google Scholar]
- 24. Enholm B., Paavonen K., Ristimaki A., Kumar V., Gunji Y., Klefstrom J., Kivinen L., Laiho M., Olofsson B., Joukov V., Eriksson U., Alitalo K. (1997) Comparison of VEGF, VEGF-B, VEGF-C and Ang-1 mRNA regulation by serum, growth factors, oncoproteins and hypoxia. Oncogene 14, 2475–2483 [DOI] [PubMed] [Google Scholar]
- 25. Goldman J., Le T. X., Skobe M., Swartz M. A. (2005) Overexpression of VEGF-C causes transient lymphatic hyperplasia but not increased lymphangiogenesis in regenerating skin. Circ. Res. 96, 1193–1199 [DOI] [PubMed] [Google Scholar]
- 26. Schindl M., Schoppmann S. F., Samonigg H., Hausmaninger H., Kwasny W., Gnant M., Jakesz R., Kubista E., Birner P., Oberhuber G. (2002) Overexpression of hypoxia-inducible factor 1alpha is associated with an unfavorable prognosis in lymph node-positive breast cancer Clin. Cancer Res. 8, 1831–1837 [PubMed] [Google Scholar]
- 27. Schoppmann S. F., Fenzl A., Schindl M., Bachleitner-Hofmann T., Nagy K., Gnant M., Horvat R., Jakesz R., Birner P. (2006) Hypoxia inducible factor-1alpha correlates with VEGF-C expression and lymphangiogenesis in breast cancer Breast. Cancer Res. Treat. 99, 135–141 [DOI] [PubMed] [Google Scholar]
- 28. Rutkowski J. M., Boardman K. C., Swartz M. A. (2006) Characterization of lymphangiogenesis in a model of adult skin regeneration. Am. J. Physiol. Heart Circ. Physiol. 291, H1402–H1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Katsuta M., Miyashita M., Makino H., Nomura T., Shinji S., Yamashita K., Tajiri T., Kudo M., Ishiwata T., Naito Z. (2005) Correlation of hypoxia inducible factor-1alpha with lymphatic metastasis via vascular endothelial growth factor-C in human esophageal cancer. Exp. Mol. Pathol. 78, 123–130 [DOI] [PubMed] [Google Scholar]
- 30. Skobe M., Brown L. F., Tognazzi K., Ganju R. K., Dezube B. J., Alitalo K., Detmar M. (1999) Vascular endothelial growth factor-C (VEGF-C) and its receptors KDR and flt-4 are expressed in AIDS-associated Kaposi's sarcoma. J. Invest. Dermatol. 113, 1047–1053 [DOI] [PubMed] [Google Scholar]
- 31. Halin C., Tobler N. E., Vigl B., Brown L. F., Detmar M. (2007) VEGF-A produced by chronically inflamed tissue induces lymphangiogenesis in draining lymph nodes. Blood 110, 3158–3167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weidemann A., Johnson R. S. (2008) Biology of HIF-1alpha. Cell Death Differ. 15, 621–627 [DOI] [PubMed] [Google Scholar]
- 33. Pouyssegur J., Dayan F., Mazure N. M. (2006) Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 441, 437–443 [DOI] [PubMed] [Google Scholar]
- 34. Scortegagna M., Cataisson C., Martin R. J., Hicklin D. J., Schreiber R. D., Yuspa S. H., Arbeit J. M. (2008) HIF-1alpha regulates epithelial inflammation by cell autonomous NFkappaB activation and paracrine stromal remodeling. Blood 111, 3343–3354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cramer T., Yamanishi Y., Clausen B. E., Forster I., Pawlinski R., Mackman N., Haase V. H., Jaenisch R., Corr M., Nizet V., Firestein G. S., Gerber H. P., Ferrara N., Johnson R. S. (2003) HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112, 645–657 [DOI] [PMC free article] [PubMed] [Google Scholar]