

Abstract
Diverse pathophysiological processes (e.g. obesity, lifespan determination, addiction and male fertility) have been linked to the expression of specific isoforms of the adenylyl cyclases (AC1-AC10), the enzymes that generate cyclic AMP (cAMP). Our laboratory recently discovered a new mode of cAMP production, prominent in certain cell types, that is stimulated by any manoeuvre causing reduction of free [Ca2+] within the lumen of the endoplasmic reticulum (ER) calcium store. Activation of this ‘store-operated’ pathway requires the ER Ca2+ sensor, STIM1, but the identity of the enzymes responsible for cAMP production and how this process is regulated is unknown. Here, we used sensitive FRET-based sensors for cAMP in single cells combined with silencing and overexpression approaches to show that store-operated cAMP production occurred preferentially via the isoform AC3 in NCM460 colonic epithelial cells. Ca2+ entry via the plasma membrane Ca2+ channel, Orai1, suppressed cAMP production, independent of store refilling. These findings are an important first step towards defining the functional significance and to identify the protein composition of this novel Ca2+/cAMP crosstalk system.
Keywords: calcium, cyclic AMP, endoplasmic reticulum
Introduction
Many intracellular organelles, including the endoplasmic reticulum (ER), actively sequester calcium ions [1]. Transient loss of Ca2+ from the ER is an inherent consequence of intracellular Ca2+ signalling. However, dysregulation of ER Ca2+ homeostasis also occurs pathologically following exposure to toxic insults, ischemia and trauma [2]. Alteration (be it of physiological or pathological origin) in free [Ca2+] within these internal Ca2+ storage compartments is often sensed and translated into downstream signalling activities, such as ER stress responses or engagement of store-dependent Ca2+ entry channels [3].
In a previous report from our laboratory, we showed that cAMP could become elevated in the cytosol of certain cell types following depletion of Ca2+ from intracellular stores (principally the ER), but this increase in cAMP did not depend whatsoever on the corresponding changes in cytosolic [Ca2+]. The increase in cAMP, rather, directly mirrored the decrease in free [Ca2+] inside the ER lumen and required a transmembrane ER Ca2+ sensor, STIM1 [4].
STIM1 has been shown to translocate from the bulk ER to specialized ER/plasma membrane junctions following store depletion [5–7]. STIM1 has also been firmly established to interact with Orai1, the pore-forming unit of a plasma membrane channel that permits Ca2+ entry from the extracellular environment [8–11]. Activation of Orai1 (and possibly other channels) via STIM1 allows a sustained Ca2+ signal, and refilling of the store. This ubiquitous, widely studied phenomenon known as ‘store-operated Ca2+ entry’ is activated by any manoeuvre that lowers the free [Ca2+] in the ER lumen [12].
In our studies on ER-dependent cAMP signalling, we found that silencing STIM1 or preventing its translocation reduced cAMP production caused by treatments that lower the levels of free Ca2+ within the ER. Because of the many parallels with store-operated Ca2+ entry, we named this process ‘Store-Operated cAMP Signalling’ (SOcAMPS). So far SOcAMPS has been described in several cell types, notably in NCM460 cells [4], a model of normal colonic crypt epithelial cells [13] and CaLu-3 cells (normal human airway epithelia cells) [14]. Although the physiological meaning of SOcAMPS in NCM460 cells is not known, in CaLu-3 this process has been shown to participate in cAMP-dependent chloride and fluid secretion induced by Ca2+-mobilizing products secreted from the bacterium Pseudomonas aeruginosa. This mechanism may contribute to airway clearance during bacterial infection. Using a model of mouse cholangiocytes, Spirli and colleagues also recently implicated SOcAMPS in the pathogenesis of polycystic liver disease [15]. Interestingly, certain other cells types, such as HeLa cells, appear to lack SOcAMPS [4]. This suggests that a specific panel of proteins must be expressed for SOcAMPS to be operational.
A pathway that links ER Ca2+ to cAMP signalling could have extremely important physiological implications, but apart from STIM1, little is known about the actual protein participants in SOcAMPS. For example, how does the principal target of STIM1, Orai1, fit into this process? Are proteins related to STIM1, such as STIM2, involved? Another key question concerns how exactly cAMP is generated following store depletion.
Our previous studies using pharmacological agents excluded the involvement of the phosphodiesterases, the enzymes that degrade cyclic nucleotides, in SOcAMP signalling, but did implicate the enzymes that produce cAMP, the adenylyl cyclases (ACs). To date, nine closely related transmembrane isoforms of AC (tmAC1-9) and one soluble adenylyl cyclase (sAC or AC10) have been cloned and characterized [16]. Classically, tmACs are activated by heterotrimeric G–proteins in response to stimulation of G-protein coupled receptor. In contrast, the sAC is directly activated by cellular metabolites (CO2, bicarbonate and ATP), consistent with the role as an intracellular metabolic sensor [17]. A panel of different AC isoforms are generally coexpressed in a given cell type.
In this study, we used genetically encoded cAMP reporters in single NCM460 cells to define whether a singular isoform of adenylyl cyclase is responsible for store-dependent cAMP production in this cell type. We overcame the limitation imposed by the lack of pharmacological inhibitors for specific AC isoforms, and lack of suitable antibodies for protein chemistry by using overexpression and knockdown approaches to show a preferential link between the AC isoform AC3 in this store-operated process. In addition, although the presence of the plasma membrane Ca2+ entry channel Orai1 (or the STIM1 relative, STIM2) did not affect SOcAMPS, Ca2+ entering through this store-operated pathway did have a strong effect in terminating cAMP production. The physiological circumstances under which SOcAMPS is expected to become the predominant second messenger pathway following loss of ER Ca2+ stores are discussed.
Materials and methods
Reagents
BAPTA-AM (1,2-bis(o-Aminophenoxy)ethane-N,N,N′,N′-tetraacetic Acid Tetra(acetoxymethyl) Ester) was purchased from Molecular Probes/Invitrogen (Carlsbad, CA, USA), and ionomycin from Calbiochem (San Diego, CA, USA). Unless otherwise noted, all other reagents were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
NCM460 cells, a model of normal human colonic crypt epithelial cells [13] were obtained by licensing agreement from INCELL, San Antonio, TX, USA, and were grown in M3:10 medium (INCELL). For most experiments, NCM460 cells stably transfected with the FRET-based cAMP probe, CFP(nd)-EPAC(dDEP/CD)-YFP(nd) × pCDNA3 (H30) Amp or ‘EpacH30’ [18], were used. HeLa cells were grown in DMEM + 10% FCS; both lines were maintained in a humidified CO2 (5%) incubator at 37°C. Cells were split every 2 days after reaching 80% confluence.
Transfection with shRNA plasmids and siRNAs
All siRNA tranfections were performed with Lipofectamine (Invitrogen) according to manufacturer's instructions, whereas plasmids were transfected using Effectene (Qiagen, Valencia, CA, USA). Briefly, 300 ng of the plasmid of interest plus 300 ng of mCherry (used as marker of transfected cells), were mixed in 180 μl of buffer reagent, to which 8 μl of Enhancer and 10 μl of Effectene reagent were added. After 20 min., the entire mixture was added to cells grown to 60–70% confluency into six-well plates.
For efficient knockdown of the different ACs we used a mix of four different HuSH 29mer shRNAs (500 ng of each plasmid per six-well plate) specific against AC3 or AC5. The pRFP-C-RS vector (Origene, Rockville, MD, USA) expressing the shRNAS also encodes the red fluorescent protein dsRED, used as a marker of transfected cells. As an alternative, we used siRNAs against AC3, AC5, AC6 and their effects were compared with that of non-effective siRNA ‘scramble’. The day after transfection, cells were trypsinized and plated on glass coverslips, a step that resulted in a high proportion of isolated single cells in each microscope field, thereby minimizing gap-junction-mediated cAMP diffusion between cells [34].
Two days after transfection, imaging experiments were performed. In addition, total RNA was collected using RNeasy kit (Qiagen). One microgram of RNA was converted into cDNA using random hexamers (Invitrogen SuperScript First-Strand Synthesis System). Total AC3, AC5 and AC6 mRNA levels were calculated by Real-time RT-PCR using a TaqMan-based assays (Applied Biosystems, Carlsbad, CA, USA) and normalized to the endogenous 18S rRNA or GAPDH levels (amplified using a standard primer sets; Applied Biosystems) in three independent experiments.
Ratio Imaging of FRET-based cAMP sensors
Glass coverslips containing cells were mounted in a home-built flow-through perfusion chamber on top of the fluorescence imaging microscope stage as described previously [34,52]. Two different FRET ratio imaging set-ups were used for this study: (i) a Nikon TE200 inverted fluorescence microscope equipped with a Photometrics QuantEM EMCCD camera, and (ii) a Nikon Eclipse TE2000-U microscope with attached Hamamatsu ORCA-ER CCD camera. Metafluor software (Molecular Devices, Sunnyvale, CA, USA) was used to control filter wheels (Sutter Instruments, Novato, CA, USA) in the excitation and emission paths and to control image acquisition (one image pair every 5–10 sec.) for both systems.
Cyclic AMP measurements were performed on single cells using the 480 nm/535 nm FRET emission ratio (440 nm excitation) of EpacH30 [18] and either a 40× Plan Fluor 1.30 NA or a 60× Plan Apo TIRF 1.45 NA oil-immersion objective. Cells were bathed in a HEPES-buffered Ringer's-like solution containing (in mM): 125 NaCl, 25 HEPES, 10 Glucose, 5 K2HPO4, 1 MgSO4 and 1 CaCl2, pH, 7.40. For Ca2+-free solutions (0 Ca2+), the CaCl2 was omitted and EGTA (25 μM) added to minimize contaminating Ca2+.
Statistics and analysis
Data are presented as mean ± S.E.M. Paired or unpaired Student's t-test was performed as appropriate to compare the means of two different groups of experiments. A value of P < 0.05 was considered statistically significant. Analysis of the change in initial slope during the 2 min. following ionomycin addition was fitted by linear regression using Kaleidagraph software, and expressed as a percentage of the change in slope of the corresponding control response.
Results
We observed previously that diverse strategies culminating in the lowering of free [Ca2+] within the ER resulted in cAMP production in NCM460 cells, measured using both a panel of FRET-based cAMP sensors and conventional cAMP immunoassays. These manoeuvres included: (i) inhibition of Ca2+ uptake by SERCA inhibitors (thapsigargin and tert-butyl hydroquinone), (ii) InsP3-dependent release of stores using native Ca2+ mobilizing agonists working through Gαq-coupled receptors (ATP and carbachol), (iii) buffering ER Ca2+ with high concentrations of membrane-permeant Ca2+ buffers (TPEN or BAPTA-AM), (iv) passive depletion of stores using high concentrations of EGTA, (v) treatment with Ca2+-mobilizing compounds such as bile acid (deoxycholic acid) or eicosapentaenoic acid and (vi) release of stores using Ca2+ ionophores such as ionomcyin.
To screen potential mediators or regulators of SOcAMPS, we developed a simplified protocol in which we released intracellular Ca2+ stores under Ca2+-free conditions using ionomycin in NCM460 cells stably expressing a FRET-based cAMP sensor, EpacH30 [18] (Fig. 1A). This resulted in a reproducible increase in cAMP production (as measured by FRET ratio change of EpacH30) that was typically ∼35–40% of the maximal ratio change obtained following saturation of the cAMP sensor using forskolin (50 μM) and IBMX (1 mM). We also observed previously that this response to store depletion could be sustained for prolonged periods (measured longer than 60 min.), provided internal Ca2+ stores were kept in an empty state [4]. It should be emphasized that this increase in cAMP was absolutely independent of the initial transient spike in Ca2+ elicited by ionomycin-induced store release [4]. When cells were loaded with the Ca2+ buffer, BAPTA-AM (20 μM for 30 min.; conditions shown to eliminate the initial spike of cytosolic Ca2+ in NCM460 cells following ionomycin treatment, as measured by fura-2 in Ca2+-free solutions), the increase in the FRET ratio following store release was not altered (Fig. 1B), consistent with our previous findings that SOcAMPS is independent of cytosolic Ca2+. In fact, manoeuvres that caused Ca2+ to become elevated within the cytoplasm, such as re-addition of bath Ca2+, strongly inhibited SOcAMPS (Fig. 1A), and this was fully reversible upon superfusion of cells with Ca2+-free solutions (not depicted). This effect of Ca2+ re-addition was slowed dramatically, but not eliminated in the BAPTA-AM pre-treated cells, consistent with the fact that Ca2+ entering from the extracellular space will eventually overwhelm the Ca2+ buffering capacity of BAPTA, resulting in elevation of free Ca2+ within the cytoplasm and inhibition of SOcAMPS. Note too, that these experiments indicated that alterations in cationic metals other than Ca2+ (e.g. Zn2+) [19] were likely not responsible for this effect as BAPTA-AM chelates many trace heavy metals. Moreover, the effect of ionomycin was not altered by pre-treatment with TPEN (N,N,N′,N′-Tetrakis-(2-pyridylmethyl)ethylenediamine; 10 μM), a membrane permeant compound that is extremely effective at scavenging heavy metals (not depicted; n = 4 expts/26 cells).
Fig. 1.
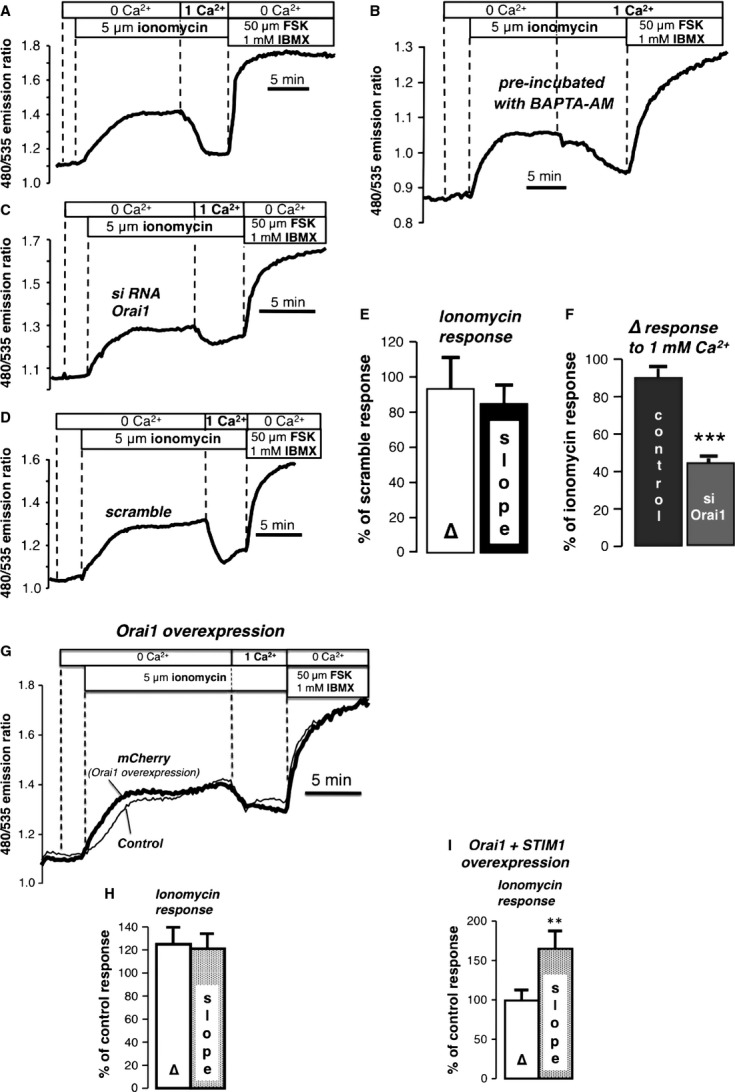
Evaluation of possible players involved in SOcAMPs in NCM460 cells stably expressing EpacH30. (A) Effect of ionomycin (5 μM) in the absence of external Ca2+. Addition of 1 mM Ca2+ resulted in a reduction in the FRET ratio (typical of 5 exp./29 cells). At the end of all experiments forskolin (FSK; 50 μM) and IBMX (1 mM) were added to obtain a supramaximal FRET response. (B) Effect of ionomycin in cells pre-loaded with BAPTA-AM (20 μM, 30 min.; typical of 6 exp./49cells). (C) Knockdown by RNAi of the Orai1 channel did not alter the response to ionomycin, however, the inhibitory action of 1 mM Ca2+ was significantly reduced as compared with scramble controls (6 exp./33 cells). (D) Response to ionomycin in scramble controls (6 exp./97 cells). (E, F) Summary of experiments in C and D: (E) Amplitude (Δ) and slope of the ionomycin response expressed as percentage of the response in scramble controls. (F) Response to 1 mM Ca2+ expressed as a % of the ionomycin response in scramble and siOrai1-treated cells. (G) Effect of overexpression of Orai1 on the response to ionomycin (5 exp./25 mCherry cells/7 control cells). (H) Summary of Orai1 overexpression experiments. Amplitude and slope of the ionomycin response expressed as a percentage of the response in control cells. (I) Summary of Orai1 + STIM1 overexpression experiments. (5 exp./9 control cells/10 mCherry cells) **P < 0.02, ***P < 0.0001.
Orai channels and STIM2 are not required for SOcAMPS
We first examined how the presence of the major Ca2+ entry channel, Orai1, affected this process. Knockdown by RNAi of the channel did not alter the FRET response to store depletion compared with scramble controls (Fig. 1C–E and Supp. Fig. 1A), but the inhibitory action of Ca2+ re-addition was dramatically attenuated in the absence of Orai1 (Fig. 1F), showing that Ca2+ entry via store-operated pathways was important for this effect. These findings are consistent with the fact that ionomycin-elicited Ca2+ transport across the plasma membrane derives mostly from activation of store-operated Ca2+ entry, with a smaller component arising from the ability of the ionophore to act as a Ca2+ carrier [20].
Overexpression of Orai1 alone or STIM1 alone is known to cause only slight alterations in store-operated Ca2+ entry. However, it has been shown that the co-expression of STIM1 and Orai1 in a specific ratio can enhance the Ca2+ entry current known as ‘ICRAC’ as much as 103-fold, creating the so called ‘monster’ ICRAC [21–23]. Overexpression of STIM1 alone [4], Orai1 alone (Fig. 1G and H) or of STIM1 + Orai1 (Fig. 1I) did not significantly enhance the amplitude of the FRET response to ionomycin, although we did note that co-expression of both Orai1 and STIM1 caused the rate of cAMP increase following store depletion to become slightly faster. Furthermore, no effect was observed when the related channel, Orai3, which is also expressed endogenously in NCM460 cells, or Orai3 + STIM1 were overexpressed (Supplemental Fig. 1B and C). The overexpresssion or knockdown of the STIM1 relative, STIM2, also had no major impact on store-dependent cAMP signalling (Supplemental Fig. 2).
The chief conclusion of the preceding is that Orai1 is not necessary for store-operated cAMP production, but Ca2+ entry through Orai1 does have a significant inhibitory action on SOcAMPS. We considered the Orai1-dependent sensitivity of SOcAMPS to Ca2+ entry as an important clue towards identifying the potential players in SOcAMPS. For example, all AC isoforms are actually inhibited by high (10–25 μM, i.e. non-physiological) [Ca2+] [24]. However, lower concentrations (0.2–0.6 μM), such as those attained during Ca2+ signalling events, can either activate or inhibit certain ACs. Ca2+ entry through store-operated Ca2+ channels is known in particular to have a strong impact on AC1 and AC8 (isoforms activated by Ca2+ via calmodulin) and AC5 and AC6 (directly inhibited by Ca2+) [25–29]. More controversial has been the role of Ca2+ and Ca2+-calmodulin on the modulation of AC3 activity, which, however, has been shown in some cell systems to be inhibited by CaM kinase II (CaMKII) [30,31].
In our previous article, RT-PCR identified seven different ACs isoforms expressed in NCM460 cells (AC3, AC4, AC5, AC6, AC7, AC9 and sAC) [4]. Isoform-specific inhibitors for the different ACs are not generally available [32,33], nor are specific antibodies that permit immunoprecipitation of many of the ACs with their potential interacting proteins. Our prior data showed that SOcAMPS was prevented by P-site inhibitors specific for tmACs, such as SQ22536, and was enhanced by forskolin, an activator of most tmAC isoforms, except AC9. We therefore focused our efforts on overexpression and silencing of AC3-AC7, but also tested sAC.
Overexpression of AC isoforms
Plasmids coding each of the isoforms of interest were cotransfected one at a time with the marker mCherry in NCM460 cells stably expressing EpacH30. To confirm that the tmAC plasmids were expressed and working properly, control experiments in HeLa cells (which do not have SOcAMPS) showed that only in mCherry positive cells was it possible to visualize enhanced cAMP production when challenging the cells with low doses of forskolin (10 nM). Using the protocol illustrated in Figure 2A, the FRET response to ionomycin was compared between mCherry-expressing and control cells in the same microscope field. Only single cells not touching other cells were analysed to avoid artefacts because of gap-junction-mediated cAMP transfer between cells [34].
Fig. 2.
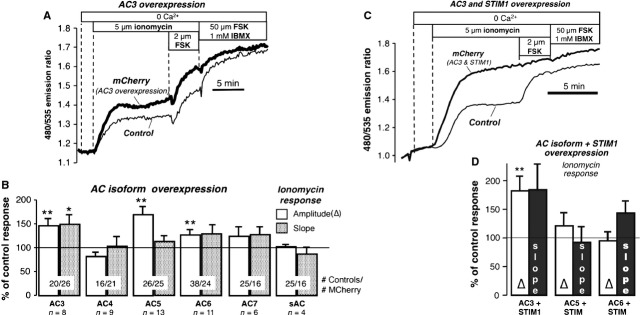
Overexpression of AC isoforms. (A) Overexpression of AC3; response to ionomycin in cells co-transfected with AC3 and mCherry (used as a marker for transfection) and in control cells. (B) Summary of experiments on AC isoforms. Amplitude and slope of the ionomycin response expressed as % of control response. AC3: 8 exp./20 control/26 mCherry cells; AC4: 9 exp./16 control/21 mCherry cells; AC5: 13 exp./26 control/25 mCherry cells; AC6: 11 exp./38 control/24 mCherry cells; AC7: 6 exp./25 control/16 mCherry cells; sAC: 4 exp./11 control/10 mCherry cells. (C) Effect of co-expression of AC3 and STIM1 on the response to ionomycin. (D) Summary of results of overexpression of AC isoforms and STIM1. The response to ionomycin is expressed as % of the response in control cells. AC3+ STIM1: 6 exp./22control/8 mCherry cells; AC5+ STIM1: 8 exp./8 control/8 mCherry cells; AC6+ STIM1: 10 exp./18 control/14 mCherry cells. *P < 0.05, **P < 0.02, ***P < 0.01.
Only overexpression of AC3 was able to consistently increase both the amplitude (P < 0.01) and slope (a measure of cAMP production velocity; P < 0.05) of the ionomycin response. Data from the other ACs are summarized in Fig. 2B, showing that the amplitude (but not the speed) was also somewhat enhanced by transfection with plasmids for AC5 and AC6. Interestingly, AC3 (but not AC5 or AC6) co-expressed with STIM1-mCherry produced an ionomycin effect that was markedly greater than AC3 alone (Fig. 2C; summary data in Fig. 2D), reminiscent of the ‘monster’ ICRAC reported for Orai1 and STIM1 co-expression.
Knockdown of AC3, but not AC5 or AC6, attenuates SOcAMPS
Based on our findings above, we next tested the effect of knocking down AC3, AC5 and AC6. We first used shRNA plasmids that also encode the red fluorescent marker, dsRed, allowing us to distinguish knockdown cells from control cells in the same microscope field. As illustrated in Fig. 3A cells expressing dsRed and shRNA against AC3 gave significantly reduced responses to ionomycin compared to both neighbouring control cells and cells expressing a dsRed + scramble construct. As summarized in Fig. 3B, this action was not observed for AC5 shRNA. It should be pointed out that the presence of dsRed produced a highly variable fluorescence artefact leading to a small downward shift in the FRET ratio of EpacH30. This might be because of green fluorescent maturation intermediates of dsRed or aggregate formation, but the comparison of ratio changes between shRNA for AC3 and the scramble control nevertheless showed a significant effect of knocking down AC3.
Fig. 3.
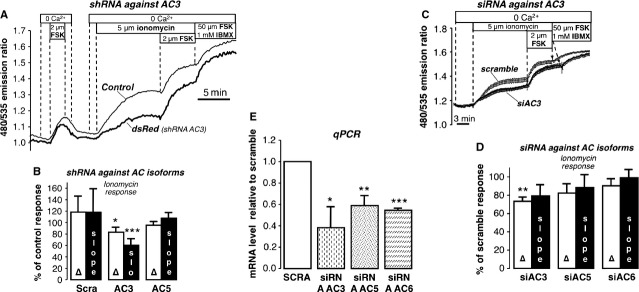
Effect of knockdown of AC isoforms. (A) Response to ionomycin in cells expressing shRNA against AC3; dsRed was used as a marker for transfection with shRNA. (B) Summary of experiments in cells expressing dsRed and shRNA against AC3 (13 exp./16 cells), AC5 (Δ: 7 exp., 12 cells; slope: 6 exp./11 cells), and treated with scramble (Δ: 10 exp./16 cells; slope: 9 exp./15 cells). Amplitude and slope of ionomycin response are expressed as % of control response. (C) Response to ionomycin in cells treated with siRNA for AC3 compared with cells treated with scramble (average of 14 scramble and 12 dsRed cells) (D) Summary of experiments with cells treated with siRNA for AC3 (6 exp./80 cells), AC5 (7 exp./68 cells) and AC6 (8 exp./91 cells) expressed as % of the response in scramble cells (scramble controls for siAC3 and siAC5: 13 exp./126 cells; scramble controls for siAC6 16 exp./202 cells). (E) Summary of qPCR experiments. siRNA AC3: 3 independent experimental samples; siRNA AC5: 3 exp.; siRNA AC6: 2 exp. *P < 0.05; **P < 0.01; ***P < 0.001.
These findings were further confirmed by examining FRET responses of populations of cells treated with siRNA for AC3, AC5 and AC6. Although all siRNA constructs were effective in knocking down their respective targets, only siRNA against AC3 was able to significantly reduce the amplitude of the ionomycin-induced ratio change (Fig. 3C–E).
Rescue of SOcAMPS phenotype in HeLa cells
We next tested whether the SOcAMPS phenotype could be rescued in HeLa cells by simply overexpressing AC3, AC5 or AC6. Although there was generally no effect of introducing these ACs into these cells (Supplemental Fig. 3), even when inhibiting PDE activity with IBMX, it should be noted that there were occasional HeLa cells that gave a credible response to ionomycin after transfection with AC3 (2/11 transfected cells in three experiments) or AC3 + STIM1 (10/45 transfected cells in n = 8 experiments), but not STIM1 alone. The reasons for this inconsistency are unknown, but this could suggest that there is an unknown factor required to reconstitute the SOcAMPS phenotype that is intermittently expressed in HeLa cells.
Effects of PTX, CaMKII and PKC inhibitors on SOcAMPs
NCM460 cells pre-incubated for 18 hrs with pertussis toxin (PTX; 200 ng/ml) yielded typical responses to ionomycin treatment (78/79 cells in n = 9 expts). This indicates that PTX-sensitive Gαi-dependent signalling is not required for the initiation of SOcAMPS.
AC3 has been reported to be a target of CaMKII and PKC [24]. Thus, we questioned whether SOcAMPS might also be affected by pharmacological modulation of these two kinases. As shown in Fig. 4A, acute treatment with the CaMKII inhibitor KN-62 (10 μM) after SOcAMPS had been initiated by ionomycin caused a slight enhancement of the response. However, treatment of NCM460 cells with KN-62 in the absence of ionomycin also caused a comparable elevation of the baseline FRET ratio. Although KN-62 did not prevent the subsequent cAMP response when ER stores were depleted with ionomycin (Fig. 4B), the amplitude of the response was partially inhibited.
Fig. 4.
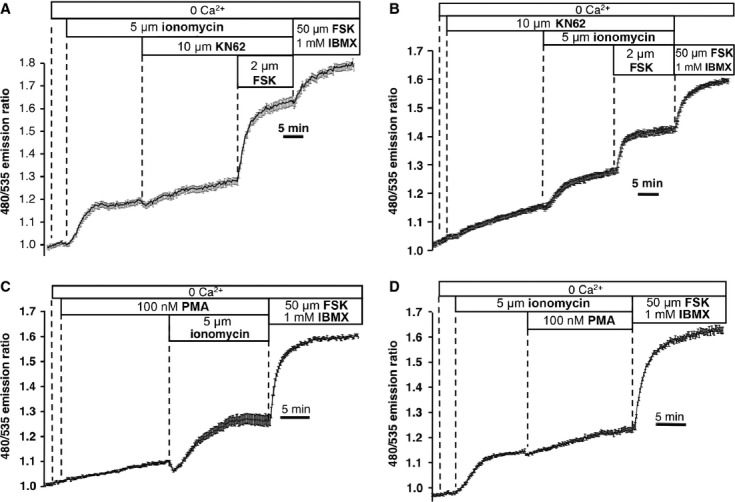
Effects of CaMKII and PKC inhibitors. (A) Treatment with KN-62 (10 μM) after ionomycin (average of 25 cells), typical of 8 exp./115 cells. (B) KN-62 added before ionomycin (average of 16 cells); typical of 4 exp./44 cells. (C) 100 nM PMA added before ionomycin (average of 20 cells); typical of 5 exp./95 cells. (D) Effect of 100 nM PMA added during the response to ionomycin (average of 18 cells); typical of 6 exp./93 cells.
Similarly, acute addition of the PKC activator PMA (phorbol 12-myristate 13-acetate; 100 nM) caused minor increases in the FRET ratio by itself, but did not prevent the subsequent response to ionomycin (Fig. 4C). PMA, like KN-62, also slightly augmented the FRET ratio when added on top of ionomycin (Fig. 4D). Long-term pre-treatment with 100 nM PMA (15 min., 3 and 12 hrs), conditions expected to down-regulate PKC activity, also did not abolish SOcAMPS (see Supplemental Fig. 4 for summary of data at 15 min. and 3 hrs time points). From these data, we can conclude that neither CaMKII nor PKC serve as the mediators linking store depletion to cAMP production, although both of these effectors can slightly modulate cAMP signalling in NCM460 cells, as might be expected from a process mediated by AC3.
Discussion
Most cells express not one, but rather a panel of multiple AC isoforms [35], the coexistence of which was formerly considered redundant. However, the striking phenotypes of isoform-specific knockout animals and of humans with polymorphisms and mutations in AC genes have revealed highly specialized physiological functions for the various ACs [16]. For example, AC5 is largely expressed in heart and brain, and when silenced in mice, prolongs lifespan and protects against stress [36]. AC6 is also expressed in heart, yet the deletion of AC6 compromises left ventricular function in a manner that is not replicable by eliminating AC5 [37]. Meanwhile, AC8 has been demonstrated to be a modulator of anxiety [38], whereas sAC is involved in male fertility, as mice lacking this gene have defects in sperm motility [39]. AC1, AC5, AC6 and AC8 have been reported to be uniquely susceptible to ‘superactivation’, a form of sensitization seen in model systems of drug withdrawal and recovery [32,40].
Of note are the dramatic phenotypes associated with knocking down AC3, which in mouse induces an obese phenotype [41]. Ablation of AC3 also leads to alterations in murine maternal behaviour [42] that are likely related to defects in olfaction [43]. Genome-wide association studies have linked AC3 to the genetic aetiology of major depressive disorder [44]. Interestingly, in mouse hippocampal and cortical neurons, AC3 is localized exclusively to the primary cilium, where it generates a cAMP signal that is essential for certain types of learning and memory such as novel object recognition and contextual fear extinction [45].
The collective evidence presented here indicates that store-operated cAMP production occurs preferentially via AC3 in NCM460 colonic epithelial cells. It is noteworthy that recent data from Spirli and colleagues implicated AC6 as the likely source of cAMP in mouse cholangiocytes during SOcAMPS [15]. However, although some of our overexpression experiments suggested that AC5 and AC6 might also participate in this process, AC3 was the only isoform to consistently yield positive results in both overexpression and knockdown studies. Concurrent expression of AC3 plus STIM1 produced an even larger change in cAMP in response to store depletion. AC3 is the dominant AC in NCM460 cells (as measured by mRNA), but is widely expressed in other cell types and tissues, including at least one cell type that does not exhibit SOcAMPS, the HeLa cell. Moreover, overexpression of AC3 (or AC3 + STIM1) in HeLa cells did not consistently rescue the SOcAMPS phenotype. Therefore, the presence of AC3 is likely not a unique factor that defines SOcAMPS, and other, as yet unidentified, components of the pathway must exist.
Lowering of free ER [Ca2+] leads to dissociation of Ca2+ from the luminal EF hand domain of STIM1, which drives the clustering of STIM1 and Orai1 into aggregates at closely apposed junctions between ER and plasma membrane. The recruitment of other plasma membrane proteins (e.g. STIM2, L-type channels, TRP family members etc.) to this complex has also been described. Because of the dependence of SOcAMPS on the expression of STIM1, we similarly envisioned a scenario in which Orai1, by virtue of its physical interaction with STIM1, might serve to stabilize a complex of AC3-STIM1-Orai1 at the plasma membrane after ER [Ca2+] was reduced. However, the expression level of Orai1 (or Orai3 or STIM2, which are also abundant in NCM cells) did not affect cAMP production following store depletion. Unfortunately, we were unable to determine whether there might be a physical linkage between AC3 and STIM1 during SOcAMPS, because of the lack of suitable antibodies for immunoprecipitation of AC3. We also could not find any evidence for clustering of a GFP-tagged AC3 [46] into punctae following store depletion (Hofer and Maiellaro, unpublished). Thus, the nature of the interaction between STIM1 and AC3 during SOcAMPS remains to be determined.
Cooper and colleagues recently suggested a functional and perhaps physical connection between exogenously expressed AC8, a Ca2+-activated AC and Orai1 [47]. This intimate arrangement allows Ca2+ entering through Orai1 to potently activate AC8 in a localized microenvironment beneath the plasma membrane. Although Orai1 does not appear to be necessary for SOcAMPS, Ca2+ entering via this store-operated Ca2+ entry channel did reproducibly attenuate store-dependent cAMP production, although not completely eliminating it. The mechanism whereby Ca2+ exerts its inhibitory action is not known, and may be indirect. For example, using total internal reflection fluorescence (TIRF) microscopy of STIM1-YFP, we found that the translocation of STIM1 is diminished by high cytosolic [Ca2+] in NCM460 cells (A.M. Hofer, unpublished data), as it is in other cell types [48,49].
The existence of a cAMP-generating pathway that is simultaneously activated by store depletion but inhibited by the concomitant store-dependent Ca2+ influx is unusual, but may provide some clues as to the function of SOcAMPS. In many cell types, Ca2+ signalling events elicited by physiological doses of agonists are oscillatory, with nominal release and refilling of the ER during each oscillatory cycle. Although the repetitive Ca2+ spiking in the cytosol may be optimally tuned to activate Ca2+ effectors, this type of ER depletion will not be of sufficient duration to elicit SOcAMPS. On the other hand, strong agonist stimulation typically results in profound ER store depletion sufficient to activate SOcAMPS, but also elicits robust Ca2+ entry through Orai channels, and this would attenuate cAMP production. Although such signals are normally terminated quite effectively by removal of the initiating stimulus, during persistent Ca2+ signalling Ca2+ entry can eventually become down-regulated, for example via feedback phosphorylation of Orai1 by Ca2+-dependent PKC [50]. Under such conditions, SOcAMPS would then become the dominant second messenger pathway. ER Ca2+ stores are also vulnerable to wide variety of environmental stressors, toxic insults, drugs, and viral and bacterial pathogens that cause unregulated, even permanent, loss of ER Ca2+. We propose that the unorthodox mode of AC3-dependent cAMP production described here may signal the initiation of adaptive responses (such as cAMP-dependent fluid secretion [51] or compensatory programmes of gene expression) in the face of persistent non-physiological loss of ER Ca2+ stores. Certain cell types (e.g. colon and airway epithelia) are at the frontline in the barrier against these noxious agents, and perhaps not coincidentally, are the same cell types that manifest the SOcAMPS phenotype.
Acknowledgments
We are thankful for the kind gift of plasmids from Dr. Kees Jalink, Netherlands Cancer Center (EpacH30), Dr. Roger Y. Tsien, UCSD (mCherry), Dr. Carole A. Parent, National Cancer Institute (AC3-GFP), Dr. Tobias Meyer, Stanford University (STIM1), Dr. Marie Dziadek, Garvan Institute, Australia (STIM2). Thanks are given to Jonathan M. Nichols and Christina N. Dragon for excellent technical assistance. We gratefully acknowledge support from the Harvard Digestive Diseases Center (5P30DK034854-24), the NIDDK (1R21DK088197-01) and the Medical Research Service of the Veteran's Administration (VA-ORD 1 I01 BX000968-01), all to AMH.
Author contributions
I.M., K.L. and A.M.H. performed experiments; I.M., K.L., S.C. and A.M.H. were involved in data analysis, experimental design and writing of the manuscript. M.P.M. provided cell models.
Conflict of interest
M.P.M. holds part ownership of InCell Corporation LLC, which sells M3:10 medium. The other authors declare no conflicts.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Fig. S1 (A) WESTERN BLOT showing effectiveness of Orai1 shRNA knockdown strategy. Bands were resolved in 5–20% tris-glycine gels and evidenced using Orai-1 primary antibody (1:500, Santa Cruz) followed by secondary anti-rabbit antibody (1:1000, Santa Cruz). (B) Bar graph summarizing effects of overexpression of Orai3 or (C) Orai3 + STIM1 in NCM460 cells. Cells expressing an Epac-based cAMP sensor plated onto glass coverslips were co-transfected with Orai3 or Orai3 + STIM1 together with mCherry (used as marker of transfected cells; 300 ng each). Forty-eight hours after transfection the coverslips were mounted in a home-built flow through perfusion chamber and imaged using a 60× oil immersion objective. Overexpression of Orai3 or Orai3 + STIM1 did not significantly alter ionomycin-induced cAMP production compared to control cells.
Fig. S2 (A) STIM2 is not required for store-operated cAMP signaling. Validation of three different strategies used for STIM2 silencing: (i) Dharmacon SmartPool (siRNA STIM2-a), (ii) Ambion silencer (siRNA STIM2-b), and (iii) Origene shRNA (shRNA STIM2). NCM460 or HeLa cells were transfected with 100nM siRNA or with 0.375 ng/μl of a plasmid expressing a single shRNA. Forty-eight hours after transfection, cells were lysed in RIPA buffer (Sigma-Aldrich) complemented with protease inhibitors (Sigma-Aldrich). Five to 10 μg of total cell lysate were resolved in 5–20% tris-glycine gels and electroblotted in PVDF membranes (Amersham Biosciences). The membranes were incubated for 2 hrs at room temperature with an anti-STIM2 polyclonal antibody (ProSci) diluted 1:500 in 5% milk/TBST. After washing with TBST membranes were incubated for 1 hr with secondary antibodies (1:2000; Santa Cruz). Peroxidase activity was detected with the enhanced chemiluminescence kit (Amersham Biosciences, ECL advance western blotting). Loading control, Glyceraldehyde-3-phopshate dehydrogenase (GAPDH; Santa Cruz 1:2000). (B) NCM460 cells expressing an Epac-based cAMP sensor plated onto glass coverslips were transfected with 100 nM of siRNAs against STIM2 (siRNA-a or siRNA-b) or their respective scramble siRNAs. Alternatively cells were co-transfected with shRNAs targeting STIM2 (or non-effective scramble) together with mCherry (used as marker of transfected cells). Forty-eight hours after transfection the coverslips were mounted in a home-built flow through perfusion chamber and cells were imaged using a 60× oil immersion objective. Initial experiments using a pool of four distinct siRNAs against STIM2 (siRNA-a, B-first bars) suggested that STIM2 attenuates SOcAMPs (data from 33 cells of scramble in 6 experiments; 73 cells siRNA STIM2 in 7 experiments; P < 0.0005). However, the scramble control was not typical, thus this series of experiments was inconclusive. Numerous subsequent experiments using either the same siRNA pool (siRNA-a) (data from 21 cells of scramble in 4 experiments; 16 cells siRNA STIM2 in 3 experiments) or two alternative strategies, a single siRNA (siRNA-b) (data from 18 cells of scramble in 4 experiments; 18 cells siRNA STIM2 in 4 experiments) or a single shRNA against STIM2 (shRNA-C) (data from 10 cells of scramble in 4 experiments; 5 cells siRNA STIM2 in 4 experiments) led to the conclusion that STIM2 is not a significant constituent of the SOcAMPs machinery in NCM cells.
Overexpression of AC3 (n = 3 expts, 3 control cells and 9/11 mCherry cells), AC5 (n = 3 expts, 12 mCherry cells and 21 control cells), or AC6 (n = 3 expts, 27 control cells and 8 mCherry cells) in HeLa cells did not generally rescue the SOcAMPS phenotype. Shown is a typical result for AC3 overexpression. HeLa cells stable expressing Epac-H90 cyclic AMP based sensor were transfected the day after plating with the 300 ng AC isoform of interest plus 300 ng of mCherry plasmid. Experiments were performed 48 hrs after transfection.
Left panel: Summary of data from Fig 4C and D in main text. Addition of PMA alone to NCM460 cells caused a minor increase in the FRET ratio (expressed as a percentage of the maximal response following treatment with forskolin + IBMX; from experiments like Fig. 4C in main text). Addition of PMA after ionomycin caused a minor elevation in the FRET ratio compared to ionomyicn alone (refer to Fig. 4D in main text). Right panel: Amplitude of ionomycin response (expressed as a percentage of the maximal FRET ratio change following treatment with forskolin + IBMX) after 15 min. and 3 hrs treatments with PMA.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev. 2006;86:369–408. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- 2.Burdakov D, Petersen OH, Verkhratsky A. Intraluminal calcium as a primary regulator of endoplasmic reticulum function. Cell Calcium. 2005;38:303–10. doi: 10.1016/j.ceca.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 3.Paschen W, Mengesdorf T. Endoplasmic reticulum stress response and neurodegeneration. Cell Calcium. 2005;38:409–15. doi: 10.1016/j.ceca.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 4.Lefkimmiatis K, Srikanthan M, Maiellaro I, et al. Store-operated cyclic AMP signalling mediated by STIM1. Nat Cell Biol. 2009;11:433–42. doi: 10.1038/ncb1850. [DOI] [PubMed] [Google Scholar]
- 5.Zhang SL, Yu Y, Roos J, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–5. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roos J, DiGregorio PJ, Yeromin AV, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–45. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liou J, Kim ML, Heo WD, et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–41. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prakriya M, Feske S, Gwack Y, et al. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–3. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 9.Vig M, Beck A, Billingsley JM, et al. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr Biol. 2006;16:2073–9. doi: 10.1016/j.cub.2006.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yeromin AV, Zhang SL, Jiang W, et al. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature. 2006;443:226–9. doi: 10.1038/nature05108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou Y, Meraner P, Kwon HT, et al. STIM1 gates the store-operated calcium channel ORAI1 in vitro. Nat Struct Mol Biol. 2010;17:112–6. doi: 10.1038/nsmb.1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smyth JT, Hwang SY, Tomita T, et al. Activation and regulation of store-operated calcium entry. J Cell Mol Med. 2010;14:2337–49. doi: 10.1111/j.1582-4934.2010.01168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moyer MP, Manzano LA, Merriman RL, et al. NCM460, a normal human colon mucosal epithelial cell line. In Vitro Cell Dev Biol Anim. 1996;32:315–7. doi: 10.1007/BF02722955. [DOI] [PubMed] [Google Scholar]
- 14.Schwarzer C, Wong S, Shi J, et al. Pseudomonas aeruginosa Homoserine lactone activates store-operated cAMP and cystic fibrosis transmembrane regulator-dependent Cl− secretion by human airway epithelia. J Biol Chem. 2010;285:34850–63. doi: 10.1074/jbc.M110.167668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spirli C, Locatelli L, Fiorotto R, et al. Altered store operated calcium entry increases cAMP production and ERK1/2 phosphorylation in Polycystin-2 defective cholangiocytes. Hepatology. 2012;55:856–68. doi: 10.1002/hep.24723. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Sadana R, Dessauer CW. Physiological roles for G protein-regulated adenylyl cyclase isoforms: insights from knockout and overexpression studies. Neurosignals. 2009;17:5–22. doi: 10.1159/000166277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buck J, Sinclair ML, Schapal L, et al. Cytosolic adenylyl cyclase defines a unique signaling molecule in mammals. Proc Natl Acad Sci USA. 1999;96:79–84. doi: 10.1073/pnas.96.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ponsioen B, Zhao J, Riedl J, et al. Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO Rep. 2004;5:1176–80. doi: 10.1038/sj.embor.7400290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qin Y, Dittmer PJ, Park JG, et al. Measuring steady-state and dynamic endoplasmic reticulum and Golgi Zn2+ with genetically encoded sensors. Proc Natl Acad Sci USA. 2011;108:7351–6. doi: 10.1073/pnas.1015686108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morgan AJ, Jacob R. Ionomycin enhances Ca2+ influx by stimulating store-regulated cation entry and not by a direct action at the plasma membrane. Biochem J. 1994;300(Pt 3):665–72. doi: 10.1042/bj3000665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mercer JC, Dehaven WI, Smyth JT, et al. Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J Biol Chem. 2006;281:24979–90. doi: 10.1074/jbc.M604589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoover PJ, Lewis RS. Stoichiometric requirements for trapping and gating of Ca2+ release-activated Ca2+ (CRAC) channels by stromal interaction molecule 1 (STIM1) Proc Natl Acad Sci USA. 2011;108:13299–304. doi: 10.1073/pnas.1101664108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soboloff J, Spassova MA, Tang XD, et al. Orai1 and STIM reconstitute store-operated calcium channel function. J Biol Chem. 2006;281:20661–5. doi: 10.1074/jbc.C600126200. [DOI] [PubMed] [Google Scholar]
- 24.Willoughby D, Cooper DM. Organization and Ca2+ regulation of adenylyl cyclases in cAMP microdomains. Physiol Rev. 2007;87:965–1010. doi: 10.1152/physrev.00049.2006. [DOI] [PubMed] [Google Scholar]
- 25.Fagan KA, Mahey R, Cooper DM. Functional co-localization of transfected Ca(2+)-stimulable adenylyl cyclases with capacitative Ca2+ entry sites. J Biol Chem. 1996;271:12438–44. doi: 10.1074/jbc.271.21.12438. [DOI] [PubMed] [Google Scholar]
- 26.Choi EJ, Wong ST, Hinds TR, et al. Calcium and muscarinic agonist stimulation of type I adenylylcyclase in whole cells. J Biol Chem. 1992;267:12440–2. [PubMed] [Google Scholar]
- 27.Boyajian CL, Garritsen A, Cooper DM. Bradykinin stimulates Ca2+ mobilization in NCB-20 cells leading to direct inhibition of adenylylcyclase. A novel mechanism for inhibition of cAMP production. J Biol Chem. 1991;266:4995–5003. [PubMed] [Google Scholar]
- 28.Yoshimura M, Cooper DM. Cloning and expression of a Ca(2+)-inhibitable adenylyl cyclase from NCB-20 cells. Proc Natl Acad Sci USA. 1992;89:6716–20. doi: 10.1073/pnas.89.15.6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wallach J, Droste M, Kluxen FW, et al. Molecular cloning and expression of a novel type V adenylyl cyclase from rabbit myocardium. FEBS Lett. 1994;338:257–63. doi: 10.1016/0014-5793(94)80279-3. [DOI] [PubMed] [Google Scholar]
- 30.Choi EJ, Xia Z, Storm DR. Stimulation of the type III olfactory adenylyl cyclase by calcium and calmodulin. Biochemistry. 1992;31:6492–8. doi: 10.1021/bi00143a019. [DOI] [PubMed] [Google Scholar]
- 31.Wayman GA, Impey S, Storm DR. Ca2+ inhibition of type III adenylyl cyclase in vivo. J Biol Chem. 1995;270:21480–6. doi: 10.1074/jbc.270.37.21480. [DOI] [PubMed] [Google Scholar]
- 32.Hanoune J, Defer N. Regulation and role of adenylyl cyclase isoforms. Annu Rev Pharmacol Toxicol. 2001;41:145–74. doi: 10.1146/annurev.pharmtox.41.1.145. [DOI] [PubMed] [Google Scholar]
- 33.Pierre S, Eschenhagen T, Geisslinger G, et al. Capturing adenylyl cyclases as potential drug targets. Nat Rev Drug Discov. 2009;8:321–35. doi: 10.1038/nrd2827. [DOI] [PubMed] [Google Scholar]
- 34.Lefkimmiatis K, Moyer MP, Curci S, et al. “cAMP sponge”: a buffer for cyclic adenosine 3′, 5′-monophosphate. PLoS One. 2009;4:e7649. doi: 10.1371/journal.pone.0007649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sunahara RK, Taussig R. Isoforms of mammalian adenylyl cyclase: multiplicities of signaling. Mol Interv. 2002;2:168–84. doi: 10.1124/mi.2.3.168. [DOI] [PubMed] [Google Scholar]
- 36.Yan L, Vatner DE, O'Connor JP, et al. Type 5 adenylyl cyclase disruption increases longevity and protects against stress. Cell. 2007;130:247–58. doi: 10.1016/j.cell.2007.05.038. [DOI] [PubMed] [Google Scholar]
- 37.Tang T, Gao MH, Lai NC, et al. Adenylyl cyclase type 6 deletion decreases left ventricular function via impaired calcium handling. Circulation. 2008;117:61–9. doi: 10.1161/CIRCULATIONAHA.107.730069. [DOI] [PubMed] [Google Scholar]
- 38.Schaefer ML, Wong ST, Wozniak DF, et al. Altered stress-induced anxiety in adenylyl cyclase type VIII-deficient mice. J Neurosci. 2000;20:4809–20. doi: 10.1523/JNEUROSCI.20-13-04809.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Esposito G, Jaiswal BS, Xie F, et al. Mice deficient for soluble adenylyl cyclase are infertile because of a severe sperm-motility defect. Proc Natl Acad Sci USA. 2004;101:2993–8. doi: 10.1073/pnas.0400050101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Avidor-Reiss T, Nevo I, Saya D, et al. Opiate-induced adenylyl cyclase superactivation is isozyme-specific. J Biol Chem. 1997;272:5040–7. doi: 10.1074/jbc.272.8.5040. [DOI] [PubMed] [Google Scholar]
- 41.Wang Z, Li V, Chan GC, et al. Adult type 3 adenylyl cyclase-deficient mice are obese. PLoS One. 2009;4:e6979. doi: 10.1371/journal.pone.0006979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Z, Storm DR. Maternal behavior is impaired in female mice lacking type 3 adenylyl cyclase. Neuropsychopharmacology. 2011;36:772–81. doi: 10.1038/npp.2010.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wong ST, Trinh K, Hacker B, et al. Disruption of the type III adenylyl cyclase gene leads to peripheral and behavioral anosmia in transgenic mice. Neuron. 2000;27:487–97. doi: 10.1016/s0896-6273(00)00060-x. [DOI] [PubMed] [Google Scholar]
- 44.Wray NR, Pergadia ML, Blackwood DH, et al. Genome-wide association study of major depressive disorder: new results, meta-analysis, and lessons learned. Mol Psychiatry. 2012;17:36–48. doi: 10.1038/mp.2010.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Z, Phan T, Storm DR. The type 3 adenylyl cyclase is required for novel object learning and extinction of contextual memory: role of cAMP signaling in primary cilia. J Neurosci. 2011;31:5557–61. doi: 10.1523/JNEUROSCI.6561-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu L, Das S, Losert W, et al. mTORC2 regulates neutrophil chemotaxis in a cAMP- and RhoA-dependent fashion. Dev Cell. 2010;19:845–57. doi: 10.1016/j.devcel.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Willoughby D, Wachten S, Masada N, et al. Direct demonstration of discrete Ca2+ microdomains associated with different isoforms of adenylyl cyclase. J Cell Sci. 2010;123:107–17. doi: 10.1242/jcs.062067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Malli R, Naghdi S, Romanin C, et al. Cytosolic Ca2+ prevents the subplasmalemmal clustering of STIM1: an intrinsic mechanism to avoid Ca2+ overload. J Cell Sci. 2008;121:3133–9. doi: 10.1242/jcs.034496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen WW, Frieden M, Demaurex N. Local cytosolic Ca2+ elevations are required for stromal interaction molecule 1 (STIM1) de-oligomerization and termination of store-operated Ca2+ entry. J Biol Chem. 2011;286:36448–59. doi: 10.1074/jbc.M111.269415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kawasaki T, Ueyama T, Lange I, et al. Protein kinase C-induced phosphorylation of Orai1 regulates the intracellular Ca2+ level via the store-operated Ca2+ channel. J Biol Chem. 2010;285:25720–30. doi: 10.1074/jbc.M109.022996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frizzell RA, Field M, Schultz SG. Sodium-coupled chloride transport by epithelial tissues. Am J Physiol. 1979;236:F1–8. doi: 10.1152/ajprenal.1979.236.1.F1. [DOI] [PubMed] [Google Scholar]
- 52.Gerbino A, Ruder WC, Curci S, et al. Termination of cAMP signals by Ca2+ and G(alpha)i via extracellular Ca2+ sensors: a link to intracellular Ca2+ oscillations. J Cell Biol. 2005;171:303–12. doi: 10.1083/jcb.200507054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 (A) WESTERN BLOT showing effectiveness of Orai1 shRNA knockdown strategy. Bands were resolved in 5–20% tris-glycine gels and evidenced using Orai-1 primary antibody (1:500, Santa Cruz) followed by secondary anti-rabbit antibody (1:1000, Santa Cruz). (B) Bar graph summarizing effects of overexpression of Orai3 or (C) Orai3 + STIM1 in NCM460 cells. Cells expressing an Epac-based cAMP sensor plated onto glass coverslips were co-transfected with Orai3 or Orai3 + STIM1 together with mCherry (used as marker of transfected cells; 300 ng each). Forty-eight hours after transfection the coverslips were mounted in a home-built flow through perfusion chamber and imaged using a 60× oil immersion objective. Overexpression of Orai3 or Orai3 + STIM1 did not significantly alter ionomycin-induced cAMP production compared to control cells.
Fig. S2 (A) STIM2 is not required for store-operated cAMP signaling. Validation of three different strategies used for STIM2 silencing: (i) Dharmacon SmartPool (siRNA STIM2-a), (ii) Ambion silencer (siRNA STIM2-b), and (iii) Origene shRNA (shRNA STIM2). NCM460 or HeLa cells were transfected with 100nM siRNA or with 0.375 ng/μl of a plasmid expressing a single shRNA. Forty-eight hours after transfection, cells were lysed in RIPA buffer (Sigma-Aldrich) complemented with protease inhibitors (Sigma-Aldrich). Five to 10 μg of total cell lysate were resolved in 5–20% tris-glycine gels and electroblotted in PVDF membranes (Amersham Biosciences). The membranes were incubated for 2 hrs at room temperature with an anti-STIM2 polyclonal antibody (ProSci) diluted 1:500 in 5% milk/TBST. After washing with TBST membranes were incubated for 1 hr with secondary antibodies (1:2000; Santa Cruz). Peroxidase activity was detected with the enhanced chemiluminescence kit (Amersham Biosciences, ECL advance western blotting). Loading control, Glyceraldehyde-3-phopshate dehydrogenase (GAPDH; Santa Cruz 1:2000). (B) NCM460 cells expressing an Epac-based cAMP sensor plated onto glass coverslips were transfected with 100 nM of siRNAs against STIM2 (siRNA-a or siRNA-b) or their respective scramble siRNAs. Alternatively cells were co-transfected with shRNAs targeting STIM2 (or non-effective scramble) together with mCherry (used as marker of transfected cells). Forty-eight hours after transfection the coverslips were mounted in a home-built flow through perfusion chamber and cells were imaged using a 60× oil immersion objective. Initial experiments using a pool of four distinct siRNAs against STIM2 (siRNA-a, B-first bars) suggested that STIM2 attenuates SOcAMPs (data from 33 cells of scramble in 6 experiments; 73 cells siRNA STIM2 in 7 experiments; P < 0.0005). However, the scramble control was not typical, thus this series of experiments was inconclusive. Numerous subsequent experiments using either the same siRNA pool (siRNA-a) (data from 21 cells of scramble in 4 experiments; 16 cells siRNA STIM2 in 3 experiments) or two alternative strategies, a single siRNA (siRNA-b) (data from 18 cells of scramble in 4 experiments; 18 cells siRNA STIM2 in 4 experiments) or a single shRNA against STIM2 (shRNA-C) (data from 10 cells of scramble in 4 experiments; 5 cells siRNA STIM2 in 4 experiments) led to the conclusion that STIM2 is not a significant constituent of the SOcAMPs machinery in NCM cells.
Overexpression of AC3 (n = 3 expts, 3 control cells and 9/11 mCherry cells), AC5 (n = 3 expts, 12 mCherry cells and 21 control cells), or AC6 (n = 3 expts, 27 control cells and 8 mCherry cells) in HeLa cells did not generally rescue the SOcAMPS phenotype. Shown is a typical result for AC3 overexpression. HeLa cells stable expressing Epac-H90 cyclic AMP based sensor were transfected the day after plating with the 300 ng AC isoform of interest plus 300 ng of mCherry plasmid. Experiments were performed 48 hrs after transfection.
Left panel: Summary of data from Fig 4C and D in main text. Addition of PMA alone to NCM460 cells caused a minor increase in the FRET ratio (expressed as a percentage of the maximal response following treatment with forskolin + IBMX; from experiments like Fig. 4C in main text). Addition of PMA after ionomycin caused a minor elevation in the FRET ratio compared to ionomyicn alone (refer to Fig. 4D in main text). Right panel: Amplitude of ionomycin response (expressed as a percentage of the maximal FRET ratio change following treatment with forskolin + IBMX) after 15 min. and 3 hrs treatments with PMA.