

Abstract
Autophagy is a catabolic process of paramount importance for cellular homeostasis during starvation. Generally, autophagy and translation are inversely regulated. Many kinds of stress lead to attenuation of translation via phosphorylation of eukaryotic translation initiation factor alpha (eIF2α). This response is conserved from yeast to man and can be either protective or detrimental depending on strength and duration of stress, and additional factors. During starvation or viral infection, phosphorylation of eIF2α is required for induction of autophagy. As exemplified here by α-hemolysin, a small pore-forming toxin (PFT) of Staphylococcus aureus and (S)-3-oxo-C12-homoserine lactone [(S)-3-oxo-C12-HSL], a quorum-sensing hormone of Pseudomonas aeruginosa, bacterial exoproducts may also impact translation and autophagy. Thereby, PFT and (S)-3-oxo-C12-HSL act differentially. Damage of the plasma membrane by PFT causes efflux of potassium, which leads to amino acid starvation and energy loss. This triggers amino acid-sensitive eIF2α-kinase GCN2, as well as energy sensor AMPK, and deactivates mTORC1. The output of this response, that is, transient metabolic reprogramming is an essential part of a defense program which enables cells to survive attack by a pore-forming agent. Thus, nutrient/energy sensors serve as sentinels of plasma membrane integrity. In contrast to PFT, (S)-3-oxo-C12-HSL does not cause acute loss of ATP or activation of GCN2, but also triggers phosphorylation of eIF2α and inhibits translation. This response appears not to depend on efflux of potassium and requires eIF2α-kinase PKR. Like α-toxin, (S)-3-oxo-C12-HSL increases lipidation of LC3 and accumulation of autophagosomes in cells. Apart from directly affecting host-cell viability, bacterial exoproducts might galvanize bystander cells to prepare for close combat with microbial offenders or inadvertently accommodate some of them.
Keywords: Membrane damage, Translation, Autophagy, Quorum-sensing hormone, Pore-forming toxins
Basic mechanisms of autophagosome formation
The term autophagy—hereafter used in place of “macroautophagy”—was first introduced during the CIBA foundation symposium on lysosomes, London, in 1963 [1]. It denotes an important catabolic process in eukaryotes, the hallmark of which is the formation of an intracellular double-bilayer membrane compartment, the autophagosome. Autophagy may occur in an apparently random manner anywhere in the cytosol, or alternatively at particulate cargo destined to be degraded or otherwise removed from a cell. The principal function of autophagy is break down and recycling of macromolecules, but there is evidence for involvement in atypical secretion as well [2–5]. Autophagic flux is subject to complex regulation by extracellular and intracellular cues to meet cellular energy requirements, provide molecular building blocks, and achieve “garbage” removal.
Although the source of the membrane required for the formation of an autophagosome remains a matter of debate, there is evidence that it may be derived from various organelles, including ER, mitochondria, nucleus, Golgi, and PM [6]. The formation of closed, vesicular autophagosomes is preceded by the occurrence of crescent-shaped double-membrane phagophores. Autophagosome biogenesis is commonly subdivided into three steps: initiation or nucleation, elongation, and maturation. Genetic studies in yeast have led to the identification of more than 30 genes involved in this process, and mammalian homologs are known for many of them [7, 8].
The class III phosphoinositide 3 kinase (PI3K) Vps34, which acts in complex with additional proteins including Beclin1 and Atg14L/Barkor catalyzes the formation of PI(3)P, which is an essential early step during phagophore formation [9]. PI3K is regulated by the ULK1-complex consisting of the serine/threonine kinases ULK1, ULK2, FIP200, mAtg10 and Atg101 [7, 10].
Elongation of phagophores and the formation of autophagosomes depend on two ubiquitination-like reactions. First, Atg7 and Atg10 catalyze conjugation of the Ub-like protein Atg12 to Atg5 [11]. The conjugation product interacts with Atg16L1, and the ternary complex finally associates with the membrane of phagophores [12]. Second, ubiquitin-like molecules of the Atg8 family (LC3, GABARAP, and GATE-16) are modified by covalent addition of phosphoethanolamine (PE). In the case of LC3 addition of PE is catalyzed by Atg7, Atg3 and Atg12/Atg5-conjugate, which function as E1-like, E2-like and E3-like enzymes, respectively [13, 14]. Lipidated LC3 (LC3II) associates with the autophagosomal membrane where it promotes tethering and hemifusion during the formation of autophagosomes [7, 15]. Therefore, LC3II is commonly used as a marker of autophagy, but its deposition is not strictly confined to autophagosomal membranes.
Selective autophagy of bacteria
Although starvation-induced autophagy appears to occur randomly in cells, autophagy of foreign bodies like invading bacteria (xenophagy) is obviously a selective process, which has become a subject of intense research. An early account of xenophagy, was the observation that rickettsiae accumulates in autophagosomal-like structures of guinea pig polymorphonuclear leukocytes [16]. The fate of microorganisms captured by autophagosomes is diverse: they may manipulate maturation of the autophagosome, escape into the cytosol, benefit from autophagy, or may be destroyed [17–22] .
Selective autophagy depends on adapter proteins, also termed SLRs (sequestosome-like receptors), because the best known adapter protein is p62/sequestosome1; other known members are NBR1, NDP52, and optineurin. SLRs comprise ubiquitin association regions (UBAs) and LC3 interacting regions (LIRs); they target bacteria to the autophagic pathway [23, 24]. Notably, p62 and NDP52 seem to serve non-redundant functions during selective autophagy [25, 26]. Modification of adapter proteins, for example, by phosphorylation adds specificity and another level of regulation to the process of selective autophagy [27, 28]. It remains unclear whether host proteins in the phagosomal membrane or bacterial proteins have to be ubiquitinated for targeting of SLRs. At any rate, ubiquitination seems not to be absolutely necessary for selective targeting of misfolded proteins to the autophagic degradation pathway. The BAG3-mediated chaperone-based targeting uses the specificity of Hsp70 chaperones to misfolded proteins as the basis for selectivity [29].
Evidence for a role of membrane damage for selective autophagy
Involvement of membrane damage and/or pore-forming toxins in selective autophagy has emerged as a common theme from several studies of different bacterial species, namely Streptococcus pyogenes, Listeria monocytogenes, Shigella flexneri, Salmonella enterica, and S. aureus [21, 30–32]. Membrane damage/PFT might promote selective autophagy in several ways: first, by allowing bacterial escape from vacuolar compartments; second, via (modification and) recognition of pore-forming agents per se by the autophagy machinery; or third, via biochemical or biophysical changes secondary to pore formation which are recognized by the autophagy machinery, three possibilities that are mutually non-exclusive. Apparently, membrane remnants or damaged vacuoles are targeted by autophagy [33, 34], and growth of Salmonella in infected cells is reportedly restricted by autophagic targeting of damaged SCV (Salmonella containing vacuoles) [35, 36].
In the case of intracellular Listeria and intracellular S. aureus, it has been demonstrated that selective autophagy correlates with the ability of these bacteria to produce a pore-forming toxin (LLO and α-hemolysin, respectively), [31, 37, 38]. GFP-LC3 and p62 were also recruited to endocytosed toxin after application of purified protein or as a liposomal complex [37, 38]. This suggests that escape from vacuoles and subsequent exposure of bacteria to the cytosol is probably not required for selective autophagy to occur at intracellular Listeria or S. aureus; toxin appears to suffice as a trigger. Because LLO has been shown to be ubiquitinated, it is possible that a fraction of modified toxin binds adapter proteins and is internalized by autophagosomes instead of being delivered to proteasomes. In line with this, LLO has been shown to colocalize with p62 and ubiquitin in aggresome-like structures [39]. Similarly, S. aureus α-toxin colocalizes with p62 in target cells. Whether ubiquitination of toxin is required for the recruitment of p62 is currently unclear, but the available data suggest that pore-forming toxins can be ubiquitinated, which might suffice to directly recruit adapter proteins [40]. Alternatively, host proteins on damaged membranes could be ubiquitinated and recruit adapter proteins. The identification of ubiquitination targets, of the enzymes involved, and cues that actually initiate the process remains an important task. Antibodies specific for ubiquitin have been shown to label membrane remnants (defined by co-staining with antibodies directed against Galectin-3) in Shigella-infected cells. The authors proposed that membrane remnants are targeted by the autophagic machinery in a p62-dependent manner [33]. More recently, it was reported that the adapter protein NDP52 directly interacts with Galectin-8 which is recruited to the SCV (Salmonella-containing vesicles) of cells infected with S. enterica [34]. Thus, sugar moieties which become exposed after damage of vacuoles play a role for early recognition by adapter proteins. Because NDP52 and p62, despite structural similarity, seem to be functionally non-redundant and localize to distinct microdomains on vacuolar membranes/bacteria targeted for autophagy, it remains unclear how p62 is targeted to bacteria and/or membrane remnants.
Another pathway implicated in selective autophagy is based on the generation of the lipid second messenger diacylglycerol; it plays a role for xenophagy of Salmonella and does not depend on Atg5 [41].
It should be noted that although deployment of p62 is a hallmark of selective autophagy, p62 is also involved in starvation-induced autophagy [42, 43], providing a molecular link between random and selective autophagy.
Perforation of the PM triggers a starvation response and autophagy
The earliest account of autophagy induction by a pore-forming toxin (PFT) was a report about the protective role of autophagy for target cells of Vibrio cholerae cytolysin, a small β-barrel PFT [44]. Subsequent work on the structurally related S. aureus α-toxin provided clues to the signaling pathways involved in autophagy induction by PFT and to the protective mechanism of autophagy: Previous studies with S. aureus α-toxin and SLO had established that membrane damage by these PFT, and consequences thereof are reversible in many cell types [45–49]. The transient nature of PM perforation is reflected by massive, but transient drop of cellular ATP levels, a fact that our group has exploited to characterize repair/recovery mechanisms. One important finding of these studies was that the removal of the oligomeric plasma membrane pore complexes by dynamin-dependent endocytosis is prerequisite for membrane repair after S. aureus α-toxin attack [50]. Second, membrane perforation turned out to trigger starvation and metabolic reprogramming, which proved to be also essential for recovery [38]. These findings were born out of an unbiased transcript profile of perforated cells obtained by serial analysis of gene expression (SAGE). This analysis revealed that membrane perforation by S. aureus α-toxin triggers expression of immediate early genes [48], whereby translation of these transcripts was delayed; moreover, eIF2α-phosphatase GADD34 was among the most abundant transcripts overexpressed under these conditions [48]. Together, this suggested that α-toxin causes an integrated stress response, via transient phosphorylation of eukaryotic translation initiation factor 2α, (eIF2α), and transient, global translational arrest. This assumption was confirmed, and GCN2 and PKR were subsequently identified as 2 kinases responsible for phosphorylation of eIF2α in response to α-toxin [38]. Importantly, pore-dead single amino acid mutants failed to activate this response, firmly establishing the link between membrane pore formation and integrated stress response. GCN2 is a sensor of amino acid starvation, which is conserved between yeast and man; uncharged tRNAs trigger autophosphorylation of GCN2. Activated GCN2 phosphorylates eukaryotic translation initiation factor 2α, leading to global translational attenuation [51, 52]. Intriguingly, GCN2 and PKR had previously been found to be required for amino acid starvation-induced autophagy [53]. Therefore, we reasoned that membrane perforation by PFT might also cause amino acid starvation, and that translational stop and autophagy might be required to overcome nutrient shortage during transient perforation.
Because exposure of susceptible cells to pore-forming toxins causes massive drop of intracellular ATP [45–49], we assumed that PFT activate AMPK. This kinase serves as the major cellular energy sensor, which regulates the activity of mTORC1, a master switch of translation and autophagy which integrates multiple signals from nutrient sensors. In fact, S. aureus α-toxin, V. cholerae cytolysin, Streptolysin O, and E. coli hemolysin induced phosphorylation of AMPK in epithelial cells [38]. Moreover, S6K, a substrate of mTORC1, became dephosphorylated. Transmission electron microscopy analyses revealed many multivesicular bodies, large, empty vacuoles, and vesicles delineated by double membranes, providing morphological evidence that PFT induced autophagy. Recently, it was found that mTORC1 activity is also regulated through availability of amino acids. Interestingly, amino acids induce relocalization of mTOR to lysosomal membranes in a p62-dependent way [42], suggesting that p62 may link selective and starvation-induced autophagy. That AMPK and GCN2 are activated by PFT suggested that damage of the plasma membrane causes cellular starvation and energy shortage, the classic inducers of autophagy. In fact, uptake of radio-labeled leucine was reduced by α-toxin [38], presumably because amino acid transporters were inhibited by the dissipation of natural ion gradients. Two recent studies appear to support our conclusion that membrane damage causes starvation and triggers nutrient sensors like GCN2/p-eIF2α and mTORC1: one paper reports that these pathways are also triggered by intracellular Salmonella and Shigella, and the authors propose that this is also due to membrane damage [54]; the second study indicates that the concept of pore-forming toxin-dependent activation of nutrient sensors holds in a Drosophila infection model [55].
Loss of potassium links plasma membrane damage to activation of metabolic sensors
Rapid release of potassium ions is a common consequence of membrane perforation by PFT, for example [56, 57]. Loss of potassium—or low concentrations of this ion in perforated cells—seems to trigger a multitude of responses in target cells of PFT, because high concentrations of potassium in the extracellular milieu prevent these responses, and because they can be also triggered by the potassium ionophore nigericine. Examples are the activation of caspases [56, 58], activation of kinases including, for example, p38 [59], AMPK, GCN2 [38], and CREB [60]. Importantly, high potassium concentrations in media did not impair activation of p38 by hydrogen peroxide, indicating that these experimental conditions do not generally paralyze pathways leading to the phosphorylation of stress kinases [38]. That GCN2 is activated by membrane perforation in a potassium-efflux-dependent manner raises the possibility that cells exploit the dependence of nutrient transport across the PM on physiological ion gradients to indirectly sense changes of ion concentrations. The link between physiologic ion gradients and regulation of translation initiation has been noted previously in a paper about the marine toxin Palytoxin (PAL) which converts the Na+/K+ exchanger to a channel. As a consequence, intracellular potassium concentrations drop and translation stops [61]. In line with this, PAL causes phosphorylation of eIF2α (Fig. 1a).
Fig. 1.
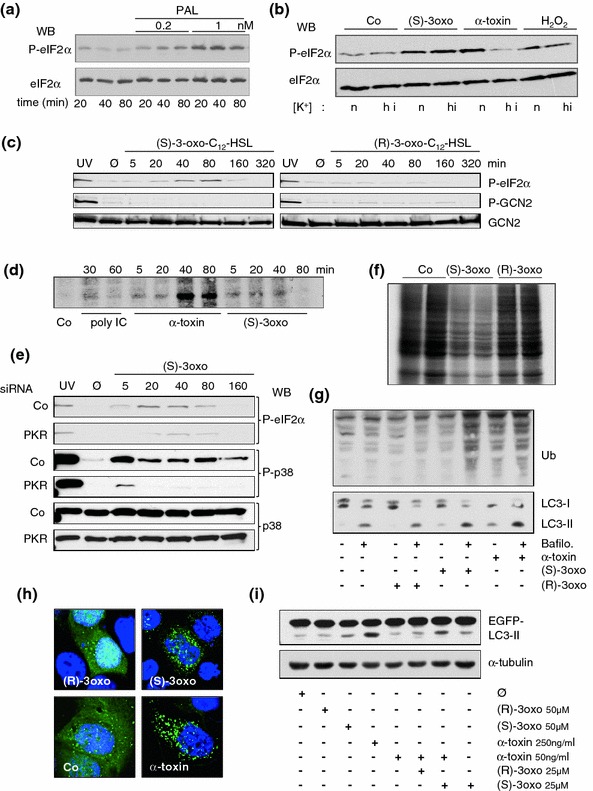
A quorum-sensing hormone of P. aeruginosa impacts translation and autophagy. Materials and methods employed here have been published previously [59, 88]. Metabolic labeling was performed as described by [89]. In the figure, (S)-3oxo and (R)-3oxo denote (S)-3-oxo-C12-HSL and (R)-3oxo-C12-HSL, respectively. a Western blot for (p)-eIF2α with whole cell lysates (HaCaT) treated with palytoxin (PAL) as indicated in the figure. b Western blot for (p)-eIF2α with whole cell lysates (HaCaT) treated with the indicated compounds in the presence of normal concentrations of potassium (n), or in media with high concentration of potassium (hi) [59]. c Western blot for p-eIF2α, p-GCN2, and GCN2 after treatment of HaCaT cells with the P. aeruginosa quorum-sensing hormone (S)-3-oxo-C12-HSL or the control compound (R)-3-oxo-C12-HSL for the indicated times. As expected, (R)-3-oxo-C12-HSL fails to cause phosphorylation of eIF2α. Note that GCN2 is not phosphorylated in response to either lactone; UV served as a positive control. Untreated cell samples (media alone) are denoted Ø. d Autoradiographic detection of P32-PKR in samples of HaCat cells treated for the indicated times with the compounds denoted underneath the panel. Co: medium alone. e Western blots for p-eIF2α, p-p38, or p38 with straight Cos7-cell lysates obtained 48 h following transfection with siRNAs and subsequent treatment with S-3-oxo-C12-HSL. f Autoradiography of an SDS-Gel visualizing incorporation of S35-Methionine into newly synthesized proteins. Note marked inhibition of protein synthesis in cells treated with S-3-oxo-C12-HSL. g Western blots for ubiquitination and LC3I/II with lysates of HaCat cells treated as indicated in the figure for 3 h. h Fluorescence microscopy images of HaCat cells transfected with EGFP-LC3 and treated for 3 h with compounds indicated in the figure. Note redistribution of diffuse green fluorescence signal into dots in cells treated with α-toxin, or S-3-oxo-C12-HSL. i Western blot for GFP with lysates of HaCat cells transfected with EGFP-LC3 treated as indicated in the figure. The untreated cell sample (media alone) is denoted Ø. Loading control with α-tubulin. Combination of (S)-3-oxo-C12-HSL and α-toxin led to a significant accumulation of LC3II
The role of MAPK for cellular responses to pore-forming toxins: potential links to autophagy
Several pathways have been implicated in the survival of cells or whole organism (Caenorhabditis elegans and insects) exposed to PFT or microorganisms releasing PFT [44, 57, 62–69]; for review, see [70–73]. Among them, the p38 MAPK pathway was the first found to protect C. elegans or mammalian cells from small pore-forming toxin attack [66]. Subsequently, we demonstrated in mammalian cells that p38 does not confer resistance to the pore-forming activity of S. aureus α-toxin, but is required for recovery from perforation. It was also demonstrated that this function was not required for recovery from membrane perforation by a large pore-forming toxin (SLO), revealing that membrane repair or metabolic recovery mechanisms are diverse [67]. At low concentrations, α-toxin does not induce a significant influx of Ca++ ions, and therefore would not be able to trigger the so-called wounded membrane response [74, 75]. Although the critical downstream targets of p38 for recovery from α-toxin are not known to date, the finding that α-toxin has to be internalized by target cells and that autophagy is required for energy homeostasis offers at least two potential explanations: Because Rab5 is a regulator of endocytosis [76], and because p38 impacts on the engagement of Rab5 with membranes [77], it is possible that p38 is required for the internalization of small PFT. MAPK p38 has also been implicated in regulation of autophagy [78, 79]. Therefore, it is possible that one way this kinase may impact on the outcome of an attack by PFT is through regulation of autophagy. JNK, another important stress-activated protein kinase (SAPK) is also activated by PFT and may therefore impact cellular responses to membrane perforation [68]. An important role of JNK for starvation-induced autophagy has been demonstrated [80].
A bacterial quorum-sensing hormone of P. aeruginosa attenuates translation and impacts autophagy
Apart from PFT, few other bacterial exoproducts including LPS [81] and NLR ligands [82–84] have been shown to induce autophagy. (S)-3-oxo-C12-HSL is a small molecule produced by the Gram-negative bacterium Pseudomonas aeruginosa. (S)-3-oxo-C12-HSL is not only involved in regulating cell-to-cell signaling in these bacteria, but impacts signaling in mammalian cells [85]. Thus, (S)-3-oxo-C12-HSL has been shown to inhibit NF-κB [86] and oxidative stress [87], and to trigger phosphorylation of p38 MAPK and eIF2α [88]. Because the latter two events are also triggered by PFT, and because eIF2α appears to be important for the induction of autophagy in response to S. aureus α-toxin, we have recently started to investigate whether (S)-3-oxo-C12-HSL might also impact on autophagy. First, we confirmed that (S)-3-oxo-C12-HSL triggered eIF2α-phosphorylation in HaCat cells. Notably, high concentration of potassium in media did not affect phosphorylation in response to (S)-3-oxo-C12-HSL, although it blocked phosphorylation of eIF2α in perforated cells (Fig. 1b). (S)-3-oxo-C12-HSL did not cause phosphorylation of eIF2α-kinase GCN2 (Fig. 1c), but led to the phosphorylation of eIF2α-kinase PKR (Fig. 1d). Therefore, signals upstream of phosphorylation of eIF2α after treatment with PFT, or (S)-3-oxo-C12-HSL are different. Although (S)-3-oxo-C12-HSL-dependent phosphorylation of PKR was weaker than that caused by α-toxin (Fig. 1d), knock down of PKR inhibited (S)-3-oxo-C12-HSL-dependent phosphorylation of eIF2α, indicating that PKR plays a critical role for cellular stress signaling in response to this compound (Fig. 1e). In line with this, activation of p38 was also markedly reduced after KD of PKR (Fig. 1e). Unlike PFT, (S)-3-oxo-C12-HSL did not cause significant loss of ATP at early time points, and only moderate dephosphorylation of S6K was discerned (data not shown). Thus, metabolic changes in response to (S)-3-oxo-C12-HSL appear to be less pronounced than those caused by PFT. However, metabolic labeling experiments revealed that (S)-3-oxo-C12-HSL inhibits translation (Fig. 1f). Many stress responses are accompanied by enhanced ubiquitination and degradation of proteins. Therefore, we investigated ubiquitination of proteins in (S)-3-oxo-C12-HSL-treated cells (Fig. 1g). In contrast to α-toxin, (S)-3-oxo-C12-HSL alone appeared not to significantly enhance steady state levels of ubiquitinated proteins, but addition of bafilomycin revealed that (S)-3-oxo-C12-HSL increased the rate of ubiquitination. In Western blot experiments with antibodies against LC3, higher levels of LC3II were observed following treatment with α-toxin or (S)-3-oxo-C12-HSL, and in both cases, this effect was enhanced by bafilomycin (Fig. 1g, lower panel). Consistently, numbers of GFP-LC3-positive puncta in transiently transfected cells were increased by (S)-3-oxo-C12-HSL (Fig. 1h). Therefore (S)-3-oxo-C12-HSL impacts autophagy. Since P. aeruginosa and S. aureus are commonly found together in wounds, or in the respiratory tract of patients suffering from cystic fibrosis, potential synergism of (S)-3-oxo-C12-HSL and S. aureus α-toxin might bear on the pathogenesis in coinfections with these bacteria. In fact, the combination of (S)-3-oxo-C12-HSL and S. aureus α-toxin at concentrations which caused little increases in LC3II, led to a significant accumulation of this product (Fig. 1i).
Conclusion
Bacterial exoproducts may impact host-cell translation and autophagy. Pore-forming toxins (PFT) represent one important class of bacterial exoproducts which affect these processes. Based on the studies of cellular responses to PFT, including S. aureus α-toxin, we found that the loss of cellular potassium from perforated cells leads to failure of nutrient transport and loss of ATP, thus activating cellular nutrient and energy sensors GCN2 and AMPK, subsequent phosphorylation of eIF2α and deactivation of mTORC1 (Fig. 2). Therefore, we propose that nutrient/energy sensors serve as sentinels of membrane integrity. In addition to the removal of membrane pores by endocytosis, phosphorylation of eIF2α and induction of autophagy by these pathways are required to prevent abysmal loss of ATP in cells perforated by S. aureus α-toxin. Thus, studying transient membrane perforation by S. aureus α-toxin has provided important novel insights into cell autonomous defense against an archaic threat to a cell, namely damage of the plasma membrane.
Fig. 2.
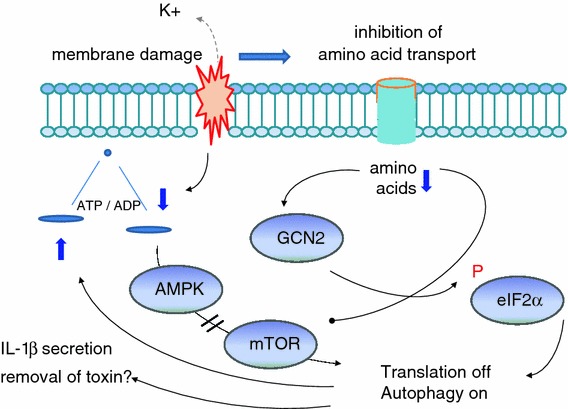
Membrane damage by pore-forming toxins induces classic starvation responses. PFT represents an important class of bacterial exoproducts which affect host-cell translation and autophagy. Loss of cellular potassium from perforated cells leads to the failure of nutrient transport and transient drop of ATP, thus activating cellular nutrient and energy sensors GCN2 and AMPK, subsequent phosphorylation of eIF2α and deactivation of mTORC1. As a consequence, transient global translational attenuation and induction of starvation-associated autophagy occur to overcome energy and nutrient crisis caused by membrane damage. Membrane perforation by α-toxin induces IL-1β secretion [56], and a role of autophagy in unconventional secretion of IL-1β has been recently documented [5]. Whether autophagy is also involved in the release of toxosomes [50], undigestible toxin oligomers associated with exosomal-like structures, remains to be investigated
Like PFT, (S)-3-oxo-C12-HSL, a quorum-sensing hormone of P. aeruginosa causes phosphorylation of eIF2α, attenuation of translation, and accumulation of autophagosomes. However, (S)-3-oxo-C12-HSL did neither cause severe loss of ATP nor phosphorylation of GCN2; and phosphorylation of p-eIF2α was insensitive to high levels of extracellular potassium. Therefore, PFT and (S)-3-oxo-C12-HSL modulate translation and impact autophagy by different pathways, which may act synergistically.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (grant to M.H.; SFB490, Projekt D3) and by intramural grants (MAIFOR) to N.K. and G.v.H.. We like to thank all current and past coworkers and collaborators, and apologize to those colleagues whose work relevant to the present issue has not been quoted here.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
References
- 1.Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8(11):931–937. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- 2.Bruns C, McCaffery JM, Curwin AJ, Duran JM, Malhotra V. Biogenesis of a novel compartment for autophagosome-mediated unconventional protein secretion. J Cell Biol. 2011;195(6):979–992. doi: 10.1083/jcb.201106098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chua CE, Lim YS, Lee MG, Tang BL. Non-classical membrane trafficking processes galore. J Cell Physiol. 2012 doi: 10.1002/jcp.24082. [DOI] [PubMed] [Google Scholar]
- 4.Duran JM, Anjard C, Stefan C, Loomis WF, Malhotra V. Unconventional secretion of Acb1 is mediated by autophagosomes. J Cell Biol. 2010;188(4):527–536. doi: 10.1083/jcb.200911154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 2011;30(23):4701–4711. doi: 10.1038/emboj.2011.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mari M, Tooze SA, Reggiori F. The puzzling origin of the autophagosomal membrane. F1000 Biol Rep. 2011;3:25. doi: 10.3410/B3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 8.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9(10):1102–1109. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- 9.Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19(12):5360–5372. doi: 10.1091/mbc.E08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6(6):764–776. doi: 10.4161/auto.6.6.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mizushima N, Sugita H, Yoshimori T, Ohsumi Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J Biol Chem. 1998;273(51):33889–33892. doi: 10.1074/jbc.273.51.33889. [DOI] [PubMed] [Google Scholar]
- 12.Kuma A, Mizushima N, Ishihara N, Ohsumi Y. Formation of the approximately 350-kDa Apg12-Apg5.Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J Biol Chem. 2002;277(21):18619–18625. doi: 10.1074/jbc.M111889200. [DOI] [PubMed] [Google Scholar]
- 13.Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282(52):37298–37302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- 14.Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, Noda T, Ohsumi Y. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408(6811):488–492. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- 15.Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007;130(1):165–178. doi: 10.1016/j.cell.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 16.Rikihisa Y. Glycogen autophagosomes in polymorphonuclear leukocytes induced by rickettsiae. Anat Rec. 1984;208(3):319–327. doi: 10.1002/ar.1092080302. [DOI] [PubMed] [Google Scholar]
- 17.Swanson MS, Isberg RR. Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect Immun. 1995;63(9):3609–3620. doi: 10.1128/iai.63.9.3609-3620.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schnupf P, Portnoy DA. Listeriolysin O: a phagosome-specific lysin. Microbes Infect. 2007;9(10):1176–1187. doi: 10.1016/j.micinf.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 19.Rich KA, Burkett C, Webster P. Cytoplasmic bacteria can be targets for autophagy. Cell Microbiol. 2003;5(7):455–468. doi: 10.1046/j.1462-5822.2003.00292.x. [DOI] [PubMed] [Google Scholar]
- 20.Meyer-Morse N, Robbins JR, Rae CS, Mochegova SN, Swanson MS, Zhao Z, Virgin HW, Portnoy D. Listeriolysin O is necessary and sufficient to induce autophagy during Listeria monocytogenes infection. PLoS One. 2010;5(1):e8610. doi: 10.1371/journal.pone.0008610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, Hamada S, Yoshimori T. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306(5698):1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 22.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119(6):753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 23.Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7(3):279–296. doi: 10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vadlamudi RK, Joung I, Strominger JL, Shin J. p62, a phosphotyrosine-independent ligand of the SH2 domain of p56lck, belongs to a new class of ubiquitin-binding proteins. J Biol Chem. 1996;271(34):20235–20237. doi: 10.1074/jbc.271.34.20235. [DOI] [PubMed] [Google Scholar]
- 25.Cemma M, Kim PK, Brumell JH. The ubiquitin-binding adaptor proteins p62/SQSTM1 and NDP52 are recruited independently to bacteria-associated microdomains to target Salmonella to the autophagy pathway. Autophagy. 2011;7(3):341–345. doi: 10.4161/auto.7.3.14046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mostowy S, Sancho-Shimizu V, Hamon MA, Simeone R, Brosch R, Johansen T, Cossart P. p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J Biol Chem. 2011;286(30):26987–26995. doi: 10.1074/jbc.M111.223610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weidberg H, Elazar Z. TBK1 mediates crosstalk between the innate immune response and autophagy. Sci Signal. 2011;4(187):pe39. doi: 10.1126/scisignal.2002355. [DOI] [PubMed] [Google Scholar]
- 28.Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, Dotsch V, Bumann D, Dikic I. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333(6039):228–233. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gamerdinger M, Kaya AM, Wolfrum U, Clement AM, Behl C. BAG3 mediates chaperone-based aggresome-targeting and selective autophagy of misfolded proteins. EMBO Rep. 2011;12(2):149–156. doi: 10.1038/embor.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.von Muhlinen N, Thurston T, Ryzhakov G, Bloor S, Randow F. NDP52, a novel autophagy receptor for ubiquitin-decorated cytosolic bacteria. Autophagy. 2010;6(2):288–289. doi: 10.4161/auto.6.2.11118. [DOI] [PubMed] [Google Scholar]
- 31.Mestre MB, Fader CM, Sola C, Colombo MI. Alpha-hemolysin is required for the activation of the autophagic pathway in Staphylococcus aureus-infected cells. Autophagy. 2010;6(1):110–125. doi: 10.4161/auto.6.1.10698. [DOI] [PubMed] [Google Scholar]
- 32.Mostowy S, Cossart P. Bacterial autophagy: restriction or promotion of bacterial replication? Trends Cell Biol. 2012;22(6):283–291. doi: 10.1016/j.tcb.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 33.Dupont N, Lacas-Gervais S, Bertout J, Paz I, Freche B, Van Nhieu GT, van der Goot FG, Sansonetti PJ, Lafont F. Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe. 2009;6(2):137–149. doi: 10.1016/j.chom.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 34.Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature. 2012;482(7385):414–418. doi: 10.1038/nature10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deretic V, Singh S, Master S, Harris J, Roberts E, Kyei G, Davis A, de Haro S, Naylor J, Lee HH, Vergne I. Mycobacterium tuberculosis inhibition of phagolysosome biogenesis and autophagy as a host defence mechanism. Cell Microbiol. 2006;8(5):719–727. doi: 10.1111/j.1462-5822.2006.00705.x. [DOI] [PubMed] [Google Scholar]
- 36.Birmingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J Biol Chem. 2006;281(16):11374–11383. doi: 10.1074/jbc.M509157200. [DOI] [PubMed] [Google Scholar]
- 37.Yoshikawa Y, Ogawa M, Hain T, Yoshida M, Fukumatsu M, Kim M, Mimuro H, Nakagawa I, Yanagawa T, Ishii T, Kakizuka A, Sztul E, Chakraborty T, Sasakawa C. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol. 2009;11(10):1233–1240. doi: 10.1038/ncb1967. [DOI] [PubMed] [Google Scholar]
- 38.Kloft N, Neukirch C, Bobkiewicz W, Veerachato G, Busch T, von Hoven G, Boller K, Husmann M. Pro-autophagic signal induction by bacterial pore-forming toxins. Med Microbiol Immunol. 2010;199(4):299–309. doi: 10.1007/s00430-010-0163-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Viala JP, Mochegova SN, Meyer-Morse N, Portnoy DA. A bacterial pore-forming toxin forms aggregates in cells that resemble those associated with neurodegenerative diseases. Cell Microbiol. 2008;10(4):985–993. doi: 10.1111/j.1462-5822.2007.01100.x. [DOI] [PubMed] [Google Scholar]
- 40.Watanabe Y, Tanaka M. p62/SQSTM1 in autophagic clearance of a non-ubiquitylated substrate. J Cell Sci. 2011;124(Pt 16):2692–2701. doi: 10.1242/jcs.081232. [DOI] [PubMed] [Google Scholar]
- 41.Shahnazari S, Yen WL, Birmingham CL, Shiu J, Namolovan A, Zheng YT, Nakayama K, Klionsky DJ, Brumell JH. A diacylglycerol-dependent signaling pathway contributes to regulation of antibacterial autophagy. Cell Host Microbe. 2010;8(2):137–146. doi: 10.1016/j.chom.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moscat J, Diaz-Meco MT. Feedback on fat: p62-mTORC1-autophagy connections. Cell. 2011;147(4):724–727. doi: 10.1016/j.cell.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, Hansen M, Moscat J, Diaz-Meco MT. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011;44(1):134–146. doi: 10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gutierrez MG, Saka HA, Chinen I, Zoppino FC, Yoshimori T, Bocco JL, Colombo MI. Protective role of autophagy against Vibrio cholerae cytolysin, a pore-forming toxin from V. cholerae. Proc Natl Acad Sci USA. 2007;104(6):1829–1834. doi: 10.1073/pnas.0601437104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walev I, Palmer M, Martin E, Jonas D, Weller U, Hohn-Bentz H, Husmann M, Bhakdi S. Recovery of human fibroblasts from attack by the pore-forming alpha-toxin of Staphylococcus aureus. Microb Pathog. 1994;17(3):187–201. doi: 10.1006/mpat.1994.1065. [DOI] [PubMed] [Google Scholar]
- 46.Walev I, Hombach M, Bobkiewicz W, Fenske D, Bhakdi S, Husmann M. Resealing of large transmembrane pores produced by streptolysin O in nucleated cells is accompanied by NF-kappaB activation and downstream events. FASEB J. 2002;16(2):237–239. doi: 10.1096/fj.01-0572fje. [DOI] [PubMed] [Google Scholar]
- 47.Dragneva Y, Anuradha CD, Valeva A, Hoffmann A, Bhakdi S, Husmann M. Subcytocidal attack by staphylococcal alpha-toxin activates NF-kappaB and induces interleukin-8 production. Infect Immun. 2001;69(4):2630–2635. doi: 10.1128/IAI.69.4.2630-2635.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Haugwitz U, Bobkiewicz W, Han SR, Beckmann E, Veerachato G, Shaid S, Biehl S, Dersch K, Bhakdi S, Husmann M. Pore-forming Staphylococcus aureus alpha-toxin triggers epidermal growth factor receptor-dependent proliferation. Cell Microbiol. 2006;8(10):1591–1600. doi: 10.1111/j.1462-5822.2006.00733.x. [DOI] [PubMed] [Google Scholar]
- 49.Thelestam M, Mollby R. Survival of cultured cells after functional and structural disorganization of plasma membrane by bacterial haemolysins and phospholipases. Toxicon. 1983;21(6):805–815. doi: 10.1016/0041-0101(83)90069-7. [DOI] [PubMed] [Google Scholar]
- 50.Husmann M, Beckmann E, Boller K, Kloft N, Tenzer S, Bobkiewicz W, Neukirch C, Bayley H, Bhakdi S. Elimination of a bacterial pore-forming toxin by sequential endocytosis and exocytosis. FEBS Lett. 2009;583(2):337–344. doi: 10.1016/j.febslet.2008.12.028. [DOI] [PubMed] [Google Scholar]
- 51.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6(5):1099–1108. doi: 10.1016/S1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 52.Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans. 2006;34(Pt 1):7–11. doi: 10.1042/BST20060007. [DOI] [PubMed] [Google Scholar]
- 53.Talloczy Z, Jiang W, Virgin HWt, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci USA. 2002;99(1):190–195. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tattoli I, Sorbara MT, Vuckovic D, Ling A, Soares F, Carneiro LA, Yang C, Emili A, Philpott DJ, Girardin SE. Amino Acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe. 2012;11(6):563–575. doi: 10.1016/j.chom.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 55.Chakrabarti S, Liehl P, Buchon N, Lemaitre B. Infection-induced host translational blockage inhibits immune responses and epithelial renewal in the Drosophila gut. Cell Host Microbe. 2012;12(1):60–70. doi: 10.1016/j.chom.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 56.Walev I, Reske K, Palmer M, Valeva A, Bhakdi S. Potassium-inhibited processing of IL-1 beta in human monocytes. EMBO J. 1995;14(8):1607–1614. doi: 10.1002/j.1460-2075.1995.tb07149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gurcel L, Abrami L, Girardin S, Tschopp J, van der Goot FG. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell. 2006;126(6):1135–1145. doi: 10.1016/j.cell.2006.07.033. [DOI] [PubMed] [Google Scholar]
- 58.Imre G, Heering J, Takeda AN, Husmann M, Thiede B, Zu Heringdorf DM, Green DR, van der Goot FG, Sinha B, Dotsch V, Rajalingam K. Caspase-2 is an initiator caspase responsible for pore-forming toxin-mediated apoptosis. EMBO J. 2012;31(11):2615–2628. doi: 10.1038/emboj.2012.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kloft N, Busch T, Neukirch C, Weis S, Boukhallouk F, Bobkiewicz W, Cibis I, Bhakdi S, Husmann M. Pore-forming toxins activate MAPK p38 by causing loss of cellular potassium. Biochem Biophys Res Commun. 2009;385(4):503–506. doi: 10.1016/j.bbrc.2009.05.121. [DOI] [PubMed] [Google Scholar]
- 60.Gonzalez MR, Bischofberger M, Freche B, Ho S, Parton RG, van der Goot FG. Pore-forming toxins induce multiple cellular responses promoting survival. Cell Microbiol. 2011;13(7):1026–1043. doi: 10.1111/j.1462-5822.2011.01600.x. [DOI] [PubMed] [Google Scholar]
- 61.Iordanov MS, Magun BE. Loss of cellular K+ mimics ribotoxic stress. Inhibition of protein synthesis and activation of the stress kinases SEK1/MKK4, stress-activated protein kinase/c-Jun NH2-terminal kinase 1, and p38/HOG1 by palytoxin. J Biol Chem. 1998;273(6):3528–3534. doi: 10.1074/jbc.273.6.3528. [DOI] [PubMed] [Google Scholar]
- 62.Babiychuk EB, Monastyrskaya K, Potez S, Draeger A. Intracellular Ca(2+) operates a switch between repair and lysis of streptolysin O-perforated cells. Cell Death Differ. 2009;16(8):1126–1134. doi: 10.1038/cdd.2009.30. [DOI] [PubMed] [Google Scholar]
- 63.Bellier A, Chen CS, Kao CY, Cinar HN, Aroian RV. Hypoxia and the hypoxic response pathway protect against pore-forming toxins in C. elegans. PLoS Pathog. 2009;5(12):e1000689. doi: 10.1371/journal.ppat.1000689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen CS, Bellier A, Kao CY, Yang YL, Chen HD, Los FC, Aroian RV. WWP-1 is a novel modulator of the DAF-2 insulin-like signaling network involved in pore-forming toxin cellular defenses in Caenorhabditis elegans. PLoS One. 2010;5(3):e9494. doi: 10.1371/journal.pone.0009494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Draeger A, Monastyrskaya K, Babiychuk EB. Plasma membrane repair and cellular damage control: the annexin survival kit. Biochem Pharmacol. 2011;81(6):703–712. doi: 10.1016/j.bcp.2010.12.027. [DOI] [PubMed] [Google Scholar]
- 66.Huffman DL, Abrami L, Sasik R, Corbeil J, van der Goot FG, Aroian RV. Mitogen-activated protein kinase pathways defend against bacterial pore-forming toxins. Proc Natl Acad Sci USA. 2004;101(30):10995–11000. doi: 10.1073/pnas.0404073101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Husmann M, Dersch K, Bobkiewicz W, Beckmann E, Veerachato G, Bhakdi S. Differential role of p38 mitogen activated protein kinase for cellular recovery from attack by pore-forming S. aureus alpha-toxin or streptolysin O. Biochem Biophys Res Commun. 2006;344(4):1128–1134. doi: 10.1016/j.bbrc.2006.03.241. [DOI] [PubMed] [Google Scholar]
- 68.Kao CY, Los FC, Huffman DL, Wachi S, Kloft N, Husmann M, Karabrahimi V, Schwartz JL, Bellier A, Ha C, Sagong Y, Fan H, Ghosh P, Hsieh M, Hsu CS, Chen L, Aroian RV. Global functional analyses of cellular responses to pore-forming toxins. PLoS Pathog. 2011;7(3):e1001314. doi: 10.1371/journal.ppat.1001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Saka HA, Gutierrez MG, Bocco JL, Colombo MI. The autophagic pathway: a cell survival strategy against the bacterial pore-forming toxin Vibrio cholerae cytolysin. Autophagy. 2007;3(4):363–365. doi: 10.4161/auto.4159. [DOI] [PubMed] [Google Scholar]
- 70.Aroian R, van der Goot FG. Pore-forming toxins and cellular non-immune defenses (CNIDs) Curr Opin Microbiol. 2007;10(1):57–61. doi: 10.1016/j.mib.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 71.Bischofberger M, Gonzalez MR, van der Goot FG. Membrane injury by pore-forming proteins. Curr Opin Cell Biol. 2009;21(4):589–595. doi: 10.1016/j.ceb.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 72.Freche B, Reig N, van der Goot FG. The role of the inflammasome in cellular responses to toxins and bacterial effectors. Semin Immunopathol. 2007;29(3):249–260. doi: 10.1007/s00281-007-0085-0. [DOI] [PubMed] [Google Scholar]
- 73.Gonzalez MR, Bischofberger M, Pernot L, van der Goot FG, Freche B. Bacterial pore-forming toxins: the (w)hole story? Cell Mol Life Sci. 2008;65(3):493–507. doi: 10.1007/s00018-007-7434-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McNeil PL, Steinhardt RA. Plasma membrane disruption: repair, prevention, adaptation. Annu Rev Cell Dev Biol. 2003;19:697–731. doi: 10.1146/annurev.cellbio.19.111301.140101. [DOI] [PubMed] [Google Scholar]
- 75.McNeil PL, Steinhardt RA. Loss, restoration, and maintenance of plasma membrane integrity. J Cell Biol. 1997;137(1):1–4. doi: 10.1083/jcb.137.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zeigerer A, Gilleron J, Bogorad RL, Marsico G, Nonaka H, Seifert S, Epstein-Barash H, Kuchimanchi S, Peng CG, Ruda VM, Del Conte-Zerial P, Hengstler JG, Kalaidzidis Y, Koteliansky V, Zerial M. Rab5 is necessary for the biogenesis of the end olysosomal system in vivo. Nature. 2012;485(7399):465–470. doi: 10.1038/nature11133. [DOI] [PubMed] [Google Scholar]
- 77.Cavalli V, Vilbois F, Corti M, Marcote MJ, Tamura K, Karin M, Arkinstall S, Gruenberg J. The stress-induced MAP kinase p38 regulates endocytic trafficking via the GDI: Rab5 complex. Mol Cell. 2001;7(2):421–432. doi: 10.1016/S1097-2765(01)00189-7. [DOI] [PubMed] [Google Scholar]
- 78.Webber JL, Tooze SA. Coordinated regulation of autophagy by p38alpha MAPK through mAtg9 and p38IP. EMBO J. 2010;29(1):27–40. doi: 10.1038/emboj.2009.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Webber JL, Tooze SA. New insights into the function of Atg9. FEBS Lett. 2010;584(7):1319–1326. doi: 10.1016/j.febslet.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 80.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30(6):678–688. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT (2007) Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 27(1):135–144 [DOI] [PMC free article] [PubMed]
- 82.Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16(1):90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 83.Deretic V. Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol Rev. 2011;240(1):92–104. doi: 10.1111/j.1600-065X.2010.00995.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, Yuan L, Soares F, Chea E, Le Bourhis L, Boneca IG, Allaoui A, Jones NL, Nunez G, Girardin SE, Philpott DJ. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11(1):55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 85.Teplitski M, Mathesius U, Rumbaugh KP. Perception and degradation of N-acyl homoserine lactone quorum sensing signals by mammalian and plant cells. Chem Rev. 2011;111(1):100–116. doi: 10.1021/cr100045m. [DOI] [PubMed] [Google Scholar]
- 86.Kravchenko VV, Kaufmann GF, Mathison JC, Scott DA, Katz AZ, Grauer DC, Lehmann M, Meijler MM, Janda KD, Ulevitch RJ. Modulation of gene expression via disruption of NF-kappaB signaling by a bacterial small molecule. Science. 2008;321(5886):259–263. doi: 10.1126/science.1156499. [DOI] [PubMed] [Google Scholar]
- 87.Horke S, Witte I, Altenhofer S, Wilgenbus P, Goldeck M, Forstermann U, Xiao J, Kramer GL, Haines DC, Chowdhary PK, Haley RW, Teiber JF. Paraoxonase 2 is down-regulated by the Pseudomonas aeruginosa quorumsensing signal N-(3-oxododecanoyl)-L-homoserine lactone and attenuates oxidative stress induced by pyocyanin. Biochem J. 2010;426(1):73–83. doi: 10.1042/BJ20091414. [DOI] [PubMed] [Google Scholar]
- 88.Kravchenko VV, Kaufmann GF, Mathison JC, Scott DA, Katz AZ, Wood MR, Brogan AP, Lehmann M, Mee JM, Iwata K, Pan Q, Fearns C, Knaus UG, Meijler MM, Janda KD, Ulevitch RJ. N-(3-oxo-acyl)homoserine lactones signal cell activation through a mechanism distinct from the canonical pathogen-associated molecular pattern recognition receptor pathways. J Biol Chem. 2006;281(39):28822–28830. doi: 10.1074/jbc.M606613200. [DOI] [PubMed] [Google Scholar]
- 89.Deng J, Harding HP, Raught B, Gingras AC, Berlanga JJ, Scheuner D, Kaufman RJ, Ron D, Sonenberg N. Activation of GCN2 in UV-irradiated cells inhibits translation. Curr Biol. 2002;12(15):1279–1286. doi: 10.1016/S0960-9822(02)01037-0. [DOI] [PubMed] [Google Scholar]