

Abstract
Bone abnormalities are frequent co-morbidities of type 1 diabetes [T1D] and are principally mediated by osteoblasts and osteoclasts which in turn are regulated by immunologic mediators. While decreased skeletal health in T1D involves alterations in osteoblast maturation and function, the affect of altered immune function on osteoclasts in T1D-associated bone and joint pathologies is less understood. Here T1D-associated osteoclast-specific differentiation and function in the presence and absence of inflammatory mediators was characterized utilizing bone marrow-derived osteoclasts [BM-OCs] isolated from non-obese diabetic [NOD] mice, a model for spontaneous autoimmune diabetes with pathology similar to individuals with T1D. Differentiation and osteoclast-mediated bone resorption were evaluated along with cathepsin K, MMP-9, and immune soluble mediator expression. The affect of LPS, a pro-inflammatory cytokine cocktail, and NOD-derived conditioned supernatants on BM-OC function was also determined. Although NOD BM-OCs cultures contained smaller osteoclasts, they resorbed more bone concomitant with increased cathepsin K, MMP-9 and pro-osteoclastogenic mediator expression. NOD BM-OCs also displayed an inhibition of LPS-induced deactivation that was not a result of soluble mediators produced by NOD BM-OCs, although a pro-inflammatory milieu did enhance NOD BM-OCs bone resorption. Together these data indicate that osteoclasts from a T1D mouse model hyper-respond to RANK-L resulting in excessive bone degradation via enhanced cathepsin K and MMP-9 secretion concomitant with an increased expression of pro-osteoclastic soluble mediators. Our data also suggest that inhibition of LPS-induced deactivation in NOD-derived BM-OC cultures is most likely due to NOD osteoclast responsiveness rather than LPS-induced expression of soluble mediators.
Keywords: Osteoclasts, Type 1 Diabetes, Bone Resorption, Inflammatory Mediators, LPS
Introduction
Bone and joint abnormalities are frequent co-morbidities of type 1 diabetes [T1D] (Khazai et al., 2009; McCabe, 2007; Paula and Rosen). T1D originates from a complex etiology with intrinsic genetic risk factors and extrinsic environmental factors. T1D-associated bone and joint pathologies likewise may originate from shared intrinsic genetic factors or from extrinsic sources of activation. Furthermore, bone and joint pathology may develop secondary to autoimmune inflammation or as a consequence of hyperglycemia. The complexity of T1D itself and added complexity of bone and joint co-morbidities necessitates well-controlled and innovative approaches to assess the totality of potential causes.
Physiological bone remodeling is a highly coordinated process that orchestrates five sequential phases: activation, resorption, reversal, formation and termination (Raggatt and Partridge, 2010). In addition to traditional bone cells including osteoclasts and osteoblasts which are responsible for bone resorption and formation, respectively, immune cells have also been implicated in the regulation of this process. Thus, alterations in immune function have been implicated in many bone diseases (Raggatt and Partridge, 2010). While decreased skeletal health in T1D involves alterations in osteoblast maturation and function, the role of osteoclasts in inflammation-induced bone and joint loss is less understood (McCabe, 2007).
Osteoclast differentiation is regulated by macrophage colony-stimulating factor [M-CSF] and the receptor activator of nuclear factor kappa B ligand [RANK-L]. Resorption of bone is initiated by binding of the osteoclasts to the mineralized bone surface via alpha v beta 3 [αvβ3] integrins forming a sealing zone membrane and a resorption lacuna (Del Fattore et al., 2008). Vesicles containing osteoclastic enzymes such as tartrate-resistant acid phosphatase [TRAP], the serine protease cathepsin K, and matrix metalloproteinase-9 [MMP-9] induce collagen degradation after bone demineralization by the vacuolar H+-ATPase (Hayman, 2008). Osteoclasts are also negatively regulated through soluble mediators including calcitonin and osteoprotegerin [OPG] (Takayanagi, 2007).
Activation of osteoclast-mediated bone resorption can be augmented by infection and inflammation, as well as hormonal alterations (Takayanagi, 2007). Lipopolysaccharide [LPS], a cell wall component of gram negative bacteria, has been found to be highly immunogenic and induces the production of pro-inflammatory cytokines by various immune cells. Osteoclasts and their precursors, which share the same lineage as macrophages and dendritic cells, express many innate immune receptors including toll-like receptors [TLRs] and thus can respond to bacterial components (Dumitrescu et al., 2004; Jiang et al., 2003; Matsuo and Irie, 2008). In a co-culture of osteoclasts and osteoblasts, LPS, the ligand for TLR4, augments bone resorption (Jiang et al., 2003). However, when supporting cells such as osteoblasts or other immune cells are absent, LPS inhibits bone resorption in osteoclast pure cultures, although the exact mechanism(s) is not known (Liu et al., 2009). In addition, pro-inflammatory cytokines including TNF-α, IL-1β, and IL-6 stimulate differentiation and activation of osteoclasts with IL-1β and TNF-α being directly involved in activating resorption (D et al., 2004; Grey et al., 1999). In both murine and human T1D, macrophages and dendritic cells have been shown to be hyperactive to TLR stimulation resulting in excessive pro-inflammatory cytokine production (Ghosh et al., 1998; Poligone et al., 2002; Sen et al., 2003). In addition, individuals with T1D are less able to clear bacterial infections thereby allowing bacterial components to accumulate further amplifying the inflammatory process (Mealey and Rose, 2008).
Since individuals with T1D respond to inflammation in such an aberrant fashion, it is entirely possible that these individuals would be more susceptible to severe bone loss than T1D-free individuals given the effects of these soluble mediators on osteoclast activation and function. Similarly, given the phylogenetic relationship of osteoclasts to macrophages and dendritic cells and the aberrant TLR-responsiveness of these cell types in T1D, we postulate that TID-derived osteoclasts have heightened sensitivity to stimulation resulting in augmented differentiation and activation. Non-obese diabetic [NOD] mice, a model for spontaneous autoimmune diabetes with pathology similar to individuals with T1D, have an osteoporotic phenotype (Botolin and McCabe, 2007). In addition, NOD mice are more susceptible to spontaneous and induced arthritis (Lawlor et al., 2001). Therefore in the present study, we characterized T1D-associated osteoclast-specific differentiation, activation and function in the presence and absence of inflammatory stimuli utilizing the NOD mouse model.
Materials and Methods
Mouse Models
NOD/LtJ [NOD], NOR/LtJ [NOR], C57Bl/6J [C57Bl/6], and Balb/cJ [Balb/c] mice were maintained in a specific pathogen-free [SPF] environment at the breeding facilities of the University of Florida. The NOD mouse spontaneously develops an autoimmune-mediated destruction of the β-cells within the islets of Langerhans of the pancreas producing very similar pathology as seen in individuals with T1D, where by 10 weeks of age severe insulitis is apparent, but mice are normoglycemic (Anderson and Bluestone, 2005). The NOR mouse model has a similar genetic background as the NOD mouse but does not develop insulitis and diabetes due to the dispersal of C57Bl/6 genome within diabetes susceptibility loci (Prochazka et al., 1992). Thus the C57Bl/6 strain serves as an additional genetic background control as well as an immunological control along with the Balb/c strain. C57Bl/6 mice are considered Th1 responders while Balb/c mice respond predominately in a Th1-directed manner (Lohoff et al., 1998) as do NOD mice (Anderson and Bluestone, 2005).
Blood glucose levels were measured at time of sacrifice with the Ascensia Contour Blood Glucose Meter (Bayer). Bone marrow was harvested from female mice of all strains at 10-12 weeks [wks] of age. Pancreata were also harvested from NOD and NOR mice. All experimental procedures were conducted in accordance with the guidelines of the University of Florida Institutional Animal Care and Use Committee.
Osteoclast Differentiation
Femora and tibiae were surgically isolated, excess tissue removed, and marrow expelled from bones using a syringe with α-MEM complete media (Sigma-Aldrich) [10% fetal bovine serum (Mediatech), 1% L-glutamine (Thermo Scientific), 1% penicillin/streptomycin/amphotericin B (Fisher)]. Cells were seeded in T75 flasks at a concentration of 1.5×106 cells/mL supplemented with 5ng/mL recombinant murine M-CSF [rmM-CSF] (Peprotech) and allowed to culture for 24hrs at 37°C and 5% CO2. Non-adherent cells were removed and 5.9×105 cells/mL of adherent cells were seeded in 24-well plates on either glass coverslips (Fisher) or 1 cm2 bovine bone slices cut with an Isomet Low Speed Saw (Buehler). All cultures were supplemented with 10ng/mL rmM-CSF and 50ng/mL recombinant murine soluble RANK-L [rmsRANK-L] (Peprotech) and allowed to culture for 6d with complete media refreshed every 3d.
TRAP Staining
After 6d or d9 of differentiation, cells plated on glass coverslips were fixed with 2% paraformaldehyde/PBS (Fisher). Cells were washed with PBS and permeablized in 0.5% Triton X-100/PBS (Fisher). Cells were washed and probed for leukocyte acid phosphatase (TRAP) [1:1:1:2:4 Fast Garnet GBC Base Solution:Sodium Nitrite Solution:Napthol AS-BI Phosphate Solution:Tartrate Solution:Acetate Solution] (Sigma Aldrich) after which cells were washed and mounted on glass slides with MOWIOL 4-88 solution (Calbiochem). TRAP positive cells [purple in color] were counted according to number of nuclei present: mononuclear cells [1 nucleus], multinucleated osteoclasts [2-10 nuclei], and giant osteoclasts [11+ nuclei] using light microscopy at 40x magnification. Whole coverslips were counted for each cell type. Percentage of total nuclei was calculated by the number of nuclei in each cell type divided by total number of nuclei counted.
Osteoclast Stimulation
After 6d of differentiation, media was refreshed with α-MEM complete media supplemented with 10ng/mL rmM-CSF and 50ng/mL rmsRANK-L. Cells were allowed to resorb bone for 72hrs in the presence or absence of the following: 1) 1ug/mL Escherichia coli LPS [LPS] (Sigma), 2) pro-inflammatory cytokine cocktail [10ng/mL recombinant human TNF-α [rhTNF-α] (R&D Systems) + 10ng/mL recombinant murine IL-1β [rmIL-1β] (Peprotech) + 100ng/mL rmIL-6 (Peprotech)], 3) NOD conditioned media [from M-CSF and RANK-L stimulated bone resorption cultures], or 4) C57Bl/6 conditioned media [from M-CSF and RANK-L stimulated bone resorption cultures]. Cultures were permeablized with 1% Triton X-100 and supernatants stored at −80C until cathepsin K ELISA, collagen type I telopeptide ELISA, MMP-9 ELISA, and Luminex cyto/chemokine analyses were performed. Bone was made devoid of cells with 10% sodium hypochlorite/PBS after which they were washed with PBS and stored in Trump’s fixative (Fisher) at 4C until scanning electron microscopy [SEM] could be performed. All bone resorption outcome measures were normalized to number of TRAP+ cells [outcome measure x (total number of TRAP+cells/total number of cells plated)], mononuclear cells [outcome measure x (total number of mononuclear cells/total number of TRAP+cells)] and multi-nucleated cells [outcome measure x (total number of mononuclear cells/total number of TRAP+cells)].
Flow Cytometry for Osteoclast Culture Purity
After 6d of differentiation media was refreshed with α-MEM complete media supplemented with 10ng/mL rmM-CSF and 50ng/mL rmsRANK-L in the presence or absence of 1ug/mL non-pure E. coli LPS for 72hr. Cells were allowed to dissociate from the bottom of UpCell (ThermoScientific) coated plates at room temperature. Suspended cells were washed with FACS Buffer [1x PBS + 5% FBS + 0.372g EDTA] and allowed to incubate with the following primary [1:200] and secondary antibodies [1:200]: goat anti-mouse calcitonin receptor [CTR] (Santa Cruz) with anti-goat Alexa Fluor 647 (Invitrogen) and biotin-conjugated rat anti-mouse RANK (eBioscience) with PerCP-Cy5.5-conjugated streptavidin (eBioscience). Cells were acquired on a FACSCalibur flow cytometer (BD Biosciences) and analyzed using FCS Express (De Novo Software).
Cell Viability Assays
Viability was assessed using a colorimetric MTT Cell Growth Assay (Millipore, Billerica, MA) at d6 and d9 post-differentiation described above. MTT assay was performed according to manufacturer’s instructions and absorbance was quantified with a spectrophotometer set at a dual wavelength reading of 570nm with a reference of 630nm. Culture media alone was used as a negative control while cells lysed with 1.0% triton were used as a positive control.
Scanning Electron Microscopy
Bone slices were sputter-coated with gold and visualized with S-4000 FE-SEM scanning electron microscope (Hitachi). Three random scanning electron micrographs [8-bit grayscale] of bone slices were acquired at 40× magnification with a 2048×1594 resolution and pits were defined as “scalloped” areas with clearly identifiable borders. Identical procedures were applied to every image from all experimental groups utilizing NIH ImageJ software to quantify the surface area resorbed. Prominent repeating elements in the frequency domain were identified and removed after which the inverse fast Fourier transformation was applied yielding the original image with reduced saw marks. The CLAHE algorithm (Karel, 1994) was used to increase contrast [block size: 256, histogram bins: 256, maximum slope: 8], and a Gaussian blur [σ: 2 pixels] was applied to the result. The rolling ball algorithm (Sternberg, 1983) was applied [radius: 100 pixels] to achieve background intensity equalization. A threshold value (85) was used to convert the result to a 1-bit image [0: normal bone, 1: region of resorption] used for quantitative analysis. Images which contained prominent artifacts spanning 5% or more of the total area were not included for analysis. Percentage of area resorbed was calculated by dividing square microns resorbed by total square microns.
Collagen Telopeptide ELISA
Collagen carboxy-terminal telopeptides were detected using an ELISA according to manufacturer instructions (Immunodiagnostic Systems). Supernatants were pre-incubated with biotin conjugated anti-telopeptide and horseradish-peroxidase [HRP] conjugated anti-telopeptide and added to an ELISA plate coated with streptavidin [SAV]. Following five washes, tetramethylbenzidine [TMB] substrate was used to develop reactivity followed by quenching with H2SO4. Colorimetric reactions were detected using an Epoch microplate spectrophotometer (Biotek) set at a dual wavelength reading of 450nm with a reference of 655nm. Gen5 Software (Biotek) and a standard curve were used to determine nM concentrations.
Cathepsin K ELISA
Active cathepsin K was detected using an ELISA according to manufacturer instructions (Alpco). Supernatants pre-incubated with HRP-conjugated anti-cathepsin K were added to an ELISA plate pre-coated with polyclonal sheep anti-cathepsin K. Following five washes, TMB substrate was used to develop reactivity followed by quenching with STOP solution. Colorimetric reactions were detected using an Epoch microplate spectrophotometer (Biotek) set at a dual wavelength reading of 450nm with a reference of 655nm. Gen5 Software (Biotek) and a standard curve were used to determine pM/L concentrations of active cathespin K.
MMP-9 ELISA
Total MMP-9 was detected using an ELISA according to manufacturer instructions (R&D Systems). Supernatants were added to an ELISA plate pre-coated with anti-MMP-9. Following four washes, HRP-anti-MMP-9 was used to detect reactivity. Following five washes, TMB substrate was used to develop reactivity followed by quenching with HCl. Colorimetric reactions were detected using an Epoch microplate spectrophotometer (Biotek) set at a dual wavelength reading of 450nm with a reference of 595nm. Gen5 Software (Biotek) and a standard curve were used to determine ng/mL concentrations.
Soluble Mediator Analysis
Cytokines and chemokines from resorption supernatants were detected and quantified using a mouse 22-cyto/chemokine multiplex (Millipore) according to the manufacturer’s instructions. Supernatant and antibody-coated beads were allowed to incubate overnight at 4C in a 96-well primed plate. Following three washes, reactivity was probed with biotinylated detection antibodies and SAV-phycoerythrin [PE]. All incubations occurred while gently shaking in the dark. Following three washes, beads were resuspended in sheath fluid and reactivity acquired using a Luminex 200 IS system with Xponent software (Millipore). Milliplex analyst software (Viagene), 5-parameter logistics and a standard curve were used to determine pg/ml concentrations. Outcome measures were normalized to number of mononuclear cells [outcome measure x (total number of mononuclear cells/total number of TRAP+cells)] and multi-nucleated cells [outcome measure x (total number of mononuclear cells/total number of TRAP+cells)].
Histological Analysis of Pancreas
Pancreata were fixed in 10% formalin (Fisher) and embedded in paraffin. 5μm sections were mounted on glass slides and deparaffinized in xylenes and rehydrated using 100% ethanol (EtOH), 95% EtOH, 75% EtOH (Fisher), and dH2O. Sections were stained with Harris hematoxylin, incubated in a bluing solution [1.5% NH4OH (EMD Chemicals) in 70% EtOH] and stained with eosin [1% aqueous Eosin Y, 1% aqueous phloxyine B (Fisher), 100% EtOH and glacial acetic acid (Mallinckrodt Chemicals)]. Sections were dehydrated with 70% EtOH, 95% EtOH, 100% EtOH, and xylene (2 times), mounted with Permount (Fisher) and observed under light microscopy. Islet infiltration was graded on a 0-2 scale, with 0=no insulitis, 1=peri-insulitis, and 2=insulitis where average insulitis score per pancreata was determined.
Statistical Analysis
One-way ANOVA with Dunns’s multiple comparisons were used to analyze and determine statistical significance (p<0.05) as appropriate.
Results
NOD-derived osteoclasts display altered differentiation
In order to determine if increased bone resorption observed in multiple T1D complications is due to alterations in osteoclast differentiation, the differentiation of bone marrow derived osteoclasts [BM-OCs] from C57Bl/6, Balb/c, NOR and NOD mice was evaluated. BM-OCs were allowed to differentiate for 6d and purity of cultures was evaluated using FACS analysis where the expression of RANK and calcitonin receptor [CTR] was used to define the osteoclast populations (Fig. 1). In addition, TRAP staining was used to determine the number of mononuclear cells, multinucleated osteoclasts, and giant osteoclasts (Fig. 2).
Figure 1. Flow cytometric analysis of osteoclast culture purity.
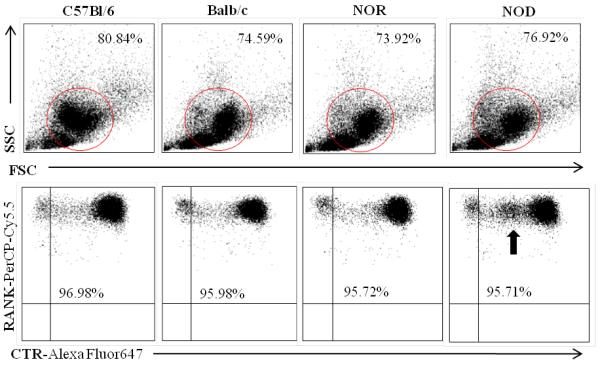
BM-OCs were stimulated with RANK-L for 72hrs on UpCell plates. Cells were probed for RANK (PerCP-Cy5.5) and calcitonin receptor [CTR] (AlexaFluor647) and acquired using a FACSCalibur flow cytometer. FCS Express software was used to determine forward and side scatter population of analyzed BM-OCs (circled in upper panel) and percentage of pure RANK+CTR+ BM-OCs (lower panel). Black arrow indicates novel population of RANK+ cells expressing low levels of CTR [RANK+CTRlo] NOD BM-OCs. Data shown as representative scatter plots of each mouse strain.
Figure 2. NOD-derived osteoclasts display altered differentiation.

(A) 6d post-differentiation, BM-OCs were imaged with light microscopy at 40x magnification. Representative images of (I) mononuclear cells, (II) multinucleated OCs, and (III) giant OCs. (B-D) Number of TRAP+ BM-OCs per coverslip were enumerated using 40x magnification. (E-G) Data shown as percentage of total nuclei TRAP+. C57Bl/6 (black bars, n=18), Balb/c (dark grey bars, n=16), NOR (light grey bars, n=16), NOD (white bars, n=16). (H) 6d and 9d post-differentiation, multinucleated and giant BM-OCs NOD mice were quantified. (I) 6d and 9d post-differentiation cell-viability of C57Bl/6 and NOD BM-OC cultures was assessed by MTT assay. *p value < 0.05. One-way ANOVA with Dunn’s multiple correction.
While BM-OC cultures from all strains generated similar percentages of RANK+CTR+ where on average cultures were 76.5% pure, NOD BM-OCs consistently had a population of cells expressing lower levels of CTR [RANK+CTRlo] (Fig. 1 arrow). In addition, while no differences in the number of mononuclear cells were observed (Fig. 2B), there were significantly fewer multinucleated and giant osteoclasts observed in NOD-derived osteoclast cultures when compared to C57Bl/6-, Balb/c-, and NOR-derived cultures (Fig. 2C,D). Because osteoclast formation is a result of cell fusion events, we also evaluated the percentage of each osteoclast phenotype type compared to the total nuclei present (Fig 2E-G). Here again, NOD-derived osteoclast cultures had a significantly lower percentage of cells which were multi-nucleated compared to those derived from all other strains (Fig. 2F,G). In order to determine if NOD BM-OC cultures simply had a delay in fusion events, differentiation of BM-OCs was also evaluated at 9d of culture, where no significant difference in the number of multi-nucleated or giant cells was observed (Fig. 2H). Similarly, to determine if there was a difference in osteoclast survival an MTT assay was performed at 6d and 9d of BM-OC culture. Again, no significant difference in cell survival was observed in NOD derived BM-OCs (Fig. 2I). Taken together, these data indicate a possible defect in the differentiation of NOD-derived osteoclasts, most likely involving the fusion of mononuclear cells into multinucleated osteoclasts.
NOD-derived osteoclasts have increased bone resorption capabilities in response to RANK-L stimulation
Although our differentiation data suggests overall smaller osteoclast size, this data does not address the bone resorbing function of these cells. In order to determine the bone resorbing capabilities of NOD-derived BM-OCs, these cells were seeded onto bovine bone slices and stimulated to resorb bone with RANK-L. To quantify the amount of bone resorbed, SEM and ImageJ analysis were performed where the total area resorbed and number of resorption pits by TRAP+ cells was determined (Fig. 3). NOD-derived BM-OCs were found to resorb more surface area in response to RANK-L stimulation compared to C57Bl/6-, Balb/c-, and NOR-derived cultures (Fig. 3A, B). In addition, the number of resorption pits was significantly higher in NOD-derived BM-OCs cultures compared to those from all other strains (Fig. 3A, E). In order to elucidate activity on a per cell basis, the total area resorbed and number of resorption pit data was stratified based on number of TRAP+ mononuclear cells (Fig. 3 C, F) and TRAP+ multinucleated cells (Fig. 3 D, G). Here again the total area resorbed and the number of resorption pits was significantly higher in NOD-derived TRAP+ mononuclear cells. On the other hand only the total area resorbed was significantly higher in NOD-derived TRAP+ multinuclear cells. Together these data suggest that irrespectively of the smaller size, more cells in NOD-derived osteoclast cultures resorbed a greater bone surface area.
Figure 3. NOD-derived osteoclasts have increased bone resorption capabilities in response to RANK-L stimulation.

BM-OCs were stimulated with RANK-L for 72hrs on bovine bone slices which were sputter-coated with gold and imaged with SEM. (A) Representative SEM and computer-quantified areas of resorption (blue) at 40x magnification. Image J software was used to determine (B- D) area of resorption and (E-G) number of resorption pits (light blue outlines = borders of resorption areas) normalized to (B, E)TRAP+ cells, (C, F) TRAP+ mononuclear cells and (D, G)TRAP+ multinucleated cells. C57Bl/6 (black bars, n=18), Balb/c (dark grey bars, n=16), NOR (light grey bars, n=16), NOD (white bars, n=16). *p value ≤ 0.05. One-way ANOVA with Dunn’s multiple correction.
NOD-derived osteoclasts degrade more type 1 collagen than controls via enhanced cathepsin K and MMP-9 secretion
During bone resorption, the organic portion of bone containing type I collagen is degraded in the resorption lacunae where vesicles containing collagen telopeptides are transcytosed out of the osteoclast and released into the extracellular milieu (Bar-Shavit, 2007). Therefore, in order to further quantify bone resorption, levels of intra- and extra-cellular collagen telopeptides were evaluated. Similar to our SEM data, RANK-L-stimulated NOD-derived BM-osteolcast cultures had significantly higher levels of collagen telopeptides than C57Bl/6-, Balb/c, and NOR-derived cultures (Fig. 4A, D, G) confirming higher bone resorption activity. Lysosomal cathepsins, such as cathepsin K, and matrix metalloproteinases such as MMP-9 can degrade type I collagen at an acidic pH created in the resorption lacunae (Vaananen and Laitala-Leinonen, 2008). Therefore, to investigate mechanisms associated with the observed increased collagen degradation, the amount of cathepsin K (Fig. 4B, E, H) and MMP-9 (Fig. 4C, F, I) in the same BM-OC cultures was quantified. Increased levels of cathepsin K and MMP-9 were detected in RANK-L-stimulated NOD-derived BM-OC cultures when compared to C57Bl/6-, Balb/c-, and NOR-derived cultures (Fig. 4B, C). Again, activity was evaluated based on nucleation, where collagen, cathepsin K and MMP-9 was significantly higher in NOD-derived TRAP+ mononuclear cells (Fig. 4D - F) with collagen, and MMP-9, but not cathepsin K being significantly higher in NOD-derived TRAP+ multinuclear cells (Fig. 4G - I). These data indicate that RANK-L stimulation of NOD-derived BM-OC results in increased cathepsin K and MMP-9 release leading to increased collagen degradation,, with the largest source of bone degradation being TRAP+ mononuclear osteoclasts.
Figure 4. NOD-derived osteoclasts degrade more type 1 collagen than controls via enhanced cathepsin K and MMP-9 secretion.

BM-OCs were stimulated with RANK-L for 72hrs on bovine bone slices. Supernatants were collected after 1% Triton X-100 solubilization and ELISA used to quantify: (A, D, G) collagen I telopeptide (B, E, H) cathepsin K (C, F, I) MMP-9 normalized to (A-C) TRAP+ cells, (D-F) TRAP+ mononuclear cells and (G-I) TRAP+ multinucleated cells. C57Bl/6 (black bars, n=18), Balb/c (dark grey bars, n=16), NOR (light grey bars, n=16), NOD (white bars, n=19). *p value ≤ 0.05. One-way ANOVA with Dunn’s multiple correction.
NOD-derived osteoclasts respond aberrantly to LPS
Inflammatory bone pathologies are often seen in individuals with T1D, including inflammatory arthritis and periodontitis-associated alveolar bone loss, where inflammatory mediators including bacterial components are abundant in these milieus (Hallmon and Mealey, 1992; Lalla et al., 2007; Mealey and Rose, 2008; Ryan et al., 2003; Somers et al., 2009; Teng, 2006). Thus, the effect of LPS on T1D-derived osteoclast function was evaluated. Osteoclasts were seeded onto bovine bone slices and stimulated to resorb bone with RANK-L in the presence of LPS. LPS-induced deactivation of osteoclast function was observed in C57Bl/6-, Balb/c-, and NOR-derived cultures indicated by a decrease in the area resorbed and collagen release as well as decreased cathepsin K and MMP-9 secretion (Fig. 5). However, this LPS-induced deactivation did not occur in the NOD-derived osteoclast cultures as shown by a lack of decrease in the area resorbed and collagen degradation (Fig. 5A, B). Interestingly, NOD-derived BM-osteoclasts also displayed an increase in LPS-responsiveness as measured by increases in cathepsin K and MMP-9 secretion compared to RANK-L stimulation alone (Fig. 5C, D). These data suggest that NOD-derived BM-OCs are not only unable to be deactivated by LPS but more importantly secrete elevated levels of bone resorbing mediators in the presence of LPS.
Figure 5. NOD-derived osteoclasts respond aberrantly to LPS.
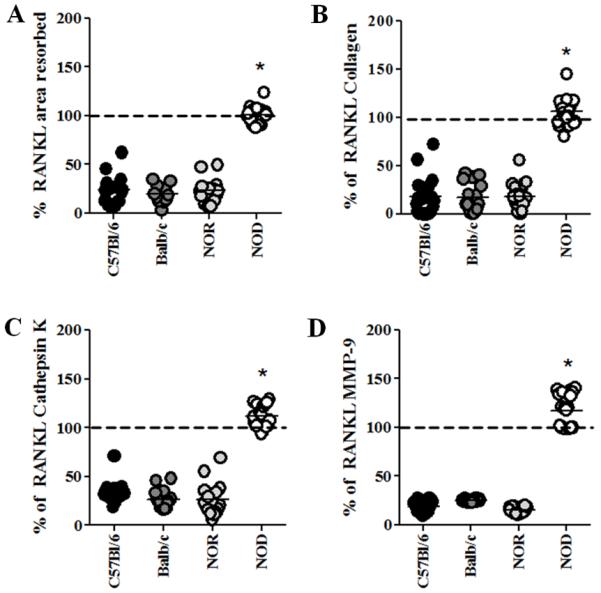
BM-OCs were stimulated with RANK-L in the presence or absence of E. coli LPS for 72hrs on bovine bone slices. Supernatants were collected after 1% Triton X-100 solubilization and bones were sputter-coated with gold and imaged with SEM. (A) Area of resorption (B) collagen I telopeptide (C) cathepsin K and (D) MMP-9 were evaluated using (A) SEM and (B-D) ELISA. Data are expressed as percent expression during RANK-L stimulation calculated by [(value in the presence of LPS/value in the absence of LPS) x 100] C57Bl/6 (black circles, n=18), Balb/c (dark grey circles, n=16), NOR (light grey circles, n=16), NOD (white circles, n=19). Dashed line = expression levels in the absence of LPS. *p value ≤ 0.05. NOD vs. all strains. One-way ANOVA with Dunn’s multiple correction.
NOD-derived osteoclasts secrete increased soluble osteoclastogenic mediators in response to LPS
Because osteoclast function is regulated by many soluble immune mediators, the cytokine and chemokine profile within BM-OC cultures from all strains was evaluated. Increased amounts of the hematopoietic growth factor GM-CSF and pro-inflammatory cytokines IL-1β and TNF-α as well as the osteoclast chemoattractants RANTES and IP-10 were elevated in BM-OCs cultures from all strains following LPS stimulation (Fig. 6). Similarly, the anti-osteoclastogenic mediator IL-10 was also elevated in BM-OCs following LPS stimulation (Fig. 6F, L). Interestingly, NOD-derived BM-OCs cultures presented with significantly higher levels of all mediators, with the exception of IL-10, compared to those found in C57Bl/6-, Balb/c-, and NOR-derived cultures when normalized to TRAP+ mononuclear cells (Fig. 6A-F). While a similar trend was observed when data was normalized for multinucleation (Fig. 6G-L), it was interesting to note that GM-CSF expression was no longer significantly higher (Fig. 6G) and IL-10 levels were significantly lower (Fig. 6L), in NOD-derived BM-OCs cultures. These data indicate an exacerbated pro-osteoclastic response to LPS by TRAP+ mononuclear NOD derived BM-OCs.
Figure 6. NOD-derived osteoclasts secrete increased soluble osteoclastogenic mediators in response to LPS.

BM-OCs were stimulated with RANK-L in the presence (+) or absence (-) of E. coli LPS for 72hrs on bovine bone slices. (A, G) GM-CSF (B, H) IL-1β (C, I) TNF-α, (D. J) RANTES, (E, K) IP-10 and (F, L) IL-10 levels were evaluated in the permeabilized supernatants using Milliplex technology. (A-F) Data shown as pg/mL normalized to TRAP+ mononuclear cells. (G-L) Data shown as pg/mL normalized to TRAP+ multinucleated cells. C57Bl/6 (black bars, n=13), Balb/c (dark grey bars, n=15), NOR (light grey bars, n=14), NOD (white bars, n=17). Open bars = absence of LPS, hatched bars = presence of LPS. *p value ≤ 0.05. One-way ANOVA with Dunn’s multiple correction.
NOD-derived osteoclasts respond aberrantly to inflammatory mediators
While pro-inflammatory cytokines such as TNF-α and IL-1β act on osteoblasts and activated T-cells to indirectly stimulate osteoclast differentiation, they also directly activate osteoclasts to resorb bone (Boyce et al., 2005; Boyce et al., 2006; Zou et al., 2001). Thus in order to determine if the higher levels of these mediators in NOD derived BM-OC cultures could be responsible for the abrogation of LPS-induced deactivation of osteoclast function, BM-OC cultures from all strains were subjected to a cocktail of pro-inflammatory cytokines and their resorptive function evaluated. As expected, increases in collagen release, cathepsin K and MMP-9 secretion were observed in BM-OC cultures from all strains in both the presence of the pro-inflammatory cytokine cocktail and in the absence of LPS (Fig. 7) when compared to those observed in the absence of the pro-inflammatory cytokine cocktail (Fig. 4). In the presence of a pro-inflammatory cytokine cocktail, NOD-derived BM-OCs display significantly increased resorptive function when compared to C57Bl/6-, Balb/c-, and NOR-cultures (Fig.7), similar to responsiveness in the absence of the pro-inflammatory cytokine cocktail (Fig. 4). In addition, the pro-inflammatory cytokine cocktail did not affect abrogation of LPS-induced deactivation in NOD-derived BM-OCs. On the other hand, C57Bl/6-, Balb/c- and NOR-derived cultures had significantly lower levels of collagen, cathepsin K and MMP-9 in the presence of LPS than in its absence even in the presence of a pro-inflammatory cytokine cocktail (Fig. 7). These data suggest that the pro-inflammatory milieu in NOD-derived BM-OCs is not solely responsible for the LPS-induced hyper-resorptive response observed.
Figure 7. Pro-inflammatory cytokines do not inhibit LPS-deactivation of OCs.
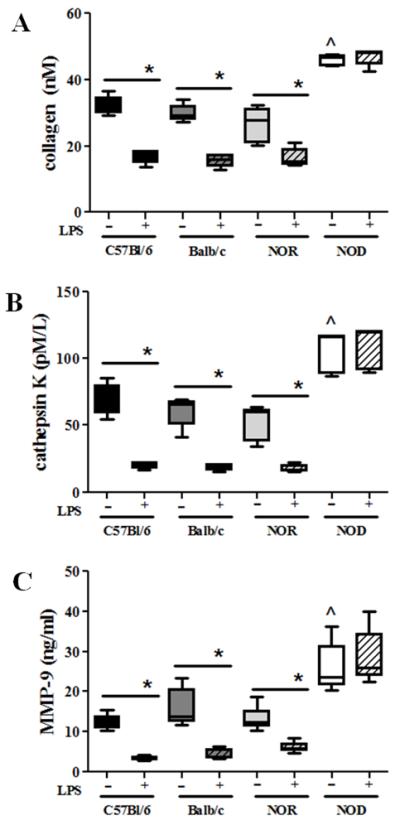
BM-OCs were stimulated with RANK-L in the presence or absence of E. coli LPS and a pro-inflammatory cytokine cocktail [TNF-α, IL-1β, and IL-6] for 72hrs on bovine bone slices. Supernatants were collected after 1% Triton X-100 solubilization and ELISA used to quantify: (A) collagen I telopeptide (B) cathepsin K (C) MMP-9. C57Bl/6 (black bars, n=5), Balb/c (dark grey bars, n=5), NOR (light grey bars, n=6), NOD (white bars, n=5). Open bars = absence of LPS, hatched bars = presence of LPS. *p value ≤ 0.05. ^p value indicates <0.05 NOD vs. all other experimental groups in the absence of LPS. One-way ANOVA with Dunn’s multiple correction
NOD-derived BM-OC conditioned media leads to increased bone resorption in control cultures
In order to determine if a soluble mediator was responsible for the abrogation of LPS-induced deactivation of osteoclast function observed in the NOD BM-OC cultures, conditioned media from LPS-free NOD BM-OC cultures was added to C57Bl/6-, Balb/c-, and NOR-derived cultures. As a control, conditioned media from LPS-free C57Bl/6 BM-OC cultures was added to Balb/c-, NOR and NOD-derived cultures. Again, collagen release, cathepsin K and MMP-9 secretion were used to evaluate osteoclast function. As expected, conditioned media from NOD BM-OC cultures caused an increase in resorption and osteoclastic enzyme production in C57Bl/6-, Balb/c-, and NOR-derived cultures when compared to conditioned media from C57Bl/6 BM-OC cultures (Fig. 8). Importantly, conditioned media from NOD BM-OC cultures were unable to inhibit LPS-induced deactivation in C57Bl/6-, Balb/c-, and NOR-derived cultures (Fig. 8). Similarly, conditioned media from C57Bl/6 BM-OC cultures were unable to inhibit LPS-induced deactivation in Balb/c-, and NOR-derived cultures. C57Bl/6 BM-OC conditioned media did not affect the inhibition of LPS-induced deactivation in NOD-derived BM-OC cultures (Fig. 8). Together these data suggest that inhibition of LPS-induced deactivation in NOD-derived BM-OC cultures is most likely due to NOD osteoclast responsiveness rather than excess or absence of an LPS-induced soluble mediator.
Figure 8. NOD-derived soluble mediators do not inhibit LPS-deactivation of OCs.
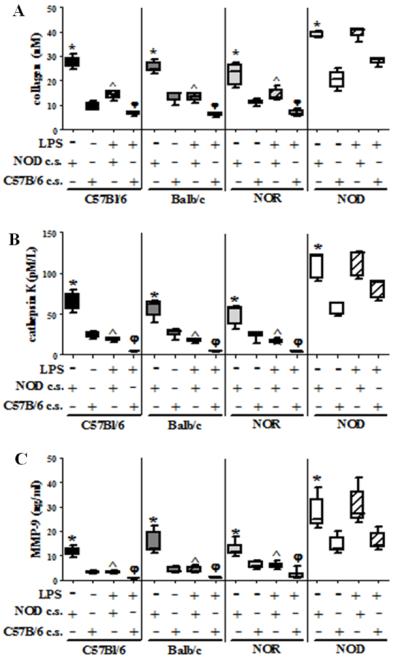
BM-OCs were stimulated with RANK-L in the presence or absence of E. coli LPS and either NOD conditioned media [NOD c.s.] or C57Bl/6 conditioned media [C57Bl/6 c.s.] for 72hrs on bovine bone slices. Supernatants were collected after 1% Triton X-100 solubilization and ELISA used to quantify: (A) collagen I telopeptide (B) cathepsin K (C) MMP-9. C57Bl/6 (black bars, n=5), Balb/c (dark grey bars, n=5), NOR (light grey bars, n=6), NOD (white bars, n=5). Open bars = absence of LPS, hatched bars = presence of LPS. *p value ≤ 0.05 NOD c.s vs C57Bl/6 c.s.; ^p value ≤ 0.05 no LPS vs LPS NOD c.s φp value ≤ 0.05 no LPS vs. LPS C57Bl/6 c.s. One-way ANOVA with Dunn’s multiple correction.
NOD -derived osteoclasts are from pre-diabetic/euglycemic mice
Bone resorption can be influenced by many factors including glucose concentration (Williams et al., 1997; Wittrant et al., 2008). Thus, in order to determine if hyperglycemia is contributing to the inhibition of LPS-induced deactivation of NOD-derived BM-OCs, the stage of T1D progression and blood glucose was evaluated. All strains had normal blood glucose at time of bone marrow harvest (Fig. 9A). Similarly, the histology of pancreata from NOD and NOR mice revealed little insulitis with approximately 75% of islets free of infiltration in the NOD model versus 90% in the NOR (Fig. 9B, C). Therefore, NOD mice were considered euglycemic and pre-diabetic at the time of marrow harvest, indicating that the events observed are not due to hyperglycemia or conditions associated with fulminant disease.
Figure 9. NOD-derived BM-OCs are from pre-diabetic/euglycemic mice.

(A) Blood glucose was measured at time of sacrifice. C57Bl/6 (black circles, n=10), Balb/c (dark grey circles, n=10), NOR (light grey circles, n=19), NOD (white circles, n=23) (B) Pancreata from NOR (n=5) and NOD (n=5) were fixed, embedded, sectioned, H&E stained and scored using light microscopy based on percentage of lymphocyte infiltration (insulitis). no insulitis = black, peri-insulitis = hatched, intra-insulitis = white. (C) Representative sections of scoring technique. (i) no insulitis (ii) peri-insulitis (iii) intra-insulitis.
Discussion
Individuals with T1D have increased incidence of inflammatory arthritis as well as osteoporosis, two diseases principally mediated by a dysregulation in bone remodeling (Somers et al., 2009; Thrailkill et al., 2005). Bone formation by osteoblasts is decreased in individuals with T1D which tips the balance of bone remodeling towards that of less bone deposition (Thrailkill et al., 2005). Here we demonstrate an osteoclast-specific contribution to altered bone remodeling, where T1D-derived osteoclasts are more osteoclastic in nature than T1D-free derived osteoclasts suggesting an additional tip in the balance to that of increased bone resorption.
While NOD BM-OC resorptive capability was heightened in response to RANK-L stimulation, NOD-derived BM-OCs were found to be smaller in size. In addition, NOD-derived BM-OCs cultures consistently contained a population of RANKL+CTRlo cells. Together these data indicate an aberrant maturation process. The fusion of mononuclear cells into multinucleated and giant cells was decreased suggesting an alteration in cell-fusion mechanism(s). Many surface receptors including DC-STAMP and MFR/SIRPα, are critical for fusion from mononucleated precursors into multinucleated cells. In addition, ADAM8 and ADAM12 which are disintegrins/metalloproteinases secreted prior to fusion, may be affected in this model (Oursler, 2010). Calcitonin, the ligand for CTR, is responsible for lowering calcium levels in the blood by inhibiting bone resorption by inducing morphological changes leading to osteoclast retraction (Naot and Cornish, 2008). In addition, while still controversial, some studies have demonstrated an effect of calcitonin in the fusion of osteoclast-precursors (Naot and Cornish, 2008). Thus it is plausible that expression or function of one or many of these molecules is altered in the NOD model leading to decreased fusion.
RANK-L stimulation of NOD-derived osteoclasts resulted in increased cathepsin K and MMP-9 secretion leading to increased collagen degradation. This suggests a hyper-responsiveness to RANK-L by NOD BM-OCs during homeostatic conditions, whereby augmented signaling leads to increased resorptive enzyme production (Cappellen et al., 2002; Mabilleau et al., 2008). In support of this theory, it has been shown by others that peripheral blood derived osteoclasts from diabetic (both type 1 and type 2) individuals were less sensitive to the soluble decoy receptor for RANKL, osteoprotegrin [OPG] than those derived from diabetes-free controls resulting in increased bone resorption, again indicating a heightened sensitivity in RANK signaling (Mabilleau et al., 2008; Poubelle et al., 2007). In addition, stratification of the resorption data indicates that TRAP+ mononuclear cells within the NOD-derived OC cultures are responsible for the augmented resorption.
Bacterial components such as LPS act on osteoblasts and activated T-cells to produce more RANK-L to stimulate osteoclasts to differentiate and resorb bone (Takahashi et al., 1994; Teng, 2006). However, LPS can also act on osteoclasts directly by inhibiting the differentiation of monocytic precursors into bone resorbing osteoclasts and bone resorption by mature osteoclasts (Liu et al., 2009; Takami et al., 2002). In addition, it has recently been demonstrated that osteoclasts have the capacity to phagocytose bacteria and act as a supporting immune cell (Li et al., 2010). While T1D-free derived osteoclasts resorb less bone in the presence of high amounts of LPS, they do produce pro-inflammatory cytokines and chemokines. This suggests a shunting of osteoclast precursors to that of an immune cell phenotype to help fight infection rather than towards mobilization to resorb bone. Interestingly, in addition to being more resorptive, NOD-derived osteoclasts were also more inflammatory in nature than the NOR, C57BL/6 or Balb/c models as indicative of increased soluble mediator secretion. Again, stratification of the data indicates that it is the TRAP+ mononuclear cells which have retained this inflammatory function.
Macrophages and monocytes (relatives of the osteoclast) from individuals with T1D secrete elevated levels of pro-inflammatory cytokines in response to LPS (Foss-Freitas et al., 2008; Salvi et al., 1997). Here NOD-derived osteoclasts also secreted elevated levels of the pro-inflammatory and pro-osteoclastic mediators, GM-CSF, RANTES, IP-10, TNF-α and IL-1β when stimulated with LPS compared to those observed in osteoclast control cultures. Where GM-CSF can mobilize osteoclast precursor release from the bone marrow, RANTES and IP-10 can act as chemokines to attract these precursors to the area of inflammation. TNF-α and IL-1β then augment osteoclast differentiation and function (Gillespie, 2007; Knowles and Athanasou, 2009; Takayanagi, 2009). In addition, our data describe for the first time that NOD derived BM-osteoclasts are refractory to LPS-induced inhibition of bone resorption. Thus one can envision that under a T1D environment, augmented secretion of these mediators can lead to a positive feedback loop where more osteoclasts are recruited to the site of inflammation where abrogation of LPS-induced inhibition leads to exacerbated bone resorption.
We found that neither addition of a pro-inflammatory cytokine cocktail nor addition of conditioned media from NOD BM-OC cultures led to inhibition of LPS-induced deactivation in our control cultures suggesting that soluble factors alone are not responsible for the aberrant LPS-responsiveness of NOD BM-OCs. Importantly, C57Bl/6 BM-OC conditioned media did not affect the inhibition of LPS-induced deactivation in NOD-derived BM-OC cultures suggesting that there is not an absence of a soluble mediator in NOD-derived BM-OC cultures responsible for LPS-induced deactivation of osteoclasts. Therefore, we hypothesize that it is the response of the NOD BM-OC to LPS and other mediators that is the cause of aberrant function rather than an excess or absence of soluble mediator expression. For instance, hyper-responsiveness in the TLR4, signaling pathway as indicated by increased translocation of NFκB has been described in macrophages and dendritic cells isolated from NOD mice (Poligone et al., 2002; Sen et al., 2003; Weaver et al., 2001). In addition, the receptors for TNF-α [TNFR] and RANK-L [RANK], which originate from the same super-family of receptors, also utilize the NFκB pathway and thus T1D-associated alteration in these signaling pathways could potentially alter the bone resorbing function of osteoclasts (Karin and Gallagher, 2009). These mechanisms are currently under investigation in our laboratory.
In addition to the inflammatory environment, bone resorption can be influenced by many factors including glucose concentration. For instance, Graves and colleagues have elegantly demonstrated in ligature, calvarial and bone fracture models (in the presence and absence of infection) that hyperglycemia and TNF-α affect fibroblast and osteoblast apoptosis which contributes to in vivo cartilage and bone loss in models of type 2 diabetes and hyperglycemia (Alblowi et al., 2009; Alikhani et al., 2007; Desta et al.,; Graves et al., 2005; He et al., 2004; Kayal et al., 2009; Kayal et al.,; Kayal et al., 2007; Liu et al., 2006; Liu et al., 2004; Lu et al., 2003; Santana et al., 2003; Siqueira et al.).While our data demonstrates an osteoclast-specific augmented function in the absence of hyperglycemic contributions, it is plausible that hyperglycemia may further contribute to osteoclast hyperactivity in T1D. Indeed, glucose is the primary energy source of the osteoclast and has been shown to augment osteoclast-mediated resorption via increases in V-ATPase expression (Larsen et al., 2005; Larsen et al., 2002). Furthermore, lack of insulin, now considered to be a bone anabolic agent, leads to decreased bone formation in patients with T1D (Thrailkill et al., 2005).
While many contributing factors to the inflammatory bone loss in patients with T1D have been surmised, the totality of osteoclast-mediated pathology was previously unknown. Dissecting osteoclast function and its role in T1D-associated bone pathologies can lead to adjunct treatment for patients with T1D that cannot respond to conventional anti-osteoclast therapies. In the present study, we have shown that a T1D genetic background leads to a hyper-reactive osteoclast phenotype which responds to bacterial components and pro-inflammatory cytokines in an aberrant fashion resulting in excessive bone degradation via enhanced cathepsin K and MMP-9 secretion concomitant with an increased expression of pro-osteoclastic soluble mediators.
Acknowledgements
This study was supported by the American Diabetes Association [1-08-JF-37and 7-11-CD-17], National Institutes of Health Training Grant [NIH/NIDCR 5T32DE007200], and the NIH Pre-doctoral NRSA Award [NIH/NIDCR 1F31DE021618-01A1].
The authors would like to acknowledge the osteoclast training by and intellectual contributions of Dr. Edgar Toro, DMD and Dr. Shannon L. Holliday from the Department of Orthodontics at the University of Florida.
Contract grant sponsor: American Diabetes Association; Contract grant number: 1-08-JF-37 and 7-11-CD-17
Contract grant sponsor: NIH/NIDCR; Contract grant number: 5T32DE007200
Contract grant sponsor: NIH/NIDCR; Contract grant number: 1F31DE021618-01A1
Footnotes
Author Contributions:
Dana L. Catalfamo – design, execution and interpretation of experiments presented in the manuscript, along with manuscript preparation.
Nadia L. Calderon – execution of data presented in Figures 4, 5, and 6 of the manuscript.
Scott W. Harden – design and training of data analysis used in Figure 3 of the manuscript.
Heather Sorenson – execution and interpretation of data presented in Figure 2 of the manuscript.
Kathleen G. Neiva – execution and interpretation of data presented in Figure 2 of the manuscript, along with manuscript editing.
Shannon M. Wallet – Principle Investigator overseeing and contributing to the design, execution and interpretation of the experiments presented in the manuscript, along with manuscript preparation and editing.
None of the authors have a conflict of interest.
References
- Alblowi J, Kayal RA, Siqueira M, McKenzie E, Krothapalli N, McLean J, Conn J, Nikolajczyk B, Einhorn TA, Gerstenfeld L, Graves DT. High levels of tumor necrosis factor-alpha contribute to accelerated loss of cartilage in diabetic fracture healing. The American journal of pathology. 2009;175(4):1574–1585. doi: 10.2353/ajpath.2009.090148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alikhani M, Alikhani Z, Boyd C, MacLellan CM, Raptis M, Liu R, Pischon N, Trackman PC, Gerstenfeld L, Graves DT. Advanced glycation end products stimulate osteoblast apoptosis via the MAP kinase and cytosolic apoptotic pathways. Bone. 2007;40(2):345–353. doi: 10.1016/j.bone.2006.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–485. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- Bar-Shavit Z. The osteoclast: a multinucleated, hematopoietic-origin, bone-resorbing osteoimmune cell. J Cell Biochem. 2007;102(5):1130–1139. doi: 10.1002/jcb.21553. [DOI] [PubMed] [Google Scholar]
- Botolin S, McCabe LR. Bone loss and increased bone adiposity in spontaneous and pharmacologically induced diabetic mice. Endocrinology. 2007;148(1):198–205. doi: 10.1210/en.2006-1006. [DOI] [PubMed] [Google Scholar]
- Boyce BF, Li P, Yao Z, Zhang Q, Badell IR, Schwarz EM, O’Keefe RJ, Xing L. TNF-alpha and pathologic bone resorption. Keio J Med. 2005;54(3):127–131. doi: 10.2302/kjm.54.127. [DOI] [PubMed] [Google Scholar]
- Boyce BF, Schwarz EM, Xing L. Osteoclast precursors: cytokine-stimulated immunomodulators of inflammatory bone disease. Curr Opin Rheumatol. 2006;18(4):427–432. doi: 10.1097/01.bor.0000231913.32364.32. [DOI] [PubMed] [Google Scholar]
- Cappellen D, Luong-Nguyen NH, Bongiovanni S, Grenet O, Wanke C, Susa M. Transcriptional program of mouse osteoclast differentiation governed by the macrophage colony-stimulating factor and the ligand for the receptor activator of NFkappa B. J Biol Chem. 2002;277(24):21971–21982. doi: 10.1074/jbc.M200434200. [DOI] [PubMed] [Google Scholar]
- D OG, Ireland D, Bord S, Compston JE. Joint erosion in rheumatoid arthritis: interactions between tumour necrosis factor alpha, interleukin 1, and receptor activator of nuclear factor kappaB ligand (RANKL) regulate osteoclasts. Ann Rheum Dis. 2004;63(4):354–359. doi: 10.1136/ard.2003.008458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Fattore A, Teti A, Rucci N. Osteoclast receptors and signaling. Arch Biochem Biophys. 2008;473(2):147–160. doi: 10.1016/j.abb.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Desta T, Li J, Chino T, Graves DT. Altered fibroblast proliferation and apoptosis in diabetic gingival wounds. Journal of dental research. 89(6):609–614. doi: 10.1177/0022034510362960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitrescu AL, Abd-El-Aleem S, Morales-Aza B, Donaldson LF. A model of periodontitis in the rat: effect of lipopolysaccharide on bone resorption, osteoclast activity, and local peptidergic innervation. J Clin Periodontol. 2004;31(8):596–603. doi: 10.1111/j.1600-051X.2004.00528.x. [DOI] [PubMed] [Google Scholar]
- Foss-Freitas MC, Foss NT, Rassi DM, Donadi EA, Foss MC. Evaluation of cytokine production from peripheral blood mononuclear cells of type 1 diabetic patients. Ann N Y Acad Sci. 2008;1150:290–296. doi: 10.1196/annals.1447.053. [DOI] [PubMed] [Google Scholar]
- Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- Gillespie MT. Impact of cytokines and T lymphocytes upon osteoclast differentiation and function. Arthritis Res Ther. 2007;9(2):103. doi: 10.1186/ar2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves DT, Naguib G, Lu H, Leone C, Hsue H, Krall E. Inflammation is more persistent in type 1 diabetic mice. Journal of dental research. 2005;84(4):324–328. doi: 10.1177/154405910508400406. [DOI] [PubMed] [Google Scholar]
- Grey A, Mitnick MA, Masiukiewicz U, Sun BH, Rudikoff S, Jilka RL, Manolagas SC, Insogna K. A role for interleukin-6 in parathyroid hormone-induced bone resorption in vivo. Endocrinology. 1999;140(10):4683–4690. doi: 10.1210/endo.140.10.7036. [DOI] [PubMed] [Google Scholar]
- Hallmon WW, Mealey BL. Implications of diabetes mellitus and periodontal disease. Diabetes Educ. 1992;18(4):310–315. doi: 10.1177/014572179201800409. [DOI] [PubMed] [Google Scholar]
- Hayman AR. Tartrate-resistant acid phosphatase (TRAP) and the osteoclast/immune cell dichotomy. Autoimmunity. 2008;41(3):218–223. doi: 10.1080/08916930701694667. [DOI] [PubMed] [Google Scholar]
- He H, Liu R, Desta T, Leone C, Gerstenfeld LC, Graves DT. Diabetes causes decreased osteoclastogenesis, reduced bone formation, and enhanced apoptosis of osteoblastic cells in bacteria stimulated bone loss. Endocrinology. 2004;145(1):447–452. doi: 10.1210/en.2003-1239. [DOI] [PubMed] [Google Scholar]
- Jiang J, Zuo J, Hurst IR, Holliday LS. The synergistic effect of peptidoglycan and lipopolysaccaride on osteoclast formation. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;96(6):738–743. doi: 10.1016/j.tripleo.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Karel Z. Contrast limited adaptive histogram equalization. Graphics gems IV: Academic Press Professional, Inc.; 1994. pp. 474–485. [Google Scholar]
- Karin M, Gallagher E. TNFR signaling: ubiquitin-conjugated TRAFfic signals control stop-and-go for MAPK signaling complexes. Immunol Rev. 2009;228(1):225–240. doi: 10.1111/j.1600-065X.2008.00755.x. [DOI] [PubMed] [Google Scholar]
- Kayal RA, Alblowi J, McKenzie E, Krothapalli N, Silkman L, Gerstenfeld L, Einhorn TA, Graves DT. Diabetes causes the accelerated loss of cartilage during fracture repair which is reversed by insulin treatment. Bone. 2009;44(2):357–363. doi: 10.1016/j.bone.2008.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayal RA, Siqueira M, Alblowi J, McLean J, Krothapalli N, Faibish D, Einhorn TA, Gerstenfeld LC, Graves DT. TNF-alpha mediates diabetes-enhanced chondrocyte apoptosis during fracture healing and stimulates chondrocyte apoptosis through FOXO1. J Bone Miner Res. 25(7):1604–1615. doi: 10.1002/jbmr.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayal RA, Tsatsas D, Bauer MA, Allen B, Al-Sebaei MO, Kakar S, Leone CW, Morgan EF, Gerstenfeld LC, Einhorn TA, Graves DT. Diminished bone formation during diabetic fracture healing is related to the premature resorption of cartilage associated with increased osteoclast activity. J Bone Miner Res. 2007;22(4):560–568. doi: 10.1359/jbmr.070115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khazai NB, Beck GR, Jr., Umpierrez GE. Diabetes and fractures: an overshadowed association. Current opinion in endocrinology, diabetes, and obesity. 2009;16(6):435–445. doi: 10.1097/MED.0b013e328331c7eb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles HJ, Athanasou NA. Canonical and non-canonical pathways of osteoclast formation. Histol Histopathol. 2009;24(3):337–346. doi: 10.14670/HH-24.337. [DOI] [PubMed] [Google Scholar]
- Lalla E, Cheng B, Lal S, Kaplan S, Softness B, Greenberg E, Goland RS, Lamster IB. Diabetes mellitus promotes periodontal destruction in children. J Clin Periodontol. 2007;34(4):294–298. doi: 10.1111/j.1600-051X.2007.01054.x. [DOI] [PubMed] [Google Scholar]
- Larsen KI, Falany M, Wang W, Williams JP. Glucose is a key metabolic regulator of osteoclasts; glucose stimulated increases in ATP/ADP ratio and calmodulin kinase II activity. Biochem Cell Biol. 2005;83(5):667–673. doi: 10.1139/o05-136. [DOI] [PubMed] [Google Scholar]
- Larsen KI, Falany ML, Ponomareva LV, Wang W, Williams JP. Glucose-dependent regulation of osteoclast H(+)-ATPase expression: potential role of p38 MAP-kinase. J Cell Biochem. 2002;87(1):75–84. doi: 10.1002/jcb.10252. [DOI] [PubMed] [Google Scholar]
- Lawlor KE, Campbell IK, O’Donnell K, Wu L, Wicks IP. Molecular and cellular mediators of interleukin-1-dependent acute inflammatory arthritis. Arthritis Rheum. 2001;44(2):442–450. doi: 10.1002/1529-0131(200102)44:2<442::AID-ANR63>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Li H, Hong S, Qian J, Zheng Y, Yang J, Yi Q. Crosstalk between the bone and immune systems: osteoclasts function as antigen-presenting cells and activate CD4+ and CD8+ T cells. Blood. 2010 doi: 10.1182/blood-2009-11-255026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Wang S, Zhang P, Said-Al-Naief N, Michalek SM, Feng X. Molecular mechanism of the bifunctional role of lipopolysaccharide in osteoclastogenesis. J Biol Chem. 2009;284(18):12512–12523. doi: 10.1074/jbc.M809789200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Bal HS, Desta T, Krothapalli N, Alyassi M, Luan Q, Graves DT. Diabetes enhances periodontal bone loss through enhanced resorption and diminished bone formation. Journal of dental research. 2006;85(6):510–514. doi: 10.1177/154405910608500606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Desta T, He H, Graves DT. Diabetes alters the response to bacteria by enhancing fibroblast apoptosis. Endocrinology. 2004;145(6):2997–3003. doi: 10.1210/en.2003-1601. [DOI] [PubMed] [Google Scholar]
- Lohoff M, Gessner A, Bogdan C, Rollinghoff M. The Th1/Th2 paradigm and experimental murine leishmaniasis. Int Arch Allergy Immunol. 1998;115(3):191–202. doi: 10.1159/000023900. [DOI] [PubMed] [Google Scholar]
- Lu H, Kraut D, Gerstenfeld LC, Graves DT. Diabetes interferes with the bone formation by affecting the expression of transcription factors that regulate osteoblast differentiation. Endocrinology. 2003;144(1):346–352. doi: 10.1210/en.2002-220072. [DOI] [PubMed] [Google Scholar]
- Mabilleau G, Petrova NL, Edmonds ME, Sabokbar A. Increased osteoclastic activity in acute Charcot’s osteoarthropathy: the role of receptor activator of nuclear factor-kappaB ligand. Diabetologia. 2008;51(6):1035–1040. doi: 10.1007/s00125-008-0992-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo K, Irie N. Osteoclast-osteoblast communication. Arch Biochem Biophys. 2008;473(2):201–209. doi: 10.1016/j.abb.2008.03.027. [DOI] [PubMed] [Google Scholar]
- McCabe LR. Understanding the pathology and mechanisms of type I diabetic bone loss. J Cell Biochem. 2007;102(6):1343–1357. doi: 10.1002/jcb.21573. [DOI] [PubMed] [Google Scholar]
- Mealey BL, Rose LF. Diabetes mellitus and inflammatory periodontal diseases. Curr Opin Endocrinol Diabetes Obes. 2008;15(2):135–141. doi: 10.1097/MED.0b013e3282f824b7. [DOI] [PubMed] [Google Scholar]
- Naot D, Cornish J. The role of peptides and receptors of the calcitonin family in the regulation of bone metabolism. Bone. 2008;43(5):813–818. doi: 10.1016/j.bone.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Oursler MJ. Recent advances in understanding the mechanisms of osteoclast precursor fusion. J Cell Biochem. 2010;110(5):1058–1062. doi: 10.1002/jcb.22640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paula FJ, Rosen CJ. Obesity, diabetes mellitus and last but not least, osteoporosis. Arquivos brasileiros de endocrinologia e metabologia. 54(2):150–157. doi: 10.1590/s0004-27302010000200010. [DOI] [PubMed] [Google Scholar]
- Poligone B, Weaver DJ, Jr., Sen P, Baldwin AS, Jr., Tisch R. Elevated NF-kappaB activation in nonobese diabetic mouse dendritic cells results in enhanced APC function. J Immunol. 2002;168(1):188–196. doi: 10.4049/jimmunol.168.1.188. [DOI] [PubMed] [Google Scholar]
- Poubelle PE, Chakravarti A, Fernandes MJ, Doiron K, Marceau AA. Differential expression of RANK, RANK-L, and osteoprotegerin by synovial fluid neutrophils from patients with rheumatoid arthritis and by healthy human blood neutrophils. Arthritis Res Ther. 2007;9(2):R25. doi: 10.1186/ar2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochazka M, Serreze DV, Frankel WN, Leiter EH. NOR/Lt mice: MHC-matched diabetes-resistant control strain for NOD mice. Diabetes. 1992;41(1):98–106. doi: 10.2337/diab.41.1.98. [DOI] [PubMed] [Google Scholar]
- Raggatt LJ, Partridge NC. Cellular and molecular mechanisms of bone remodeling. J Biol Chem. 2010;285(33):25103–25108. doi: 10.1074/jbc.R109.041087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan ME, Carnu O, Kamer A. The influence of diabetes on the periodontal tissues. J Am Dent Assoc. 2003;134(Spec No):34S–40S. doi: 10.14219/jada.archive.2003.0370. [DOI] [PubMed] [Google Scholar]
- Salvi GE, Collins JG, Yalda B, Arnold RR, Lang NP, Offenbacher S. Monocytic TNF alpha secretion patterns in IDDM patients with periodontal diseases. J Clin Periodontol. 1997;24(1):8–16. doi: 10.1111/j.1600-051x.1997.tb01178.x. [DOI] [PubMed] [Google Scholar]
- Santana RB, Xu L, Chase HB, Amar S, Graves DT, Trackman PC. A role for advanced glycation end products in diminished bone healing in type 1 diabetes. Diabetes. 2003;52(6):1502–1510. doi: 10.2337/diabetes.52.6.1502. [DOI] [PubMed] [Google Scholar]
- Sen P, Bhattacharyya S, Wallet M, Wong CP, Poligone B, Sen M, Baldwin AS, Jr., Tisch R. NF-kappa B hyperactivation has differential effects on the APC function of nonobese diabetic mouse macrophages. J Immunol. 2003;170(4):1770–1780. doi: 10.4049/jimmunol.170.4.1770. [DOI] [PubMed] [Google Scholar]
- Siqueira MF, Li J, Chehab L, Desta T, Chino T, Krothpali N, Behl Y, Alikhani M, Yang J, Braasch C, Graves DT. Impaired wound healing in mouse models of diabetes is mediated by TNF-alpha dysregulation and associated with enhanced activation of forkhead box O1 (FOXO1) Diabetologia. 53(2):378–388. doi: 10.1007/s00125-009-1529-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somers EC, Thomas SL, Smeeth L, Hall AJ. Are individuals with an autoimmune disease at higher risk of a second autoimmune disorder? Am J Epidemiol. 2009;169(6):749–755. doi: 10.1093/aje/kwn408. [DOI] [PubMed] [Google Scholar]
- Sternberg SR. Biomedical Image Processing. Computer. 1983;16(1):22–34. [Google Scholar]
- Takahashi N, Udagawa N, Tanaka S, Murakami H, Owan I, Tamura T, Suda T. Postmitotic osteoclast precursors are mononuclear cells which express macrophage-associated phenotypes. Dev Biol. 1994;163(1):212–221. doi: 10.1006/dbio.1994.1137. [DOI] [PubMed] [Google Scholar]
- Takami M, Kim N, Rho J, Choi Y. Stimulation by toll-like receptors inhibits osteoclast differentiation. J Immunol. 2002;169(3):1516–1523. doi: 10.4049/jimmunol.169.3.1516. [DOI] [PubMed] [Google Scholar]
- Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol. 2007;7(4):292–304. doi: 10.1038/nri2062. [DOI] [PubMed] [Google Scholar]
- Takayanagi H. Osteoimmunology and the effects of the immune system on bone. Nat Rev Rheumatol. 2009;5(12):667–676. doi: 10.1038/nrrheum.2009.217. [DOI] [PubMed] [Google Scholar]
- Teng YT. Protective and destructive immunity in the periodontium: Part 1--innate and humoral immunity and the periodontium. J Dent Res. 2006;85(3):198–208. doi: 10.1177/154405910608500301. [DOI] [PubMed] [Google Scholar]
- Thrailkill KM, Lumpkin CK, Jr., Bunn RC, Kemp SF, Fowlkes JL. Is insulin an anabolic agent in bone? Dissecting the diabetic bone for clues. Am J Physiol Endocrinol Metab. 2005;289(5):E735–745. doi: 10.1152/ajpendo.00159.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaananen HK, Laitala-Leinonen T. Osteoclast lineage and function. Arch Biochem Biophys. 2008;473(2):132–138. doi: 10.1016/j.abb.2008.03.037. [DOI] [PubMed] [Google Scholar]
- Weaver DJ, Jr., Poligone B, Bui T, Abdel-Motal UM, Baldwin AS, Jr., Tisch R. Dendritic cells from nonobese diabetic mice exhibit a defect in NF-kappa B regulation due to a hyperactive I kappa B kinase. J Immunol. 2001;167(3):1461–1468. doi: 10.4049/jimmunol.167.3.1461. [DOI] [PubMed] [Google Scholar]
- Williams JP, Blair HC, McDonald JM, McKenna MA, Jordan SE, Williford J, Hardy RW. Regulation of osteoclastic bone resorption by glucose. Biochem Biophys Res Commun. 1997;235(3):646–651. doi: 10.1006/bbrc.1997.6795. [DOI] [PubMed] [Google Scholar]
- Wittrant Y, Gorin Y, Woodruff K, Horn D, Abboud HE, Mohan S, Abboud-Werner SL. High d(+)glucose concentration inhibits RANKL-induced osteoclastogenesis. Bone. 2008;42(6):1122–1130. doi: 10.1016/j.bone.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, Hakim I, Tschoep K, Endres S, Bar-Shavit Z. Tumor necrosis factor-alpha mediates RANK ligand stimulation of osteoclast differentiation by an autocrine mechanism. J Cell Biochem. 2001;83(1):70–83. doi: 10.1002/jcb.1202. [DOI] [PubMed] [Google Scholar]