

Abstract
Arsenic trioxide (arsenite, AsIII) has shown a remarkable clinical efficacy, whereas its side effects are still a serious concern. Therefore, it is critical to understand the effects of AsIII on human-derived normal cells for revealing the mechanisms underlying these side effects. We examined the effects of AsIII on primary cultured chorion (C) and amnion (A) cells prepared from human fetal membranes. A significant dose-dependent AsIII-mediated cytotoxicity was observed in the C-cells accompanied with an increase of lactate dehydrogenase (LDH) release. Higher concentrations of AsIII were required for the A-cells to show cytotoxicity and LDH release, suggesting that the C-cells were more sensitive to AsIII than the A-cells. The expression levels of aquaporin 9 (AQP9) were approximately 2 times higher in the C-cells than those in the A-cells. Both intracellular arsenic accumulation and its cytotoxicity in the C-cells were significantly abrogated by sorbitol, a competitive AQP9 inhibitor, in a dose-dependent manner. The protein expression levels of multidrug resistance-associated protein (MRP) 2 were downregulated by AsIII in the C-cells, but not in the A-cells. No significant differences in the expression levels of MRP1 were observed between C- and A-cells. The protein expression of P-glycoprotein (P-gp) was hardly detected in both cells, although a detectable amount of its mRNA was observed. Cyclosporine A, a broad-spectrum inhibitor for ABC transporters, and MK571, a MRP inhibitor, but not PGP-4008, a P-gp specific inhibitor, potently sensitized both cells to AsIII-mediated cytotoxicity. These results suggest that AQP9 and MRP2 are involved in controlling arsenic accumulation in these normal cells, which then contribute to differential sensitivity to AsIII cytotoxicity between these cells.
Keywords: Arsenite, Aquaporin 9, Multidrug resistance protein 2, P-glycoprotein, Fetal membranes
Introduction
Administration of arsenic trioxide (arsenite, AsIII), an arsenic derivative, has demonstrated a remarkable efficacy in the treatment of relapsed and refractory acute promyelocytic leukemia (APL) patients. The successful clinical efficacy in the treatment of APL patients has led to investigations exploring potential treatment applications for other malignancies, including solid tumors (Dilda and Hogg, 2007; Litzow, 2008). In order to understand the mode of action of AsIII and provide an effective treatment protocol for individual APL patients, studies have been conducted on the pharmacokinetics of AsIII in APL patients using biological samples such as urine, blood and cerebrospinal fluid (Shen et al., 1997; Fujisawa et al, 2007; Yoshino et al., 2009; Kiguchi et al., 2010). In fact, we recently demonstrated that not only inorganic arsenic but also methylated arsenic metabolites accumulated in red blood cells during the consecutive administration of AsIII to APL patients (Yoshino et al., 2009). Furthermore, we have demonstrated for the first time that these arsenic metabolites also existed in cerebrospinal fluid (Kiguchi et al., 2010), in which the concentrations of arsenic reached levels necessary for differentiation induction (Chen et al., 1997; Soignet et al., 1998). These findings on the pharmacokinetics of AsIII in APL patients provide a new insight into clinical applications of AsIII, and may contribute to better therapeutic protocols (Yuan et al., 2011).
Although a remarkable clinical efficacy of AsIII-based regimens against APL has been reported (Shen et al., 1997; Soignet et al., 1998), and AsIII has been suggested as a promising candidate for the treatment of refractory solid tumors (Dilda and Hogg, 2007; Litzow, 2008), side effects of AsIII are still a serious concern and hamper its clinical applications. It is thus critical to investigate the effects of AsIII on normal cells and/or tissues for clinical implications. However, very few studies to date have been conducted to investigate the effects of AsIII on normal cells, because of difficulty in obtaining human-derived normal cells (Chattopadhyay et al., 2002; Ferrario et al., 2009).
Recently, we have established a unique in vitro system, comprising the primary cultured chorion (C−) cells and amnion (A−) cells prepared from human fetal membranes obtained at the month of normal parturition, for studying biological responses to external stimuli in normal cells (Yuan et al., 2006, 2008, 2009). So far, we have demonstrated that the C-cells are more vulnerable to oxidative stress than the A-cells (Yuan et al., 2006, 2008, 2009), suggesting that the in vitro system is a good model system to study the role of oxidative stress induced by various external stimuli including anticancer drugs. It is well known that oxidative stress is involved in the mechanisms underlying the therapeutic efficacy of AsIII and plays a major role in the toxicity of AsIII (Ninomiya et al., 2006). In fact, the model system has been proposed to be used for studying the toxicological as well as pharmacological relevance of AsIII (Yuan et al., 2011).
It is quite logical to consider that intracellular arsenic accumulation (As[i]) is critical for the control of various biological functions and that these levels are tightly associated with AsIII uptake and efflux (Liu et al., 2002; Leslie et al., 2004; Lee et al., 2006; Leung et al., 2007; Shinkai et al., 2009). Accumulating evidence indicates that aquaglyceroporins (AQPs), which are members of the aquaporin superfamily and responsible for transporting small uncharged molecules such as glycerol and urea as well as water, play a pivotal role in the uptake of AsIII (Lee et al., 2006; Leung et al., 2007; Shinkai et al., 2009). In particular, a close correlation between the expression levels of AQP9 and AsIII efficacy has been established in leukemia samples (Leung et al., 2007). Regarding efflux, several adenosine triphosphate (ATP) binding cassette (ABC) transporters, including multidrug resistance-associated proteins 1 and 2 (MRP1/2), multidrug resistance protein 1 (MDR1; also known as P-glycoprotein, P-gp) are involved in the efflux of AsIII (Liu et al., 2002; Leslie et al., 2004; Lee et al., 2006). All of the evidence for AsIII transport was obtained from studies using human-derived malignant cells and/or primary mouse hepatocytes as well as in vivo studies on mice. However, the correlation between the effects of AsIII and the expression levels of these transporters in human-derived normal cells has not yet been conducted.
In this study, we investigated the effects of AsIII on C- and A-cells by assessing cell viability, apoptosis induction and lactate dehydrogenase (LDH) release. Furthermore, we investigated the role of AsIII-associated transporters in these cells using various inhibitors, such as sorbitol and phloretin, both of which are inhibitors for AQP9 (Tsukaguchi et al., 1998; Shinkai et al., 2009); cyclosporine A (CsA), a broad-spectrum inhibitor for ABC transporters (Qadir et al., 2005); MK571, a MRP inhibitor (Matsson et al., 2009); and PGP-4008, a P-gp specific inhibitor (Lee et al., 2003). Results demonstrated for the first time that AQP9 and MRP2 are involved in controlling As[i] in primary cultured normal cells, which then contribute to differential sensitivity to AsIII cytotoxicity between these cells.
Materials and Methods
Materials
Sodium arsenite (AsIII) was purchased from Tri Chemical Laboratories (Yamanashi, Japan). LDH cytotoxicity detection kits were purchased from Roche (Mannheim, Germany). An RNA extraction kit, ISOGEN was obtained from Wako Pure Chemical Industries (Osaka, Japan). CsA, a broad-spectrum inhibitor for ABC transporters, was kindly provided by Novartis Pharma Co. (Basel, Switzerland). 3-[[3-[2-(7-chloroquinolin-2-yl)vinyl]phenyl]-(2-dimethylcarbamoylethylsulfanyl)methylsulfan yl] propionic acid (MK571), an inhibitor of MRPs, was purchased from Merck (Darmstadt, Germany). N-(1-benzyl-2,3-dihydro-1H-pyrrolo[2,3-b]quinolin-4-yl)-2-phenylacetamide (PGP-4008), a specific inhibitor for P-gp, was obtained from Sigma (MD, USA). Sorbitol and phloretin, inhibitors for AQP9, were purchased from Sigma and Wako Pure Chemical Industries, respectively.
Preparation of primary cultured C- and A-cells from human fetal membranes
Fetal membranes were prepared aseptically from placenta obtained during normal parturition by Cesarean section as described previously (Ohyama et al., 1998). Primary cultured C- and A-cells were prepared from fetal membranes, which were freshly isolated according to the methods described previously (Ohyama et al., 1998). This study has been approved by the IRB committee of Tokyo University of Pharmacy and Life Sciences. An informed consent was obtained from patients at the time of surgery.
XTT assay
The cytotoxicity of AsIII to C- and A-cells was investigated by XTT dye-reduction assays according to the method previously described with slight modifications (Scudiero et al., 1988). Briefly, the cells were seeded in 96-well plates coated with type I collagen (Iwaki, Tokyo, Japan) at a density of 5×104 cells per well in 0.1 mL cell culture medium and allowed to become confluent. Cultures in triplicate were treated with various concentrations of AsIII in the presence or absence of inhibitors for transporters at the concentrations indicated. In experiments using transporter inhibitors, both cells were preincubated with transporter inhibitors at the indicated concentrations for 30 min. After treatment with AsIII for an additional 48 h, 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl) -5-[(phenylamino)carbonyl]-2H-tetrazolium hydroxide (XTT) (Sigma, MD, USA) and phenazine methosulfate (Wako Pure Chemical Industries, Osaka, Japan) were added into each well at final concentrations of 0.2 mg/mL and 1 mM, respectively. After incubation at 37 °C for 2 h, the plates were mixed, and the absorbance at 450 nm was measured with a microplate reader (Safire, Tecan, Switzerland). The relative cell viability was expressed as the ratio of the absorbance of each treatment group against those of the corresponding untreated control group. Data are shown as means and SD from three independent experiments.
Lactate dehydrogenase (LDH) assay
After treatment with various concentrations of AsIII for 48 h, LDH leakage from both C- and A-cells was measured using a LDH cytotoxicity detection kit. Twenty-five microliters of culture supernatants were collected by centrifugation at 450×g for 5 min at 4 °C, and transferred into wells of a 96-well plate. After discarding the remaining culture supernatants, the cells were lysed with 200 μL of 0.1% Triton-X100. Twenty-five microliters of cell lysates were subsequently loaded into the 96-well plate. LDH activities in both the culture supernatants and the cell lysates were determined by adding 50 μL of reaction reagent from the kit and 25 μL of PBS to each well, followed by incubation at 37 °C for 15 min. The reaction was stopped by the addition of 100 μL of 1 N HCl, and the absorbance at 490 nm was measured with a microplate reader (Safire, Tecan, Switzerland). Cell damage was calculated as a percentage of LDH leakage from damaged cells using the following formula: LDH leakage (%) = (Sup)/(Sup + Cell) × 100 where Sup and Cell refer to absorption of culture supernatant and cell lysate, respectively. Data are shown as means and SD from three independent experiments.
Reverse transcription-polymerase chain reaction (RT-PCR) analysis
Total RNA and complementary DNA were prepared as described previously (Yuan et al., 2006). Total RNA was extracted from cells using an RNA extraction kit, ISOGEN. Complementary DNA was synthesized from 1 g of RNA using 100 pmol random hexamers and 100 U Moloney murine leukemia virus reverse transcriptase (Invitrogen, CA, USA) in a total volume of 20 μL, according to the manufacturer’s instructions. PCR was performed according to the method described previously (Andus et al., 1993) using a Takara Thermal Cycler MP (TAKARA SHUZO, Osaka, Japan). DNA sequence of PCR primers and optimal conditions for PCR were shown in Table 1. PCR primers were purchased from Sigma-Aldrich (Tokyo, Japan). PCR products and Ready-Load™ 100 bp DNA Ladder (Invitrogen, CA, USA) marker were electrophoresed, respectively, on a 2.0% UltraPure™ agarose gel (Invitrogen, CA, USA), and visualized by ethidium bromide staining, followed by viewing under UV Light Printgraph (ATTO Corp, Tokyo, Japan). The signal intensity of the specific mRNAs was calculated from the density of the gray level on a digitized image using a program, NIH ImageJ 1.43u.
Table 1-1.
PCR primers and optimal numbers of PCR cycle
| Target mRNA | DNA sequence of PCR primer | Optimal cycles | |
|---|---|---|---|
| HO-1 | sense | 5′-CCAGCAACAAAGTGCAAGATTC -3′ | 27 |
| antisense | 5′-CTGCAGGAACTGAGGATGCTG -3′ | ||
| AQP9 | sense | 5′-CTCAGTG TCATCATGTAGTG-3′ | 39 |
| antisense | 5′-GACTATCGTCAAGATGCCG-3′ | ||
| ABCC1 (MRP1) | sense | 5′-CTGACAAGCTAGACCATGAATGT-3 | 30 |
| antisense | 5′-TCACACCAAGCCGGCGTCTTT-3′ | ||
| ABCC2 (MRP2) | sense | 5′-CTTCGGAAATCCAAGATCCTGG-3′ | 39 |
| antisense | 5′-AGGCAAGTC CAGCATCTCTGG-3′ | ||
| ABCB1 (P-gp) | sense | 5′-ATATCAGCAGCCCACATCAT-3′ | 39 |
| antisense | 5′-GAAGCACTGGGATGTCCGGT-3′ | ||
| β-actin | sense | 5′-CCTTCCTGG GCATGGAGTCCTG-3′ | 21 |
| Antisense | 5′-GGAGCAATGATCTTGATCTTC-3′ |
Western blot analysis
For preparation of protein samples, cells were washed twice with cold PBS, and lysed in lysis buffer (50 mM Tris-HCl pH 6.8, 150 mM NaCl, 1 % Triton X-100, 1 mM EDTA, 2 μg/mL leupeptin, 2 μg/mL aprotinin, 1 μg/mL pepstatin, and 1 mM PMSF) on ice for 30 min. After centrifugation at 13, 000 g for 15 min at 4 °C, the supernatants were collected and mixed with 4×SDS buffer (50 mM Tris-HCl pH 6.8, 8% sodium dodecyl sulfate, 24% 2-mercaptoethanol, 40% glycerol, and 0.8% bromphenol blue) in a 1:3 ratio at 4 °C for overnight. Protein concentrations were determined by Bradford’s method using the protein assay dye reagent (Bio-Rad, CA, USA) according to the manufacturer’s instructions, and using BSA as standard. Protein samples were separated on a SDS polyacrylamide gel electrophoresis, followed by transferring to a nitrocellulose membrane according to the method described previously (Yuan et al., 2009). Protein bands were detected using the following primary antibodies: mouse anti-human AQP9 (G-3), mouse anti-human MRP1 (QCRL-1) (Santa Cruz Biotechnology, CA, USA); mouse anti-human MRP2 (M2-III6), mouse anti-human P-gp (C219) (Abcam, MA, USA); and mouse anti-human β-actin (AC-15) (Sigma, MO, USA). Blotted protein bands were detected with respective horseradish peroxidase-conjugated secondary antibodies (GE Healthcare, Buckinghamshire, UK) and an enhanced chemiluminescence (ECL) Western bolt analysis system (GE Healthcare, Buckinghamshire, UK). The signal intensity of the immunoreactive proteins was calculated from the density of the gray level on a digitized image using a program, NIH ImageJ 1.43u.
Analysis of intracellular arsenic accumulation (As[i])
After exposure of C- and A-cells to nontoxic concentrations of 17 μM AsIII in the presence or absence of inhibitors for transporters at the concentrations indicated, cells were washed three times with PBS. Cells were preincubated with transporter inhibitors at concentrations indicated for 30 min. Then, cells were harvested, and the weight of cell pellets were measured and used to normalize cellular arsenic concentrations. The As[i] was normalized by cell weights and given as nanomolar of arsenic per gram of cell weights. The analysis of total arsenic was performed by inductively coupled plasma-mass spectrometry (ICP-MS) (PerkinElmer SCIEX, ON, Canada) according to the methods reported previously (Yoshino et al., 2009).
Statistical analysis
Data were analyzed using Student’s t-test and p<0.05 was considered as statistically significant.
Results
AsIII-induced cytotoxicity in primary cultured C- and A-cells
Cytotoxic effects of AsIII were observed in the C-cells after exposure to AsIII ranging from 5 to 80 μM for 48 h accompanied with an increase of LDH leakage in a dose-dependent manner (Fig. 1A and B). When the concentrations of AsIII increased up to 20 μM, statistically significant differences were observed between AsIII-exposed group and control group (Fig. 1A). On the other hand, in the A-cells, the cytotoxicity of AsIII was only observed when treated with 80 μM of AsIII (Fig. 1A and B).
Fig. 1.

AsIII-induced cytotoxicity in primary cultured C- and A-cells.
After treatment with various concentrations of AsIII (0, 5, 10, 20, 40 or 80 μM) for 48 h, cell viability was determined by XTT assay (A), and LDH leakage (B) of C- and A-cells were analyzed, as described under Materials and Methods. Significant differences were observed between control group and AsIII treatment group (*, p<0.05 and †, p<0.001). Data are shown as the mean and SD from three independent experiments.
Expression profile of HO-1 gene in primary cultured C- and A-cells treated with AsIII
After exposure of these primary normal cells to nontoxic concentrations of AsIII (17 μM), we analyzed the expression profile of HO-1 gene by RT-PCR. An approximately 2-fold increase in the expression levels of HO-1 gene was observed in the A-cells after 0.5-h exposure. Furthermore, the expression levels of HO-1 gene markedly increased more than 8 times in AsIII-treated A-cells compared to those in control cells at 2-h post-exposure, and the increase continued up to 24 h (Fig. 2). In contrast, the same phenomena were not observed in the C-cells, although a slight increase in HO-1 expression was detected in the cells (Fig. 2).
Fig. 2.
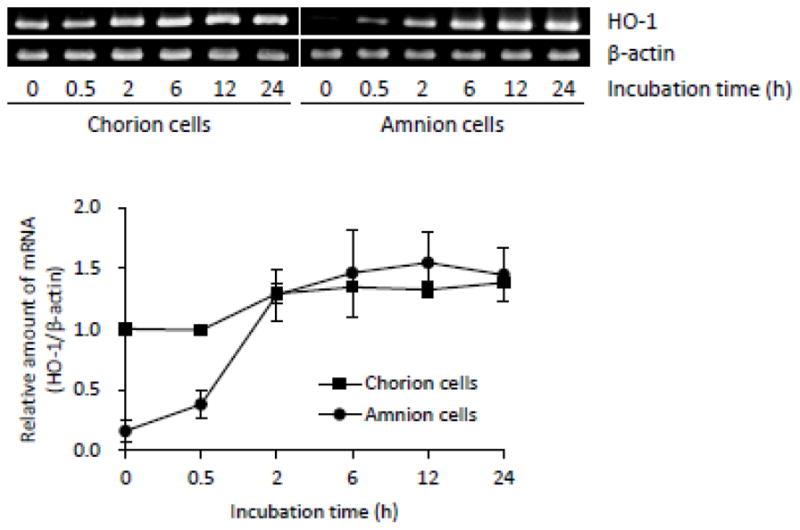
Expression profile of HO-1 mRNA in primary cultured C- and A-cells treated with AsIII.
After exposure to 17 μM AsIII for 0, 0.5, 2, 6, 12, or 24 h, the expression level of HO-1 mRNA was analyzed by RT-PCR, followed by an agarose gel electrophoresis as described under Materials and Methods. The relative expression levels were expressed as the ratios of the expression levels of HO-1 against those of β-actin, and were compared with those in C-cells at 0-time incubation. Experiments were carried out in three independent experiments, and representative electrophoretic profiles are shown. Significant differences were observed between control group (0-time incubation) and AsIII treatment group (from 2-h post-exposure in C-cells, p<0.05; from 0.5-h post-exposure in A-cells, p<0.05)
Intracellular arsenic accumulation (As[i]) in primary cultured C- and A-cells treated with AsIII
After the exposure of these primary cells to 17 μM AsIII for 0, 0.25, 0.5, 1, 2, 4, 6, 12 or 24 h, As[i] was measured by ICP-MS. As shown in Fig. 3, the levels of the As[i] increased with time in both cells, and almost reached a plateau at 6-h post-exposure. Furthermore, the levels of the As[i] were much higher in the C-cells than those in the A-cells, and statistically significant differences in the levels were confirmed at 1-h post-exposure and thereafter. These data clearly demonstrated that the accumulation of AsIII in these cells almost reached a plateau at 6-h post-exposure, and that there was the largest difference in the levels of the As[i] between these cells at the same time point. Thus, the following experiments on arsenic accumulation were conducted by exposing these cells to 17 μM of AsIII for 6 h.
Fig. 3.
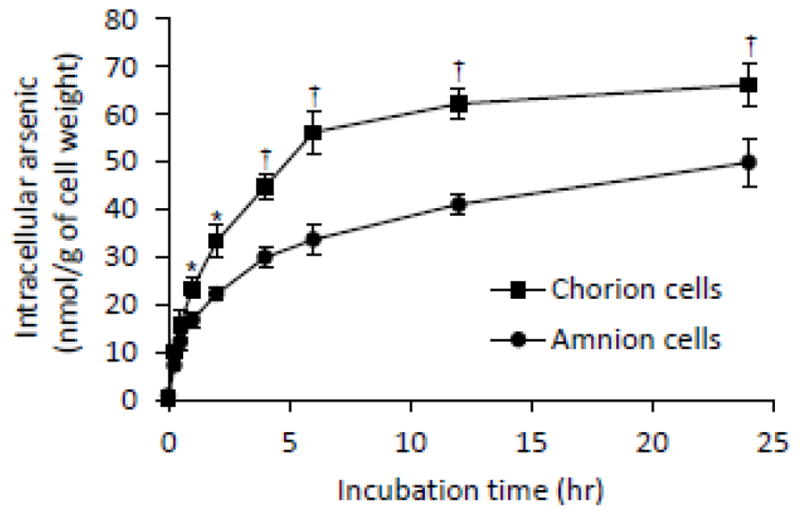
Intracellular arsenic accumulation (As[i]) in primary cultured C- and A-cells treated with AsIII.
Both cells were treated with 17 μM AsIII for 0, 0.25, 0.5, 1, 2, 4, 6, 12, or 24 h, followed by assessing the As[i] using ICP-MS. Data are shown as the mean and SD from three independent experiments. Significant differences between C- and A-cells are shown (*, p<0.05 and †, p<0.001).
Expression levels of AsIII-associated transporters in primary cultured C- and A-cells treated with AsIII
After the treatment of these primary cells with 17 μM AsIII for 0, 6 or 48 h, the expression profiles of AQP9, ABCC1 (MRP1), ABCC2 (MRP2) and ABCB1 (P-gp) were assessed by RT-PCR and Western blot analysis. Since As[i] and cell viability were determined at 6-h and 48-h post-exposure, respectively, we focused on the expression profiles of AsIII-associated transporter genes at the two time points. The levels of endogenous AQP9 gene expression were approximately 2 times higher in the C-cells than those in the A-cells at 0-time incubation and during the treatment with AsIII (Fig. 4). Furthermore, the protein expression level of AQP9 was slightly but significantly upregulated by treatment with AsIII in the C-cells at 48-h post-exposure, but not in the A-cells (Fig. 4C and D). Regarding ABCC1, no significant difference in its mRNA and protein expression level was confirmed between C- and A-cells, although a slight, but significant upregulation of its mRNA expression level was observed in both cells at 48-h post-exposure (Fig. 4). Intriguingly, an inductive effect of AsIII on the expression of ABCC2 mRNA was observed and continued up to 48 h in the A-cells (Fig. 4A and B). However, in the C-cells, the expression levels of ABCC2 mRNA exhibited a transient, significant upregulation at 6-h, followed by a substantial decrease at 48-h post-exposure (Fig. 4A and B). Unlike ABCC2 mRNA expression pattern in both cells, a time-dependent decrease in its protein expression levels was observed in the C-cells, whereas no alteration was observed in the A-cells during the exposure period (Fig. 4C and D). Furthermore, the expression levels of endogenous ABCC2 protein were significant higher in the A-cells than those in the C-cells (Fig. 4C and D). Unexpectedly, the expression of ABCB1 protein was hardly detected in both cells regardless of AsIII treatment, although a significant time-dependent increase in its mRNA expression was observed in these cells (Fig. 4).
Fig. 4.

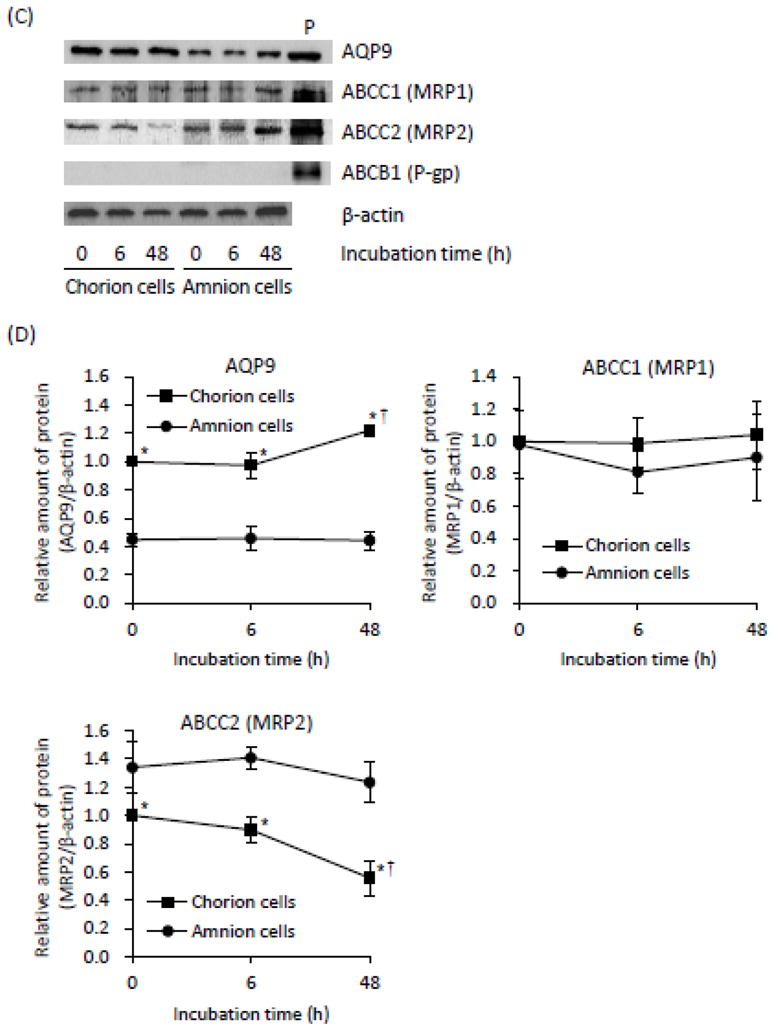
Expression levels of AsIII-associated transporters in primary cultured C- and A-cells treated with AsIII
Expression levels of mRNAs (A), the relative expression levels of each mRNA (B), the expression levels of proteins (C), and the relative expression levels of each protein (D) of AsIII-associated transporters were analyzed, respectively, as described under Materials and Methods. The relative expression levels of each gene were expressed as described in Fig. 2. Both cells were treated with 17 μM AsIII. A representative electrophoretic profile of three separate experiments is shown. P represents positive control used in this study, where NB4, an APL cell line, for AQP9; a MRP1-expressing cell line for MRP1; HEK293 cells stably expressing MRP2 cDNA for MRP2; and P-gp-expressing daunorubicin-resistant MOLT-4 cells for P-gp protein. Data are shown as the mean and SD from three independent experiments. Significant differences in the expression levels of each gene are shown (* p<0.05 C-cell vs. A-cell; † p<0.05 vs. control)
Effects of sorbitol and phloretin on As[i] and AsIII-induced cytotoxicity in primary cultured C- and A-cells
Both primary cells were exposed to 17 μM AsIII in the presence or absence of sorbitol (10, 30, 50 or 100 mM) and phloretin (50, 100, 200 or 500 μM), respectively, for 6 h, followed by the assessment of As[i]. In the C-cells, a significant, dose-dependent decrease in the As[i] was observed in the presence of sorbitol (Fig. 5A). Moreover, the levels of the As[i] were reduced by 40% in the presence of 100 mM sorbitol compared to those treated with AsIII alone. However, no such effects were observed in the A-cells (Fig. 5A). Consistent with the decreased As[i], AsIII-induced cytotoxicity in the C-cells was significantly abrogated by the presence of sorbitol in a dose-dependent manner when the cells were exposed to AsIII ranging from 5 to 80 μM in the presence of 30 and 100 mM sorbitol for 48 h (Fig. 5B). Again, no alterations of AsIII-induced cytotoxicity were observed in the A-cells exposed to AsIII ranging from 5 to 40 μM with or without 30 and 100 mM sorbitol for 48 h (Fig. 5B). Consistent with XTT results (Fig. 1), AsIII-induced cytotoxicity was observed in the cells treated with as high as 80 μM AsIII regardless of the presence or absence of sorbitol (Fig. 5B). Unexpectedly, the levels of As[i] were hardly altered by the addition of phloretin in both cells, although the addition of 500 μM phloretin slightly but significantly reduced the level of As[i] only in C-cells (Fig. 5C). Furthermore, no alternations of AsIII-induced cytotoxicity were observed in both cells when treated with AsIII ranging from 5 to 80 μM for 48 h, regardless of the presence or absence of 100 μM and 200 μM phloretin (Fig. 5D). Moreover, it was confirmed that cell viability was not altered by sorbitol or phloretin alone in both cells.
Fig. 5.


Effects of sorbitol on As[i] and AsIII-induced cytotoxicity in primary cultured C- and A-cells.
(A and C): Both cells were treated with 17 μM of AsIII in the presence or absence of sorbitol (10, 30, 50 or 100 mM) and phloretin (50, 100, 200 or 500 μM), respectively, for 6 h, followed by the assessment of As[i] using ICP-MS. (B and D): Cell viability was determined by XTT assay after treatment with AsIII ranging from 5 to 80 μM in the presence or absence of sorbitol (30 or 100 mM) and phloretin (100 or 200 μM), respectively, for 48 h. Data are shown as the mean and SD from three independent experiments. Significant differences were observed between control groups (AsIII-only treatment) and the treated groups (AsIII plus sorbitol) (*, p<0.05 and †, p<0.001).
Effects of CsA on As[i] and AsIII-induced cytotoxicity in primary cultured C- and A-cells
Both primary cells were exposed to 17 μM AsIII in the presence or absence of CsA at various concentrations (1, 3, 10, 30 μM) for 6 h, followed by the assessment of As[i]. In both cells, the addition of CsA significantly increased the As[i] in a dose-dependent manner (Fig. 6A). Moreover, the As[i] increased from 40.2 to 68.8 nmol/g of cell weight and from 25.5 to 43.3 nmol/g of cell weight in the C-cells and the A-cells, respectively, by the addition of 30 μM of CsA (Fig. 6A). Consistent with the increased As[i] in both cells, AsIII-induced cytotoxicity was significantly enhanced by the addition of CsA in a dose-dependent manner when treated with AsIII ranging from 5 to 80 μM in the presence of 3 or 10 μM of CsA for 48 h (Fig. 6B). More potent enhancing effect of CsA were observed in the C-cells when compared with those in the A-cells. Furthermore, it was confirmed that cell viability was not altered by the presence of CsA alone in both cells.
Fig. 6.

Effects of CsA on As[i] and AsIII-induced cytotoxicity in primary cultured C- and A-cells.
(A) Both cells were treated with 17 μM of AsIII in the presence or absence of CsA (1, 3, 10, or 30 μM) for 6 h, followed by the assessment of As[i] using ICP-MS. (B) Cell viability was determined by XTT assay after treatment with AsIII ranging from 5 to 80 μM in the presence or absence of 3 or 10 μM CsA for 48 h. Data are shown as the means and SD from three independent experiments. Significant differences were observed between control groups (AsIII-only treatment) and the other treatment groups (AsIII plus CsA) (*, p<0.05 and †, p<0.001).
Effects of MK571 on As[i] and AsIII-induced cytotoxicity in primary cultured C- and A-cells
Both primary cells were exposed to 17 μM AsIII in the presence or absence of MK571 at various concentrations (10, 30, 50, 100 μM) for 6 h, followed by the assessment of As[i]. The levels of the As[i] were prominently increased by the addition of MK571 in a dose-dependent manner in the C-cells (Fig. 7A). In contrast, the levels of As[i] in the A-cells were only slightly, but significantly, increased by MK571 (Fig. 7A). The As[i] increased from 36.1 to 70.4 nmol/g of cell weight in the C-cells, and from 19.6 to 25.7 nmol/g of cell weight in the A-cells respectively, by the addition of 100 μM of MK571 (Fig. 7A). Consistent with the increased As[i] in the C-cells, AsIII-induced cytotoxicity was significantly enhanced by MK571 in a dose-dependent manner when treated with AsIII ranging from 5 to 80 μM in the presence of 10 or 50 μM of MK571 for 48 h (Fig. 7B). Intriguingly, the addition of MK571 did not alter cell viability of the A-cells when exposed to AsIII ranging from 5 to 40 μM (Fig. 7B). However, the addition of 50 μM of MK571 slightly but significantly enhanced AsIII-induced cytotoxicity in the cells exposed to AsIII at a concentration of 80 μM (Fig. 7B). Moreover, it was confirmed that cell viability was not altered by MK571 alone in both cells.
Fig. 7.

Effects of MK571 on As[i] and AsIII-induced cytotoxicity in primary cultured C- and A-cells.
(A) Both cells were treated with 17 μM of AsIII in the presence or absence of MK571 (10, 30, 50, or 100 μM) for 6 h, followed by the assessment of As[i] using ICP-MS. (B) Cell viability was determined by XTT assay after treatment with AsIII ranging from 5 to 80 μM in the presence or absence of 10 or 50 μM MK571 for 48 h. Data are shown as the means and SD from three independent experiments. Significant differences were observed between control groups (AsIII-only treatment) and the other treatment groups (AsIII plus MK571) (*, p<0.05 and †, p<0.001).
Effects of PGP-4008 on As[i] and AsIII-induced cytotoxicity in primary cultured C- and A-cells
Both primary cells were exposed to 17 μM AsIII in the presence or absence of PGP-4008 at various concentrations (5, 10, 20 μM) for 6 h, followed by the assessment of As[i]. The levels of As[i] were not altered by the addition of PGP-4008 in both cells (Fig. 8A). At the same time, no alteration of AsIII-induced cytotoxicity was observed in both cells when treated with AsIII ranging from 5 to 80 μM for 48 h, regardless of the presence or absence of 3 or 10 μM of PGP-4008 (Fig. 8B). In order to verify whether these concentrations of PGP-4008 could functionally inhibit P-gp function, we initially evaluated P-gp efflux function using rhodamine 123 in P-gp-expressing daunorubicin-resistant MOLT-4 cells, a human T-lymphoblastoid leukemia cell line. Results demonstrated that these concentrations are enough to inhibit P-gp function (data not shown). Furthermore, it was confirmed that cell viability was not altered by PGP-4008 alone in both cells.
Fig. 8.

Effects of PGP-4008 on As[i] and AsIII-induced cytotoxicity in primary cultured C- and A-cells.
(A) Both cells were treated with 17 μM of AsIII in the presence or absence of PGP-4008 (5, 10, or 20 μM) for 6 h, followed by the assessment of As[i] using ICP-MS. (B) Cell viability was determined by XTT assay after treatment with AsIII ranging from 5 to 80 μM in the presence or absence of 3 or 10 μM PGP-4008 for 48 h. Data are shown as the mean and SD from three independent experiments.
Discussion
In this study, we demonstrated that a significant AsIII-mediated cytotoxicity was observed in C-cells in a dose-dependent manner, but not in A-cells, suggesting that C-cells were more sensitive to AsIII than A-cells. We also demonstrated that the levels of the intracellular arsenic accumulation (As[i]) were much higher in the C-cells than those in the A-cells, indicating that the sensitivity to AsIII correlates with the As[i]. Furthermore, our results suggest that the immediate and marked upregulation of HO-1 expression levels observed only in the A-cells contribute to the maintenance of cellular homeostasis. This idea is supported by our previous observations that an increased level of HO-1 gene expression is responsible for higher tolerance to oxidative stress in the A-cells (Yuan et al., 2008). Of note, it has been reported that the expression of HO-1 plays an important role in arsenite-resistant human lung adenocarcinoma cells (Lee and Ho, 1994). We further demonstrated that no nuclear condensation and DNA fragmentation (data not shown) was observed in both cells, indicating no involvement of apoptosis induction in these cells. Collectively, our results suggested that AsIII-induced cell death in primary cultured normal cells is predominantly associated with a necrosis-like phenotype as assessed by LDH release, and that AsIII-mediated side effects can be partially attributed to the necrosis-like cell death induction.
Accumulating evidence suggests that AQP9 is a primary route of AsIII uptake into leukemia cells, suggesting that monitoring its expression levels in APL patients before or during treatment with AsIII may provide an index for therapeutic efficacy (Leung et al., 2007; Yuan et al., 2011). On the other hand, it has been established that MRP1/2 and P-gp are involved in the efflux of AsIII (Liu et al., 2002; Leslie et al., 2004; Lee et al., 2006; Yuan et al., 2011). In order to investigate whether these transporters regulate the As[i] in normal cells, we characterized the expression profiles of these transporter genes, and the contribution of these genes to As[i] as well as AsIII-triggered cytotoxicity in both C- and A-cells using inhibitors for these transporters. Our results clearly demonstrated that the levels of endogenous AQP9 gene expression were much higher in the C-cells than those in the A-cells. These findings coincide well with a previous study indicating that the expression levels of AQP9 were much stronger in C-cells than that in A-cells, in which AQP9 was proposed to be responsible for the regulation of intramembranous amniotic fluid (Wang et al., 2004). Furthermore, both As[i] and AsIII-triggered cytotoxicity in the C-cells were significantly attenuated by sorbitol, a competitive AQP9 inhibitor (Shinkai et al., 2009), in a dose-dependent manner, but a similar phenomenon was not observed in the A-cells. Intriguingly, both As[i] and AsIII-induced cytotoxicity were hardly altered by the addition of phloretin, another AQP9 inhibitor, in both cells, results of which are similar to those reported by other group (McDermott et al., 2010). Furthermore, our experimental data also demonstrated that the inhibitory efficiency towards AQP9 of sorbitol (100 mM) is much higher than that of phloretin (200 μM) in an APL cell line, NB4 cells (data not shown), in which AQP9 is highly expressed (Leung et al., 2007; Wang et al., 2008). These results might be attributed to different cell types used and could explain why the levels of As[i] and AsIII-induced cytotoxicity were altered by the addition of sorbitol, but not phloretin in our experimental system. Collectively, our results suggest that AQP9 is more functionally implicated in AsIII uptake and AsIII-induced cytotoxicity in the C-cells rather than in the A-cells. Although the exact molecular mechanisms underlying AsIII uptake in the A-cells are not clear at present, it is possible that the uptake is through an alternative pathway involving AQP3, another member of the aquaporin transporter superfamily, and glucose transporter 1. This hypothesis is based on the studies demonstrating that these two transporters have been identified in A-cells (Mann et al., 2002; Gude et al., 2005), and involved in arsenic uptake (Liu et al., 2004, 2006; Lee et al., 2006; Yuan et al., 2011). Understandably, further investigation of a contribution of the alterative pathway is need.
We also demonstrated that the expression level of ABCC2 mRNA was upregulated in the A-cells, whereas it was downregulated in the C-cells after 48-h post-exposure. Furthermore, Western blot analysis demonstrated that the expression levels of endogenous ABCC2 protein were significant higher in the A-cells than those in the C-cells, and that a time-dependent decrease in ABCC2 protein expression levels was observed in the C-cells, but not in the A-cells. These results thus suggested that a differential expression pattern of the gene may contribute to the differences in the AsIII sensitivity between these two cell types. It should be noted that while the upregulation of ABCC2 mRNA expression level was observed in the A-cells, no alternation in its protein expression level was observed during the exposure period. It is possible that a post-transcriptional regulation plays a key role in the discordance between mRNA and protein expression. Details of the study are currently underway. We further demonstrated that AsIII-triggered cytotoxicity as a result of As[i] in the C-cells was significantly augmented by CsA and MK571 in a dose-dependent manner, suggesting that MRP2 appear to be functionally implicated in As[i] and cytotoxicity in the cells. It is noteworthy that in the A-cells, an increase in the As[i] was much higher in the presence of CsA compared to MK571. Consequently, CsA was much more efficient in potentiating AsIII-mediated cytotoxicity compared with MK571, implicating a close correlation between sensitivity to AsIII and As[i]. Moreover, several previous reports have demonstrated that CsA not only increases the production of reactive oxygen species in rat glial cells and porcine renal endothelial cells (Mun et al., 2008; de Arriba et al., 2009), but also alters the expression levels of antioxidant enzymes by decreasing the activity of catalase and glutathione peroxidase in rabbit kidney tissue (Durak et al., 1998). On the basis of these results and current findings, we suggest that not only modulating effects on multidrug resistant transporters but also reactive oxygen species induction effects of CsA might sensitize the A-cells to the cytotoxic effects of AsIII.
The current study also demonstrated that no significant differences in the expression levels of MRP1 were observed between C- and A-cells, indicating that MRP2, instead of MRP1, plays a critical role in AsIII-efflux system in both cells. Unexpectedly, the expression of ABCB1 protein was hardly detected in both cells, yet the expression of mRNA was detected. While more detailed analysis of the molecular event is obviously needed, our results suggested that P-gp was not functionally involved in arsenic efflux in these normal cells. This is clearly supported by our experimental data showing that neither As[i] nor AsIII-mediated cytotoxicity was augmented by the addition of PGP-4008, a specific inhibitor for P-gp.
In conclusion, we propose for the first time that AQP9 and MRP2 are functionally involved in controlling arsenic accumulation in these normal cells, which in turn contribute to differential sensitivity to AsIII cytotoxicity between these cells. Although studies regarding the action of arsenic have been conducted using normal human fetal brain explants and human cord blood cells (Chattopadhyay et al., 2002; Ferrario et al., 2009), the correlation between biological functions of arsenic and these transporters has not yet been clarified in human-derived normal cells. To the best of our knowledge, this is the first correlation of AsIII-associated transporters with cytotoxic effects of AsIII in primary normal cells. Given that these arsenic transporters play a pivotal role in AsIII-mediated cytotoxic effects on normal cells and/or tissues, monitoring expression levels of AsIII-associated transporters has important implications for predicting not only clinical efficacy but also appearance of side effects in patients with cancer.
Table 1-2.
Conditions for PCR
| Target mRNA | Denaturation reaction | Annealing reaction | Extension reaction | |||
|---|---|---|---|---|---|---|
|
| ||||||
| Temp. (°C) | Time (sec) | Temp. (°C) | Time (sec) | Temp. (°C) | Time (sec) | |
| HO-1 | 94 | 60 | 65 | 60 | 72 | 60 |
| AQP9 | 94 | 30 | 60 | 30 | 72 | 40 |
| ABCC1 (MRP1) | 94 | 60 | 66 | 60 | 72 | 90 |
| ABCC2 (MRP2) | 94 | 60 | 68 | 60 | 72 | 90 |
| ABCB1 (P-gp) | 94 | 60 | 55 | 30 | 72 | 30 |
| β-actin | 94 | 60 | 50 | 60 | 72 | 60 |
Abbreviations: HO-1, hemeoxygenase-1; AQP9, aquaporin 9; MRP, multidrug resistance-associated protein; P-gp, P-glycoprotein.
Research highlights.
Examination of effect of AsIII on primary cultured chorion (C) and amnion (A) cells
Dose-dependent AsIII-mediated cytotoxicity in C-cells, not in A-cells
Intracellular arsenic accumulation and cytotoxicity regulated by AQP9 and ABCC2
Prediction of AsIII side effects by monitoring these transporters
Acknowledgments
The authors thank Dr. Kunio OHYAMA for encouraging suggestions and arranging sample supply for this study. The authors also thank Dr. Mariko UMEMURA and Mr. Takashi YAMAZAKI for their technical assistance. This work was supported in part by grants from the Ministry of Education, Culture, Sports, Science and Technology and by the Promotion and Mutual Aid Corporation for Private Schools of Japan.
Footnotes
Conflict of interest statement
The authors declare that there is no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could a3ect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andus T, Targan SR, Deem R, Toyoda H. Measurement of tumor necrosis factor alpha mRNA in small numbers of cells by quantitative polymerase chain reaction. Reg Immunol. 1993;5:11–17. [PubMed] [Google Scholar]
- Chattopadhyay S, Bhaumik S, Purkayastha M, Basu S, Nag Chaudhuri A, Das Gupta S. Apoptosis and necrosis in developing brain cells due to arsenic toxicity and protection with antioxidants. Toxicol Lett. 2002;136:65–76. doi: 10.1016/s0378-4274(02)00282-5. [DOI] [PubMed] [Google Scholar]
- Chen GQ, Shi XG, Tang W, Xiong SM, Zhu J, Cai X, Han ZG, Ni JH, Shi GY, Jia PM, Liu MM, He KL, Niu C, Ma J, Zhang P, Zhang TD, Paul P, Naoe T, Kitamura K, Miller W, Waxman S, Wang ZY, de The H, Chen SJ, Chen Z. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): I. As2O3 exerts dose-dependent dual effects on APL cells. Blood. 1997;89:3345–3353. [PubMed] [Google Scholar]
- de Arriba G, de Hornedo JP, Rubio SR, Fernández MC, Martínez SB, Camarero MM, Cid TP. Vitamin E protects against the mitochondrial damage caused by cyclosporin A in LLC-PK1 cells. Toxicol Appl Pharmacol. 2009;239:241–250. doi: 10.1016/j.taap.2009.05.028. [DOI] [PubMed] [Google Scholar]
- Dilda PJ, Hogg PJ. Arsenical-based cancer drugs. Cancer Treat Rev. 2007;33:542–564. doi: 10.1016/j.ctrv.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Durak I, Karabacak HI, Büyükkoçak S, Cimen MY, Kaçmaz M, Omeroglu E, Oztürk HS. Impaired antioxidant defense system in the kidney tissues from rabbits treated with cyclosporine. Protective effects of vitamins E and C. Nephron. 1998;78:207–211. doi: 10.1159/000044912. [DOI] [PubMed] [Google Scholar]
- Ferrario D, Collotta A, Carfi M, Bowe G, Vahter M, Hartung T, Gribaldo L. Arsenic induces telomerase expression and maintains telomere length in human cord blood cells. Toxicology. 2009;260:132–141. doi: 10.1016/j.tox.2009.03.019. [DOI] [PubMed] [Google Scholar]
- Fujisawa S, Ohno R, Shigeno K, Sahara N, Nakamura S, Naito K, Kobayashi M, Shinjo K, Takeshita A, Suzuki Y, Hashimoto H, Kinoshita K, Shimoya M, Kaise T, Ohnishi K. Pharmacokinetics of arsenic species in Japanese patients with relapsed or refractory acute promyelocytic leukemia treated with arsenic trioxide. Cancer Chemother Pharmacol. 2007;59:485–493. doi: 10.1007/s00280-006-0288-4. [DOI] [PubMed] [Google Scholar]
- Gude NM, Stevenson JL, Murthi P, Rogers S, Best JD, Kalionis B, King RG. Expression of GLUT12 in the fetal membranes of the human placenta. Placenta. 2005;26:67–72. doi: 10.1016/j.placenta.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Kiguchi T, Yoshino Y, Yuan B, Yoshizawa S, Kitahara T, Akahane D, Gotoh M, Kaise T, Toyoda H, Ohyashiki K. Speciation of arsenic trioxide penetrates into cerebrospinal fluid in patients with acute promyelocytic leukemia. Leuk Res. 2010;34:403–405. doi: 10.1016/j.leukres.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Lee BD, French KJ, Zhuang Y, Smith CD. Development of a syngeneic in vivo tumor model and its use in evaluating a novel P-glycoprotein modulator, PGP–4008. Oncol Res. 2003;14:49–60. doi: 10.3727/000000003108748603. [DOI] [PubMed] [Google Scholar]
- Lee TC, Ho IC. Expression of heme oxygenase in arsenic-resistant human lung adenocarcinoma cells. Cancer Res. 1994;54:1660–1664. [PubMed] [Google Scholar]
- Lee TC, Ho IC, Lu WJ, Huang JD. Enhanced expression of multidrug resistance-associated protein 2 and reduced expression of aquaglyceroporin 3 in an arsenic-resistant human cell line. J Biol Chem. 2006;281:18401–18407. doi: 10.1074/jbc.M601266200. [DOI] [PubMed] [Google Scholar]
- Leslie EM, Haimeur A, Waalkes MP. Arsenic transport by the human multidrug resistance protein 1 (MRP1/ABCC1). Evidence that a tri-glutathione conjugate is required. J Biol Chem. 2004;279:32700–32708. doi: 10.1074/jbc.M404912200. [DOI] [PubMed] [Google Scholar]
- Leung J, Pang A, Yuen WH, Kwong YL, Tse EW. Relationship of expression of aquaglyceroporin 9 with arsenic uptake and sensitivity in leukemia cells. Blood. 2007;109:740–746. doi: 10.1182/blood-2006-04-019588. [DOI] [PubMed] [Google Scholar]
- Litzow MR. Arsenic trioxide. Expert Opin Pharmacother. 2008;9:1773–1785. doi: 10.1517/14656566.9.10.1773. [DOI] [PubMed] [Google Scholar]
- Liu J, Liu Y, Powell DA, Waalkes MP, Klaassen CD. Multidrug-resistance mdr1a/1b double knockout mice are more sensitive than wild type mice to acute arsenic toxicity, with higher arsenic accumulation in tissues. Toxicology. 2002;170:55–62. doi: 10.1016/s0300-483x(01)00532-7. [DOI] [PubMed] [Google Scholar]
- Liu Z, Carbrey JM, Agre P, Rosen BP. Arsenic trioxide uptake by human and rat aquaglyceroporins. Biochem Biophys Res Commun. 2004;316:1178–1185. doi: 10.1016/j.bbrc.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Liu Z, Sanchez MA, Jiang X, Boles E, Landfear SM, Rosen BP. Mammalian glucose permease GLUT1 facilitates transport of arsenic trioxide and methylarsonous acid. Biochem Biophys Res Commun. 2006;351:424–430. doi: 10.1016/j.bbrc.2006.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann SE, Ricke EA, Yang BA, Verkman AS, Taylor RN. Expression and localization of aquaporin 1 and 3 in human fetal membranes. Am J Obstet Gynecol. 2002;187:902–907. doi: 10.1067/mob.2002.127168. [DOI] [PubMed] [Google Scholar]
- Matsson P, Pedersen JM, Norinder U, Bergström CA, Artursson P. Identification of novel specific and general inhibitors of the three major human ATP-binding cassette transporters P-gp, BCRP and MRP2 among registered drugs. Pharm Res. 2009;26:1816–1831. doi: 10.1007/s11095-009-9896-0. [DOI] [PubMed] [Google Scholar]
- McDermott JR, Jiang X, Beene LC, Rosen BP, Liu Z. Pentavalent methylated arsenicals are substrates of human AQP9. Biometals. 2010;23:119–127. doi: 10.1007/s10534-009-9273-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mun KC, Lee KT, Choi HJ, Jin KB, Han SY, Park SB, Kim HC, Ha EY, Kim YH. Effects of cyclosporine on the production of the reactive oxygen species in the glial cells. Transplant Proc. 2008;40:2742–2743. doi: 10.1016/j.transproceed.2008.08.007. [DOI] [PubMed] [Google Scholar]
- Ninomiya M, Kajiguchi T, Yamamoto K, Kinoshita T, Emi N, Naoe T. Increased oxidative DNA products in patients with acute promyelocytic leukemia during arsenic therapy. Haematologica. 2006;91:1571–1572. [PubMed] [Google Scholar]
- Ohyama K, Oka K, Emura A, Tamura H, Suga T, Bessho T, Hirakawa S, Yamakawa T. Suppression of apoptotic cell death progressed in vitro with incubation of the chorion laeve tissues of human fetal membrane by glucocorticoid. Biol Pharm Bull. 1998;21:1024–1029. doi: 10.1248/bpb.21.1024. [DOI] [PubMed] [Google Scholar]
- Qadir M, O’Loughlin KL, Fricke SM, Williamson NA, Greco WR, Minderman H, Baer MR. Cyclosporin A is a broad-spectrum multidrug resistance modulator. Clin Cancer Res. 2005;11:2320–2326. doi: 10.1158/1078-0432.CCR-04-1725. [DOI] [PubMed] [Google Scholar]
- Scudiero DA, Shoemaker RH, Paull KD, Monks A, Tierney S, Nofziger TH, Currens MJ, Seniff D, Boyd MR. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res. 1988;48:4827–4833. [PubMed] [Google Scholar]
- Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, Zhu J, Tang W, Sun GL, Yang KQ, Chen Y, Zhou L, Fang ZW, Wang YT, Ma J, Zhang P, Zhang TD, Chen SJ, Chen Z, Wang ZY. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood. 1997;89:3354–3360. [PubMed] [Google Scholar]
- Shinkai Y, Sumi D, Toyama T, Kaji T, Kumagai Y. Role of aquaporin 9 in cellular accumulation of arsenic and its cytotoxicity in primary mouse hepatocytes. Toxicol Appl Pharmacol. 2009;237:232–236. doi: 10.1016/j.taap.2009.03.014. [DOI] [PubMed] [Google Scholar]
- Soignet SL, Maslak P, Wang ZG, Jhanwar S, Calleja E, Dardashti LJ, Corso D, DeBlasio A, Gabrilove J, Scheinberg DA, Pandolfi PP, Warrell RP. Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N Engl J Med. 1998;339:1341–1348. doi: 10.1056/NEJM199811053391901. [DOI] [PubMed] [Google Scholar]
- Tsukaguchi H, Shayakul C, Berger UV, Mackenzie B, Devidas S, Guggino WB, van Hoek AN, Hediger MA. Molecular characterization of a broad selectivity neutral solute channel. J Biol Chem. 1998;273:24737–24743. doi: 10.1074/jbc.273.38.24737. [DOI] [PubMed] [Google Scholar]
- Wang L, Zhou GB, Liu P, Song JH, Liang Y, Yan XJ, Xu F, Wang BS, Mao JH, Shen ZX, Chen SJ, Chen Z. Dissection of mechanisms of Chinese medicinal formula Realgar-Indigo naturalis as an effective treatment for promyelocytic leukemia. Proc Natl Acad Sci USA. 2008;105:4826–4831. doi: 10.1073/pnas.0712365105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Chen J, Beall M, Zhou W, Ross MG. Expression of aquaporin 9 in human chorioamniotic membranes and placenta. Am J Obstet Gynecol. 2004;191:2160–2167. doi: 10.1016/j.ajog.2004.05.089. [DOI] [PubMed] [Google Scholar]
- Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood. 2008;111:2505–2515. doi: 10.1182/blood-2007-07-102798. [DOI] [PubMed] [Google Scholar]
- Yoshino Y, Yuan B, Miyashita S, Iriyama N, Horikoshi A, Shikino O, Toyoda H, Kaise T. Speciation of arsenic trioxide metabolites in blood cells and plasma of a patient with acute promyelocytic leukemia. Anal Bioanal Chem. 2009;393:689–697. doi: 10.1007/s00216-008-2487-9. [DOI] [PubMed] [Google Scholar]
- Yuan B, Ohyama K, Bessho T, Toyoda H. Contribution of inducible nitric oxide synthase and cyclooxygenase-2 to apoptosis induction in smooth chorion trophoblast cells of human fetal membrane tissues. Biochem Biophys Res Commun. 2006;341:822–827. doi: 10.1016/j.bbrc.2006.01.042. [DOI] [PubMed] [Google Scholar]
- Yuan B, Ohyama K, Bessho T, Uchide N, Toyoda H. Imbalance between ROS production and elimination results in apoptosis induction in primary smooth chorion trophoblast cells prepared from human fetal membrane tissues. Life Sci. 2008;82:623–630. doi: 10.1016/j.lfs.2007.12.016. [DOI] [PubMed] [Google Scholar]
- Yuan B, Ohyama K, Takeichi M, Toyoda H. Direct contribution of inducible nitric oxide synthase expression to apoptosis induction in primary smooth chorion trophoblast cells of human fetal membrane tissues. Int J Biochem Cell Biol. 2009;41:1062–1069. doi: 10.1016/j.biocel.2008.09.031. [DOI] [PubMed] [Google Scholar]
- Yuan B, Yoshino Y, Kaise T, Toyoda H. Application of arsenic trioxide therapy for patients with leukemia. In: Sun HZ, editor. Biological Chemistry of As, Sb and Bi. John Wiley & Sons; New York: 2011. pp. 263–292. [Google Scholar]