

Abstract
Molecularly targeted therapies have emerged as the leading theme in cancer therapeutics. Multi-cytotoxic drug regimens have been highly successful, yet many studies in targeted therapeutics have centered on a single agent. We investigated whether the Src/Abl kinase inhibitor dasatinib displays synergy with other agents in molecularly heterogeneous breast cancer cell lines. MCF-7, SKBR-3, and MDA-MB-231 display different signaling and gene signatures profiles due to expression of the estrogen receptor, ErbB2, or neither. Cell proliferation was measured following treatment with dasatinib ± cytotoxic (paclitaxel, ixabepilone) or molecularly targeted agents (tamoxifen, rapamycin, sorafenib, pan PI3K inhibitor LY294002, and MEK/ERK inhibitor U0126). Dose-responses for single or combination drugs were calculated and analyzed by the Chou-Talalay method. The drugs with the greatest level of synergy with dasatinib were rapamycin, ixabepilone, and sorafenib, for the MDA-MB-231, MCF-7, and SK-BR-3 cell lines respectively. However, dasatinib synergized with both cytotoxic and molecularly targeted agents in all three molecularly heterogeneous breast cancer cell lines. These results suggest that effectiveness of rationally designed therapies may not entirely rest on precise identification of gene signatures or molecular profiling. Since a systems analysis that reveals emergent properties cannot be easily performed for each cancer case, multi-drug regimens in the near future will still involve empirical design.
1. INTRODUCTION
Combination cytotoxic therapy has produced cures in adult and pediatric leukemias, lymphomas, and solid tumors as well as longstanding disease control for a number of other cancers.[1] However, cytotoxic therapy may result in severe toxicity and compromise in quality of life. By being more selective and less toxic, molecularly targeted therapy provides a new paradigm in rational cancer therapy.[2] Three principles drive the development of molecularly targeted therapy. Firstly, tumor cells depend on or are “addicted” to the activities of an oncogene, which provides an “Achilles’ heel” for a drug to target.[3] A second principle states that a combination of drugs is more effective than any single agent by preventing chemoresistance.[4] An emerging third principle advances that each cancer has its own signature.[5] The design of multi-drug regimens incorporating molecularly targeted agents amidst a growing range of novel therapeutics and in the context of unique cancer profiles poses a great challenge.[6] Also complicating the design of molecularly targeted therapy has been the revelations of previously unappreciated pathways, such as feedback upregulation of PI 3-kinase by rapamycin or activation of c-Raf by B-Raf inhibitors.[7; 8]
Dasatinib is an oral, Src/Abl tyrosine kinase inhibitor, first approved in 2006 by the Food and Drug Administration for use in patients with resistance or intolerance to prior therapy including imatinib in patients with Ph+ chronic myeloid leukemia (CML). Dasatinib also targets Src family kinases (SFK), which drives many different signaling pathways.[9] Aberrant SFK activity promotes the survival, proliferation, and metastases of many different human cancers, such as breast, colorectal, and prostate cancers.[10] We have previously reported that dasatinib inhibits cell progression by inducing G1 arrest and blocks migration in the highly invasive, triple-negative (ER-, PR-, Her2-) MDA-MB-231 breast cancer cell line.[11] By blocking the actions of either non-receptor or receptor tyrosine kinases, dasatinib exerts anti-cancer actions by promoting apoptosis or inhibiting proliferation, angiogenesis, invasion, or bone resorption.[12]
Despite supportive preclinical data, two single agent phase II studies showed limited responses to dasatinib in patients with advanced Her2-positive, hormone-receptor-positive, or triple negative breast cancers.[13; 14] We hypothesized that combining a specific molecular targeted drug with dasatinib can enhance efficacy in inhibiting cell growth of a specific breast cancer cell line according to the molecular profile of the given breast cancer cell type. To test this hypothesis, we evaluated dasatinib-containing regimens on breast cancer cell lines with different molecular profiles. Breast cancer cells may be distinguished by the presence or absence of estrogen receptor (ER), progesterone receptor (PR), or ErbB2 (Her2). We studied three cell lines with different receptor profiles: MDA-MB-231 (ER-, PR-, Her2-), MCF-7 (ER+, PR+, Her2-), and SK-BR-3 (ER-, PR-, Her2+). These three cell lines also possess different oncogene mutations (Table 1). MDA-MB-231 cells are highly sensitive to dasatinib, while MCF-7 cells are moderately sensitive and SK-BR-3 cells are resistant to dasatinib. [11; 15] These cell lines display different genetic, [16; 17] epigenetic,[18] and protein[19] expression patterns as well as single drug response profiles.[20] Surprisingly, our results showed that synergy between dasatinib and both cytotoxic and molecularly targeted agents were found in all cell lines. These results suggest that molecularly targeted agents, such as the multi-kinase inhibitor dasatinib, can have a broader role in cancer therapeutics and that the design of clinical trials of combination therapies will remain empiric.
Table 1.
Characteristics of signaling molecules in MDA-MB-231, MCF-7, and SK-BR-3 cell lines.
| Cell line | ER, PR expression | Her2 amplification | PI3K mutation | PTEN mutation | Ras mutation | Raf mutation |
|---|---|---|---|---|---|---|
| MDA-MB-231 | - | - | - | + | + | - |
| MCF-7 | + | - | + | - | - | - |
| SK-BR-3 | - | + | - | - | - | - |
2. MATERIALS AND METHODS
2.1 Cell Culture
MDA-MB-231 and MCF-7 cells were cultured in MEM media (Invitrogen) supplemented with 10% FBS, 2 mM glutamine (Invitrogen), 100 U/ml penicillin, 100 ug/ml streptomycin, 0.1 mM non-essential amino acids, and 10 mM HEPES buffer solution. SK-BR-3 cells were cultured in DMEM media (Invitrogen) supplemented with 10% FBS, 2 mM glutamine, 100 U/ml penicillin, and 100 ug/ml streptomycin. Cells were incubated in 37 C in 5% CO2. The cell lines were validated by short tandem repeat DNA fingerprinting, as reported elsewhere.[21]
2.2 Cell Proliferation Assay
Dose-responses were measured through MTT assay (ATCC #30-1010K) following the manufacturer’s instructions. Cells were seeded at a density of 3500 cells per well in 96-well plates for 24 hrs prior to treatment in growth medium with 10% FCS. After 24, 48, or 72 hrs of incubation with the indicated drugs or compounds, an MTT assay was performed with absorbance measured at 600 nm using a microplate reader (FLUOstar OPTIMA). Data analysis was performed with Excel software (Microsoft, Redmond, WA). Surface response models were generated using MATLAB software (The MathWorks, Inc, Natick, MA).
2.3 Cell Cycle Distribution
Cells were plated 5 × 105 cells in 100 mm dishes for 24 hrs prior to treatment in growth medium containing 10% FCS. Drugs were administered for 48 hrs. Cells were detached from the dish surface through trypsinization, fixed with 70% cold EtOH, and stored at -20 C. Cells were stained with 100 ug/ml RNase A (Sigma), 10 ug/ml propidium iodide (Sigma), and 0.1% Triton X-100 in PBS for 30 min at room temperature. Fluorescence was measured using a flow cytometer (BD LSR II). Cell cycle distribution was analyzed with FlowJo software (TreeStar, San Carlos, CA).
2.4 Mathematical Analysis
The dose-response of the drug combinations were modeled using the Median-Effect Equation, a generalized equation unifying the Hill, Scatchard, Michaelis-Menten, and Hasselbalch equations to succinctly describe the dose-effect relationship.[22] The Median-Effect Equation provides a way to assess the dose-response of a substance in a given population of cells:
where D is the dose, fa is fraction affected, fu is fraction unaffected, IC50 is median-effect dose, and m is the slope or kinetic order. Taking the logarithm converts this equation into a linear form that can be easily applied to linear regression analysis:
From obtaining a small collection of data points and using regression analysis, IC50 values can be accurately and rapidly interpolated from dose-response curves modeled using the Median-Effect Equation.
Combination index (CI) values were calculated through the Chou-Talalay method.[22] The CI provides a quantitative value for synergy and is given by:
for two mutually exclusive drugs. CI values less than 1.0 indicate synergy, with values closer to zero representing increasing synergy.
A surface response model provides a drug response landscape between two drugs and is useful in predicting the effects of a combination of drugs at various ratios. In this study, drug combinations were performed in a 1-to-1 ratio. Since a 1-to-1 ratio of drugs may not generally be the most efficacious combination, response surface models were generated to simulate the dose-response at all ratios of a two drug combination. The drug response landscapes were modeled using the following equation:[23]
The maximal effect is represented by Emax, which is 1 since ideally 100% of the cells become affected with increasing drug concentration. The coefficient α is a factor for the interaction between the two drugs, with α > 0 indicating synergy, α = 0 additivity, and α < 0 antagonism. This equation assumes that the response of each drug has the same kinetic order, n, which measures the steepness of the effect. However, each drug in actuality possesses a different kinetic order. Therefore, the model will provide a more accurate representation of the interaction between drugs with relatively similar kinetic orders. Matlab code for generating surface response plots is provided in Supplemental Text.
3. RESULTS
To test our hypothesis, we chose to study the effects of dasatinib in combination with cytotoxic drugs or molecularly targeted agents on three breast cancer cell lines, which are well-characterized in the variability in their gene expression, protein expression, and cancer phenotype.[24; 25] Single agent dose-response and combination dose-responses were experimentally derived and are shown in Figures 1 and 2. Table 2 summarizes the calculated IC50 values for single agents. Not surprisingly, the cytotoxic drug paclitaxel was the most potent for all three cell lines studied. The IC50 and CI values of combinations with dasatinib are shown in Table 3. The combination of dasatinib and rapamycin was observed to be strongly synergistic in both MDA-MB-231 and MCF-7 cell lines.
Figure 1. MTT assay of MDA-MB-231, MCF-7, and SK-BR-3 cells treated with single drugs for 48 hours.
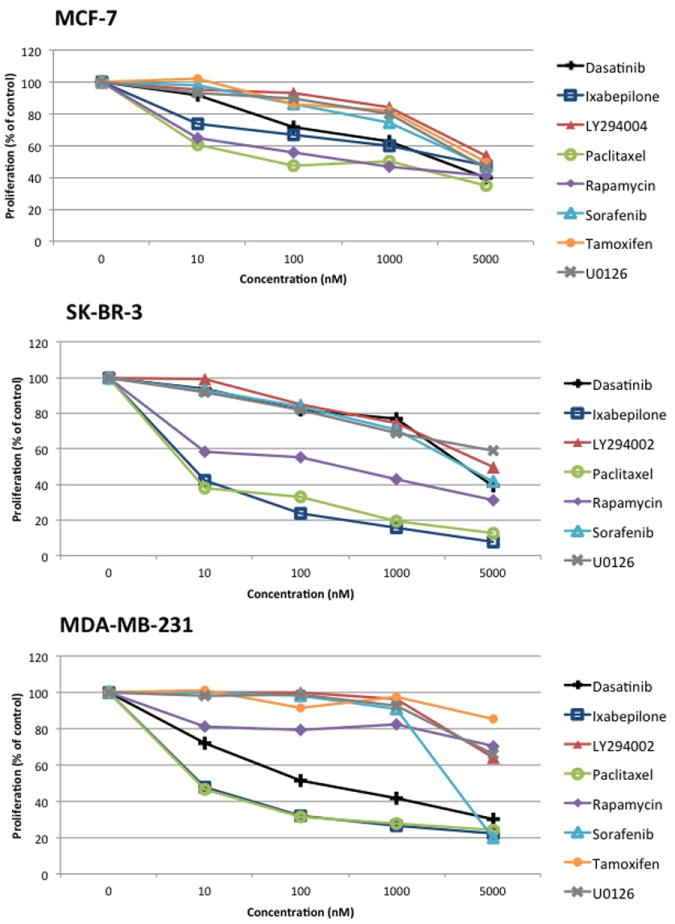
Percent proliferation is calculated relative to untreated controls. Data points represent averages from triplicates from a representative experiment, which were done at least three times independently.
Figure 2. MTT assay of MDA-MB-231, MCF-7, and SK-BR-3 cells treated with drug combinations with dasatinib for 48 hours.
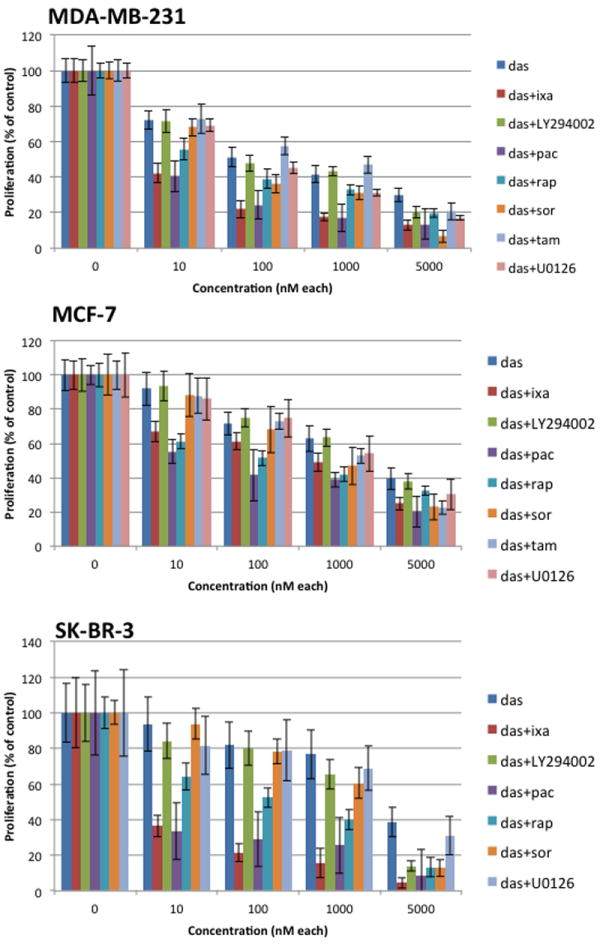
Drug combinations were administered in a 1:1 ratio. Percent proliferation was calculated relative to untreated controls. Data points represent averages from triplicates. Error bars indicate standard deviation.
Table 2.
IC50 values of single drugs in MDA-MB-231, MCF-7, and SK-BR-3 cell lines
| Dasatinib* | Ixabepilone | LY294002 | Paclitaxel | Rapamycin | Sorafenib | Tamoxifen | U0126 | |
|---|---|---|---|---|---|---|---|---|
| MB-231 | ||||||||
| IC50 (nM) | 230 | 3.7 | 6900 | 1.9 | 6.9E10 | 2700 | 8.0E7 | 5.6E4 |
| MCF-7 | ||||||||
| IC50 (nM) | 2100 | 5300 | 1.8E4 | 160 | 480 | 4000 | 9900 | 9600 |
| SK-BR-3 | ||||||||
| IC50 (nM) | 4000 | 3.8 | 3500 | 2.2 | 130 | 3800 | --- | 1.2E4 |
The IC50 for dasatinib was averaged throughout all of the combination experiments. Each combination experiment was plated in triplicate with single dose dasatinib, single dose of another drug, and 1:1 combination of dasatinib plus the other drug.
Table 3.
IC50 and CI values of 1:1 drug combinations for MDA-MB-231, MCF-7, and SK-BR-3 cell lines.
| das* | das+ixa | das+LY294002 | das+pac | das+rap | das+sor | das+tam | das+U0126 | |
|---|---|---|---|---|---|---|---|---|
| MB-231 | ||||||||
| IC50 (nM) | 230 | 1.5 | 140 | 1.3 | 24 | 50 | 240 | 80 |
| CI at IC50 | NA | 0.41 | 0.73 | 0.71 | 0.11 | 0.29 | 0.76 | 0.35 |
| MCF-7 | ||||||||
| IC50 (nM) | 2100 | 270 | 2000 | 30 | 130 | 550 | 680 | 1000 |
| CI at IC50 | NA | 0.19 | 0.74 | 0.21 | 0.32 | 0.87 | 0.44 | 0.81 |
| SK-BR-3 | ||||||||
| IC50 (nM) | 4000 | 3.0 | 660 | 1.3 | 88 | 670 | --- | 2100 |
| CI at IC50 | NA | 0.80 | 0.45 | 0.62 | 0.70 | 0.38 | --- | 0.70 |
The IC50 for dasatinib was averaged throughout all of the combination experiments. Each combination experiment was plated in triplicate with single dose dasatinib, single dose of another drug, and 1:1 combination of dasatinib plus the other drug.
3.1 Effects of dasatinib-based treatment on ER+ breast cancer cells
Derived from invasive breast ductal carcinoma, MCF-7 cells display estrogen and progesterone receptors.[26] MCF-7 cells were found to be moderately sensitive to dasatinib with an IC50 of 2100 nM. In combinations with other drugs, dasatinib was most synergistic with the cytotoxic molecules ixabepilone and paclitaxel with CI of 0.19 and 0.21 respectively. The molecularly targeted agents sorafenib, PI3K inhibitor, and MEK/ERK inhibitor were only slightly synergistic when combined with dasatinib (Figures 2 and 3).
Figure 3. Compilation of CI values @ IC50 for different drug combinations in MCF-7, MB-231, and SK-BR-3 cells.
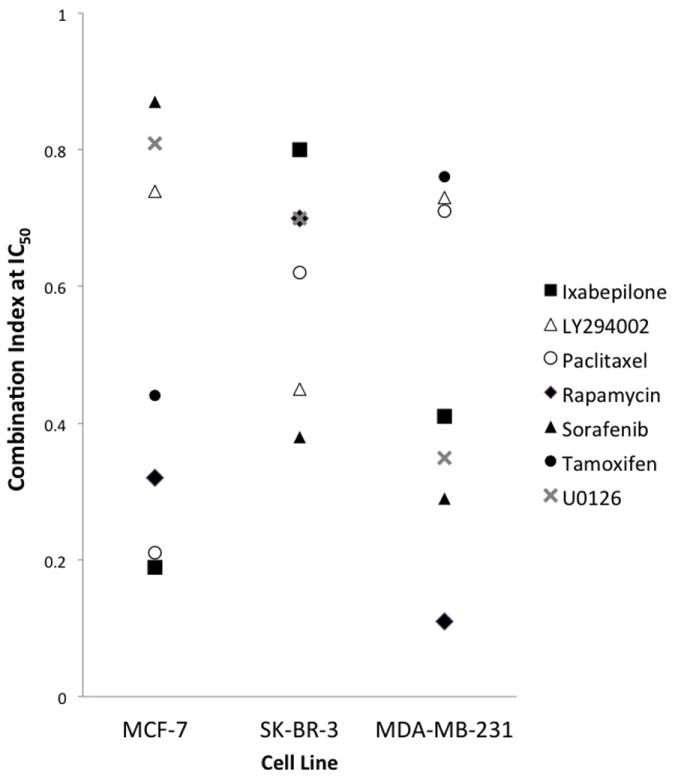
The CI values provide the following interpretations: <0.1 very strong synergism; 0.1 to 0.3 strong synergism, 0.3 to 0.7 synergism, 0.7 to 0.85 moderate synergism, 0.85 to 0.9 slight synergism, 0.9 to 1.10 nearly additive.
MCF-7 cells will respond to estrogen with cell proliferation. As a result, the estrogen receptor antagonist tamoxifen was expected to impose a large inhibitory growth response. However, the observed IC50 value for single agent tamoxifen was large (IC50 = 9900 nM) and had only a marginal inhibitory effect in the proliferation of MCF-7 cells. This observation can be attributed to the absence of the hormone estrogen in our cultured growth media. Therefore, tamoxifen was essentially inhibiting a receptor for a hormone that was nonexistent in the surrounding environment. The fact that synergy was observed in the combination of dasatinib and tamoxifen is intriguing because of crosstalk between Src and ER signaling pathways, a mechanism demonstrated previously but perhaps underappreciated.[27]
3.2 Effects of dasatinib-based treatment on Her2+ breast cancer cells
The SK-BR-3 cell line displays high levels of epidermal growth factor receptor, erbB2 (Her2), and c-Myc.[28] Once known to be an ominous subtype of breast cancer, Her2+ breast cancers have now become radically exploited by their receptor characteristics and successfully treated by targeted anti-Her2 antibodies, such as trastuzumab. Before the advent of Her2-specific targeted drugs, very few successful therapies existed for Her2+ breast cancers. Our findings indicate other potential therapeutic options for Her2+ breast cancers. The cytotoxic molecules, ixabepilone and paclitaxel, were very potent inhibitors of cell proliferation of SK-BR-3 cells with IC50 of 3.8 nM and 2.2 nM respectively. Of the three cell lines examined in this study, dasatinib inhibited growth least effectively in SK-BR-3 cells with an IC50 of 4000 nM. This finding is consistent with studies that have shown that SK-BR3 cells are resistant to dasatinib for growth inhibition. Nonetheless, synergy was observed in all combinations with dasatinib (Table 3). Synergy was greatest when dasatinib was combined with the signaling pathway inhibitor sorafenib, resulting in a CI of 0.38. Moderate synergism was seen for combinations with ixabepilone, paclitaxel, rapamycin, and LY294002, while tamoxifen was not tested since SK-BR-3 cells lack estrogen receptors. Although the combination of dasatinib and U0126 was calculated to be slightly synergistic, Figure 2 shows that there was no statistically significant difference between single dasatinib and dasatinib+U0126 at all tested drug doses.
3.3 Effects of dasatinib-based treatment on triple-negative breast cancer cells
Originating from a breast cancer patient with malignant pleural effusion,[29] MDA-MB-231 cells lack expression for ER, PR, and Her2. Triple-negative breast cancers are a relatively aggressive and invasive subtype of breast cancer with relatively few available targeted treatment therapies to date. For instance, tamoxifen (IC50 ≫ 5000 nM) had no inhibitory effect on the growth and proliferation of MDA-MB-231 cells due to their lack of estrogen receptors. MDA-MB-231 cells were found to be sensitive to dasatinib with an IC50 of 230 nM. As with the other cell lines in this study, Table 3 shows that all drug combinations with dasatinib synergistically inhibited cell proliferation in MDA-MB-231 cells. Dasatinib displayed strong synergistic interactions with other signaling pathway inhibitors (rapamycin, sorafenib) and moderate synergism with cytotoxic molecules (ixabepilone, paclitaxel). Flow cytometric analysis of cell cycle distribution following dasatinib + rapamycin or dasatinib + sorafenib treatment demonstrated no synergy in increasing the number of cells in G1 arrest (Supplemental Figure 1). This is probably due to a maximal response in G1 arrest induced by dasatinib
3.4 Graphical and mathematical analysis of drug synergy with dasatinib
The CI values were plotted side-by-side for MD-MB-231, MCF-7, and SK-BR-3 cell lines to look for any general trends of synergism (Figure 3). While none of the drug combinations exhibited comparable levels of synergism across all three lines, there were similar CI values (within 0.1) between the MCF-7 and SK-BR-3 lines for several combinations, including paclitaxel, the PI3K inhibitor LY294002, sorafenib and tamoxifen, a fact that may suggest similarities in aberrant signaling pathways between the lines that should be investigated further.
Interestingly, the strongest synergy (CI = 0.11) was observed for the combination of dasatinib and rapamycin in MDA-MB-231 cells. In addition, the surface response model for this combination possessed the greatest alpha value (α ≫ 0), with α > 0 signifying synergy as shown in Supplemental Table 1. Single therapy rapamycin on MDA-MB-231 cell proliferation was found to be unresponsive (IC50 ≫ 5000 nM), which agrees with previous studies.[30; 31] This suggests that mTOR plays a minimal effect on the growth and proliferation of triple-negative breast cancers. However, the combination of dasatinib plus rapamycin drastically lowered the IC50 down to 24 nM, indicating that mTOR signaling may instead play more of a role as a salvage pathway for growth and proliferation of triple-negative breast cancers. For the combination of dasatinib and rapamycin, calculating the CI value is useful in providing insight into the feedback interaction mechanism between the Abl/Src and mTOR signaling pathways. mTOR signaling may be further induced as the Abl/Src signaling becomes compromised. Thus, simultaneously inhibiting both pathways explains the synergistic effects of dasatinib plus rapamycin.
Mathematical analysis through the CI and surface response models are helpful in determining the additive or multiplicative benefits of adding another drug to an existing therapy. MDA-MB-231 cells were observed to be very sensitive to the cytotoxic drugs ixabepilone and paclitaxel, both with IC50 < 5nM. Adding incremental amounts of dasatinib to an existing ixabepilone based therapy showed little benefit as opposed to adding incremental amounts of ixabepilone to dasatinib (Figure 4). These data suggest that it might be clinically more effective to design therapy based on a molecular targeted agent with few side effects and then add a cytotoxic drug that carries more toxicity. Following the law of diminishing returns (also known as the law of increasing relative cost), mathematical analysis can predict the most efficacious drug combinations as well as the most beneficial ratios of drugs.[32]
Figure 4. Surface response model illustrate profound changes in dose-response of ixabepilone with dasatinib in MDA-MB-231 cells.
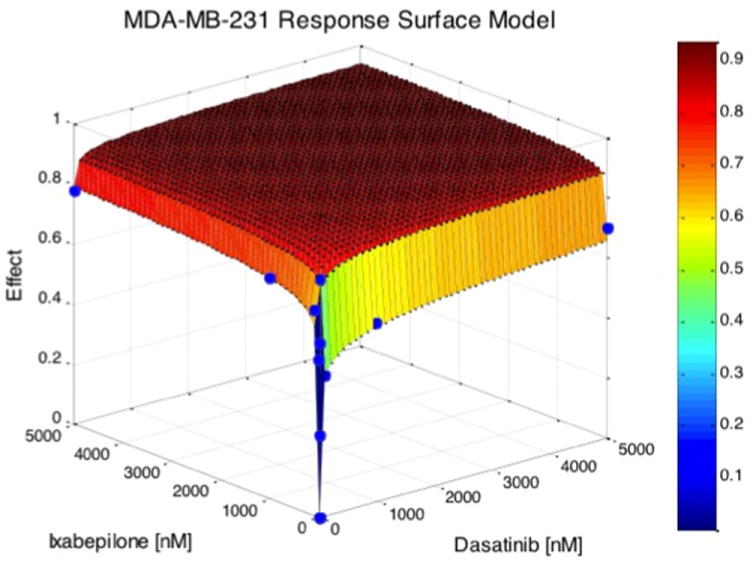
Two-dimensional projections of the surface response contours were generated to better visualize the dose-response relationship for the drug combinations tested (Supplemental Figure 2). In these plots, different colors correspond to different degrees of growth inhibition. For instance, when dasatinib is dosed at 2000 nM, then the addition of a PI 3-Kinase inhibitor does not add to growth inhibition in MD-MB-231 cells (Supplemental Figure 2E). Because the number of drug combinations and their dosages becomes astronomical, experimental determination of surface response contours can facilitate clinical trial design of combinations.
4. DISCUSSION
Despite major advances in the identification of oncogenes and components of signaling pathways and molecular classification of cancers, molecularly targeted therapy has had limited success in oncology. Genetic heterogeneity of tumors and their plasticity will provide obstacles to new multi-drug therapies. Here, we have shown that dasatinib synergizes with both molecularly targeted agents and cytotoxic drugs in molecularly heterogeneous breast cancer cells. Recent studies suggest that dasatinib also synergizes with activators of p53[33], or inhibitors of Chk1[34], androgen receptor[35], and HSP90[36]. The CI value provides a measure of synergy at a certain observed level of effect. In this study, the observed effects were compared at the median-effect dose. Response surface models were also generated to substantiate the observed synergy calculated through the combination index. Our data suggest that molecularly targeted therapies can still be discovered via traditional empirical methods and their use need not be limited by specific tumor molecular profile.
A principle in systems biology, such as highly optimized theory, predicts the existence of critical hubs.[37] As applied to cancer therapeutics, this principle does not take into consideration drug resistance due to cancer cells displaying genetic instability or recruiting salvage pathways.[38; 39; 40] While resistance due to acquired mutations in either the kinase or ATP binding domains may be overcome by synthesizing new inhibitors, a more vexing problem is the recruitment of salvage pathways. For example, the Src kinase Lyn mediates growth and survival of imatinib-resistant CML cells by both providing tyrosine kinase activity and upregulating Bcl-2 anti-apoptotic pathways.[41] We observed that dasatinib when combined with PI 3-kinase pathways inhibitors (PI 3-Kinase or mTOR) or MEK/ERK inhibitors showed synergy. Thus, the plasticity of a cancer cell belies the principle of therapeutic targeting of a specific set of hubs. Whether multi-drug regimens incorporating molecularly targeted agents can more successfully evade acquired resistance remains to be demonstrated.
Clinical testing of novel combination therapies must overcome multiple obstacles, one of which rests on compelling biologic rationale for their use.[6] A number of trials involving dasatinib in combination with other agents are ongoing for ER+ breast cancer, Her2+ breast cancer, prostate cancer, melanoma, non-small cell lung cancer, colorectal cancer, and head/neck cancer.[12] Although identification of a Src oncogenic signaling pathway can predict dasatinib sensitivity,[42] it inhibits a much wider range of receptor and non-receptor tyrosine kinases. Our results challenge the notion that effectiveness of rationally designed therapies must be guided by the precise identification of gene signatures or cancer fingerprints. The rationale for combining a particular set of molecularly targeted agents may be elusive, unpredictable, or even counter-intuitive.[43; 44] Since systems analysis that reveals emergent properties cannot yet be easily performed for each cancer case, design of multi-drug regimens will still involve empiricism in the foreseeable future. One objective of systems biology is to map signaling pathways and identify vital nodes or hubs as potential targets for small molecule inhibitors. Experimental evidence bearing out rational combinations of molecularly targeted agents against oncogene addiction pathways has been restricted to a few particularly strong cases, still far from supporting a generalizable concept. The demonstration of antagonistic drug combinations that prevent drug resistance and treatment failure provides another example of counter-intuitive multi-drug design. A brute-force exploration of all possible combinations is ideal but cost-prohibitive, necessitating the use of algorithmically guided screens. Zinner et al have employed hill-climbing algorithms based on multi-generational iterative searches of the combinatorial space to increase the efficiency and lower the cost of finding such combinations.[44]
Progress in treating cancers has come from empiric design of combination therapies. Their mechanism of action was to prevent cell division based on the use of genotoxic drugs to target either DNA or microtubules. With the discovery of oncogenes, the therapeutic goal has shifted to blocking oncogene addiction to growth, especially those involving kinases. With few exceptions, chiefly imatinib in chronic phase CML or gastrointestinal stromal tumors and trastuzumab in Her2+ breast cancer, molecularly targeted therapy has had only limited or transient non-curative responses. This may be largely due to the multivariate components of signaling networks driving surivival/apoptosis decision points.[45; 46] An immediate benefit, however, is that development of molecularly targeted agents expands the number of anti-cancer drugs and broadens the mechanisms of actions. With dasatinib as an example, this class of drugs shows synergy with a wide range of other molecularly targeted agents as well as cytotoxic drugs. Co-culturing breast cancer cells with stromal cells may reveal further dasatinb-based synergy.[47]The sheer number of new drugs entering trials is staggering, and their combinatorial permutations numbing. Emerging cancer therapies will remain combinational and largely empirically designed, yet greater computational power and newer in silico methods[48; 49] will help us find optimal combinations more economically, effectively, and expeditiously. These methods include new or improved search algorithms based upon hill-climbing methods[44] used to find combinations in a noisy combinatorial landscape that maximize a pre-defined fitness function. Such tools will be useful in directing the exploration of new drug cocktails when exhaustive screens of all combinations are impossible.[50]
Supplementary Material
Acknowledgments
Bristol-Myers Squibb Pharmaceuticals provided dasatinib, ixabepilone, and paclitaxel. This work was supported in part by a grant from Bristol-Myers Squibb and NIH RO1CA108922 to S.J.C.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chabner BA, Roberts TG., Jr Timeline: Chemotherapy and the war on cancer. Nat Rev Cancer. 2005;5:65–72. doi: 10.1038/nrc1529. [DOI] [PubMed] [Google Scholar]
- 2.Druker BJ, David A. Karnofsky Award lecture. Imatinib as a paradigm of targeted therapies. J Clin Oncol. 2003;21:239s–245s. doi: 10.1200/JCO.2003.10.589. [DOI] [PubMed] [Google Scholar]
- 3.Weinstein IB, Begemann M, Zhou P, Han EK, Sgambato A, Doki Y, Arber N, Ciaparrone M, Yamamoto H. Disorders in cell circuitry associated with multistage carcinogenesis: exploitable targets for cancer prevention and therapy. Clin Cancer Res. 1997;3:2696–2702. [PubMed] [Google Scholar]
- 4.Coldman AJ, Goldie JH. Variation in growth parameters and their effect on the acquisition of drug resistance. Prog Clin Biol Res. 1986;223:103–111. [PubMed] [Google Scholar]
- 5.Liotta L, Petricoin E. Molecular profiling of human cancer. Nat Rev Genet. 2000;1:48–56. doi: 10.1038/35049567. [DOI] [PubMed] [Google Scholar]
- 6.Woodcock J, Griffin JP, Behrman RE. Development of novel combination therapies. N Engl J Med. 2011;364:985–987. doi: 10.1056/NEJMp1101548. [DOI] [PubMed] [Google Scholar]
- 7.Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007;26:1932–1940. doi: 10.1038/sj.onc.1209990. [DOI] [PubMed] [Google Scholar]
- 8.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kopetz S, Shah AN, Gallick GE. Src continues aging: current and future clinical directions. Clin Cancer Res. 2007;13:7232–7236. doi: 10.1158/1078-0432.CCR-07-1902. [DOI] [PubMed] [Google Scholar]
- 10.Kim LC, Song L, Haura EB. Src kinases as therapeutic targets for cancer. Nat Rev Clin Oncol. 2009;6:587–595. doi: 10.1038/nrclinonc.2009.129. [DOI] [PubMed] [Google Scholar]
- 11.Pichot CS, Hartig SM, Xia L, Arvanitis C, Monisvais D, Lee FY, Frost JA, Corey SJ. Dasatinib synergizes with doxorubicin to block growth, migration, and invasion of breast cancer cells. Br J Cancer. 2009;101:38–47. doi: 10.1038/sj.bjc.6605101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Montero JC, Seoane S, Ocana A, Pandiella A. Inhibition of Src Family Kinases and Receptor Tyrosine Kinases by Dasatinib: Possible Combinations in Solid Tumors. Clin Cancer Res. 2011 doi: 10.1158/1078-0432.CCR-10-2616. [DOI] [PubMed] [Google Scholar]
- 13.Finn RS, Bengala C, Ibrahim N, Roche H, Sparano J, Strauss LC, Fairchild J, Sy O, Goldstein LJ. Dasatinib as a Single Agent in Triple-Negative Breast Cancer: Results of an Open-Label Phase 2 Study. Clin Cancer Res. 2011;17:6905–6913. doi: 10.1158/1078-0432.CCR-11-0288. [DOI] [PubMed] [Google Scholar]
- 14.Mayer EL, Baurain JF, Sparano J, Strauss L, Campone M, Fumoleau P, Rugo H, Awada A, Sy O, Llombart-Cussac A. A Phase 2 Trial of Dasatinib in Patients with Advanced HER2-Positive and/or Hormone Receptor-Positive Breast Cancer. Clin Cancer Res. 2011;17:6897–6904. doi: 10.1158/1078-0432.CCR-11-0070. [DOI] [PubMed] [Google Scholar]
- 15.Finn RS, Dering J, Ginther C, Wilson CA, Glaspy P, Tchekmedyian N, Slamon DJ. Dasatinib, an orally active small molecule inhibitor of both the src and abl kinases, selectively inhibits growth of basal-type/“triple-negative” breast cancer cell lines growing in vitro. Breast Cancer Res Treat. 2007;105:319–326. doi: 10.1007/s10549-006-9463-x. [DOI] [PubMed] [Google Scholar]
- 16.Heiser LM, Wang NJ, Talcott CL, Laderoute KR, Knapp M, Guan Y, Hu Z, Ziyad S, Weber BL, Laquerre S, Jackson JR, Wooster RF, Kuo WL, Gray JW, Spellman PT. Integrated analysis of breast cancer cell lines reveals unique signaling pathways. Genome Biol. 2009;10:R31. doi: 10.1186/gb-2009-10-3-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lapuk A, Marr H, Jakkula L, Pedro H, Bhattacharya S, Purdom E, Hu Z, Simpson K, Pachter L, Durinck S, Wang N, Parvin B, Fontenay G, Speed T, Garbe J, Stampfer M, Bayandorian H, Dorton S, Clark TA, Schweitzer A, Wyrobek A, Feiler H, Spellman P, Conboy J, Gray JW. Exon-level microarray analyses identify alternative splicing programs in breast cancer. Mol Cancer Res. 2010;8:961–974. doi: 10.1158/1541-7786.MCR-09-0528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loss LA, Sadanandam A, Durinck S, Nautiyal S, Flaucher D, Carlton VE, Moorhead M, Lu Y, Gray JW, Faham M, Spellman P, Parvin B. Prediction of epigenetically regulated genes in breast cancer cell lines. BMC Bioinformatics. 2010;11:305. doi: 10.1186/1471-2105-11-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Creighton CJ, Fu X, Hennessy BT, Casa AJ, Zhang Y, Gonzalez-Angulo AM, Lluch A, Gray JW, Brown PH, Hilsenbeck SG, Osborne CK, Mills GB, Lee AV, Schiff R. Proteomic and transcriptomic profiling reveals a link between the PI3K pathway and lower estrogen-receptor (ER) levels and activity in ER+ breast cancer. Breast Cancer Res. 2010;12:R40. doi: 10.1186/bcr2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang K, Gray JW, Parvin B. Sparse multitask regression for identifying common mechanism of response to therapeutic targets. Bioinformatics. 2010;26:i97–105. doi: 10.1093/bioinformatics/btq181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pichot CS, Arvanitis C, Hartig SM, Jensen SA, Bechill J, Marzouk S, Yu J, Frost JA, Corey SJ. Cdc42-interacting protein 4 promotes breast cancer cell invasion and formation of invadopodia through activation of N-WASp. Cancer Res. 2010;70:8347–8356. doi: 10.1158/0008-5472.CAN-09-4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 23.Kern SE, Xie G, White JL, Egan TD. A response surface analysis of propofolremifentanil pharmacodynamic interaction in volunteers. Anesthesiology. 2004;100:1373–1381. doi: 10.1097/00000542-200406000-00007. [DOI] [PubMed] [Google Scholar]
- 24.Charafe-Jauffret E, Ginestier C, Monville F, Finetti P, Adelaide J, Cervera N, Fekairi S, Xerri L, Jacquemier J, Birnbaum D, Bertucci F. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene. 2006;25:2273–2284. doi: 10.1038/sj.onc.1209254. [DOI] [PubMed] [Google Scholar]
- 25.Lacroix M, Leclercq G. Relevance of breast cancer cell lines as models for breast tumours: an update. Breast Cancer Res Treat. 2004;83:249–289. doi: 10.1023/B:BREA.0000014042.54925.cc. [DOI] [PubMed] [Google Scholar]
- 26.Levenson AS, Jordan VC. MCF-7: the first hormone-responsive breast cancer cell line. Cancer Res. 1997;57:3071–3078. [PubMed] [Google Scholar]
- 27.Wong CW, McNally C, Nickbarg E, Komm BS, Cheskis BJ. Estrogen receptor-interacting protein that modulates its nongenomic activity-crosstalk with Src/Erk phosphorylation cascade. Proc Natl Acad Sci U S A. 2002;99:14783–14788. doi: 10.1073/pnas.192569699. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Kraus MH, Popescu NC, Amsbaugh SC, King CR. Overexpression of the EGF receptor-related proto-oncogene erbB-2 in human mammary tumor cell lines by different molecular mechanisms. EMBO J. 1987;6:605–610. doi: 10.1002/j.1460-2075.1987.tb04797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cailleau R, Young R, Olive M, Reeves WJ., Jr Breast tumor cell lines from pleural effusions. J Natl Cancer Inst. 1974;53:661–674. doi: 10.1093/jnci/53.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y, Zheng Y, Foster DA. Phospholipase D confers rapamycin resistance in human breast cancer cells. Oncogene. 2003;22:3937–3942. doi: 10.1038/sj.onc.1206565. [DOI] [PubMed] [Google Scholar]
- 31.Chang SB, Miron P, Miron A, Iglehart JD. Rapamycin inhibits proliferation of estrogen-receptor-positive breast cancer cells. J Surg Res. 2007;138:37–44. doi: 10.1016/j.jss.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 32.Alvan G, Paintaud G, Wakelkamp M. The efficiency concept in pharmacodynamics. Clinical pharmacokinetics. 1999;36:375–389. doi: 10.2165/00003088-199936050-00005. [DOI] [PubMed] [Google Scholar]
- 33.Zauli G, Voltan R, Bosco R, Melloni E, Marmiroli S, Rigolin GM, Cuneo A, Secchiero P. Dasatinib plus Nutlin-3 shows synergistic antileukemic activity in both p53 wild-type and p53 mutated B chronic lymphocytic leukemias by inhibiting the Akt pathway. Clin Cancer Res. 2011;17:762–770. doi: 10.1158/1078-0432.CCR-10-2572. [DOI] [PubMed] [Google Scholar]
- 34.Mitchell C, Hamed HA, Cruickshanks N, Tang Y, Bareford MD, Hubbard N, Tye G, Yacoub A, Dai Y, Grant S, Dent P. Simultaneous exposure of transformed cells to SRC family inhibitors and CHK1 inhibitors causes cell death. Cancer Biol Ther. 2011;12 doi: 10.4161/cbt.12.3.16218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cai H, Babic I, Wei X, Huang J, Witte ON. Invasive prostate carcinoma driven by c-Src and androgen receptor synergy. Cancer Res. 2011;71:862–872. doi: 10.1158/0008-5472.CAN-10-1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCaig AM, Cosimo E, Leach MT, Michie AM. Dasatinib inhibits B cell receptor signalling in chronic lymphocytic leukaemia but novel combination approaches are required to overcome additional pro-survival microenvironmental signals. Br J Haematol. 2011 doi: 10.1111/j.1365-2141.2010.08507.x. [DOI] [PubMed] [Google Scholar]
- 37.Carlson JM, Doyle J. Highly optimized tolerance: robustness and design in complex systems. Phys Rev Lett. 2000;84:2529–2532. doi: 10.1103/PhysRevLett.84.2529. [DOI] [PubMed] [Google Scholar]
- 38.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 39.Shah NP, Sawyers CL. Mechanisms of resistance to STI571 in Philadelphia chromosome-associated leukemias. Oncogene. 2003;22:7389–7395. doi: 10.1038/sj.onc.1206942. [DOI] [PubMed] [Google Scholar]
- 40.Sawyers CL. Shifting paradigms: the seeds of oncogene addiction. Nat Med. 2009;15:1158–1161. doi: 10.1038/nm1009-1158. [DOI] [PubMed] [Google Scholar]
- 41.Dai Y, Rahmani M, Corey SJ, Dent P, Grant S. A Bcr/Abl-independent, Lyn-dependent form of imatinib mesylate (STI-571) resistance is associated with altered expression of Bcl-2. J Biol Chem. 2004;279:34227–34239. doi: 10.1074/jbc.M402290200. [DOI] [PubMed] [Google Scholar]
- 42.Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A, Olson JA, Jr, Marks JR, Dressman HK, West M, Nevins JR. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353–357. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 43.Bollenbach T, Quan S, Chait R, Kishony R. Nonoptimal microbial response to antibiotics underlies suppressive drug interactions. Cell. 2009;139:707–718. doi: 10.1016/j.cell.2009.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zinner RG, Barrett BL, Popova E, Damien P, Volgin AY, Gelovani JG, Lotan R, Tran HT, Pisano C, Mills GB, Mao L, Hong WK, Lippman SM, Miller JH. Algorithmic guided screening of drug combinations of arbitrary size for activity against cancer cells. Mol Cancer Ther. 2009;8:521–532. doi: 10.1158/1535-7163.MCT-08-0937. [DOI] [PubMed] [Google Scholar]
- 45.Janes KA, Albeck JG, Gaudet S, Sorger PK, Lauffenburger DA, Yaffe MB. A systems model of signaling identifies a molecular basis set for cytokine-induced apoptosis. Science. 2005;310:1646–1653. doi: 10.1126/science.1116598. [DOI] [PubMed] [Google Scholar]
- 46.Sharma SV, Gajowniczek P, Way IP, Lee DY, Jiang J, Yuza Y, Classon M, Haber DA, Settleman J. A common signaling cascade may underlie “addiction” to the Src, BCR-ABL, and EGF receptor oncogenes. Cancer Cell. 2006;10:425–435. doi: 10.1016/j.ccr.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martinez-Outschoorn UE, Lin Z, Ko YH, Goldberg AF, Flomenberg N, Wang C, Pavlides S, Pestell RG, Howell A, Sotgia F, Lisanti MP. Understanding the metabolic basis of drug resistance: Therapeutic induction of the Warburg effect kills cancer cells. Cell Cycle. 2011;10 doi: 10.4161/cc.10.15.16584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Calzolari D, Bruschi S, Coquin L, Schofield J, Feala JD, Reed JC, McCulloch AD, Paternostro G. Search algorithms as a framework for the optimization of drug combinations. PLoS Comput Biol. 2008;4:e1000249. doi: 10.1371/journal.pcbi.1000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stamatakos GS, Kolokotroni EA, Dionysiou DD, Georgiadi E, Desmedt C. An advanced discrete state-discrete event multiscale simulation model of the response of a solid tumor to chemotherapy: Mimicking a clinical study. Journal of theoretical biology. 2010;266:124–139. doi: 10.1016/j.jtbi.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 50.Feala JD, Cortes J, Duxbury PM, Piermarocchi C, McCulloch AD, Paternostro G. Systems approaches and algorithms for discovery of combinatorial therapies. Wiley interdisciplinary reviews. Systems biology and medicine. 2010;2:181–193. doi: 10.1002/wsbm.51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.