

Abstract
Insulin stimulates glucose transport in adipocytes by triggering translocation of GLUT4 glucose transporters to the plasma membrane (PM) and several Rabs including Rab10 have been implicated in this process. To delineate the molecular regulation of this pathway, we conducted a TBC/RabGAP overexpression screen in adipocytes. This identified TBC1D13 as a potent inhibitor of insulin-stimulated GLUT4 translocation without affecting other trafficking pathways. To determine the potential Rab substrate for TBC1D13 we conducted a yeast two-hybrid screen and found that the GTP bound forms of Rabs 1 and 10 specifically interacted with TBC1D13 but not with eight other TBC proteins. Surprisingly, a comprehensive in vitro> screen for TBC1D13 GAP activity revealed Rab35 but not Rab10 as a specific substrate. TBC1D13 also displayed in vivo GAP activity towards Rab35. Overexpression of constitutively active Rab35 but not constitutively active Rab10 reversed the block in insulin-stimulated GLUT4 translocation observed with TBC1D13 overexpression. These studies implicate an important role for Rab35 in insulin-stimulated GLUT4 translocation in adipocytes.
INTRODUCTION
Rab GTPases form the largest branch of the Ras super family of small monomeric GTP-binding proteins with at least 60 members predicted in the human genome (1). These proteins play a major role in regulating vesicle trafficking and maintaining organelle structure and identity (2–4). Rabs are localized to specific sub-cellular membranes where they recruit Rab effector proteins in a nucleotide-dependent manner. Rab effectors mediate functions that include, but are not limited to; vesicle tethering, attachment to the cytoskeleton, and maintenance of organelle structure (2, 3, 5). Rab GTPase activating proteins (RabGAPs) and Rab guanine nucleotide exchange factors (RabGEFs) control the nucleotide binding state of Rabs. The RabGAP family contains 52 members, as defined by the presence of the Tre2/Bub2p/Cdc16 (TBC) or RabGAP domain (6), which mirrors the large number of Rabs encoded in the mammalian genome. Hence, one model is that each RabGAP controls a specific Rab. To test this model it is important to begin to define the Rab specificity of different RabGAPs as well as the vesicle transport steps they regulate.
The insulin regulation of GLUT4 trafficking in muscle and fat cells is a particularly interesting vesicle transport process as it plays a key role in regulating whole body metabolism and defects in this process contribute to insulin resistance and possibly type 2 diabetes (7, 8). In the basal state, GLUT4 is excluded from the plasma membrane by active sequestration in a vesicular compartment referred to as GLUT4 storage vesicles or GSVs. GLUT4 is also found in tubulo-vesicular structures in the trans-Golgi network (TGN) and endosomes (9) and it traffics between endosomes and the TGN possibly en route to GSVs (10). Insulin-stimulation leads to the rapid redistribution of GLUT4 from these compartments to the PM where it facilitates the uptake of glucose into the cell.
A proteomic survey of GLUT4 vesicles immuno-purified from adipocytes identified a number of Rabs including Rab8, Rab10, Rab11, and Rab14 (11–13). It is not surprising that multiple Rabs have been implicated in GLUT4 trafficking in view of the large number of compartments that GLUT4 traffics through. This could call for the activity of multiple Rabs as well as multiple RabGAPs and GEFs in order to regulate the sequential movement of GLUT4 through its itinerant compartments. Consistent with this, Rab cascades whereby the activity of individual Rabs that act in series are coupled via the coordinated regulation of GAPs and GEFs (14–16). Thus far, the RabGAP TBC1D4/AS160 and the Rab GEF Dennd4C have been implicated in GLUT4 trafficking in adipocytes and these are thought to control the activity of Rab10 (11, 17–20). However, in view of the complexity of GLUT4 trafficking it is likely that additional GAPs and GEFs play a role (11, 18, 19, 21–23).
In the present study we sought to identify additional RabGAPs that regulate GLUT4 trafficking and link them to their cognate Rabs. To this end we initiated a qualitative screen to identify RabGAPs that regulate insulin-stimulated GLUT4 translocation. This initial screen identified the RabGAP TBC1D13 as a potent suppressor of GLUT4 translocation. TBC1D13 interacted with Rab10 and Rab1 in a GTP-dependent manner but it did not display GAP activity toward either of these Rabs. Rather, TBC1D13 stimulated Rab35 GTP hydrolysis in vitro and in vivo. Overexpression of constitutively active Rab35 but not Rab10 overcame the TBC1D13-mediated block in insulin-stimulated GLUT4 translocation. These studies reveal an important role for Rab35 and TBC1D13 in GLUT4 trafficking in adipocytes.
RESULTS
TBC1D13 specifically inhibits insulin-stimulated GLUT4 translocation
GLUT4 translocation to the PM in adipocytes involves a complex intracellular trafficking program whereby the transporter navigates through multiple compartments likely requiring the activity of multiple Rab GTPases. In view of the critical role of RabGAPs in regulating the activity of Rab GTPases we implemented a screen to identify RabGAPs that regulate GLUT4 trafficking with the view that this might bring us closer to a molecular understanding of this process. Several RabGAPs were electroporated into adipocytes that were expressing HA-tagged GLUT4 via retroviral infection thus enabling analysis of PM levels of GLUT4 (18, 24–26). TBC1D4 and TBC1D1 were used as positive and negative controls, respectively. Consistent with other studies (18), overexpression of wild type TBC1D4 had no effect on insulin-stimulated GLUT4 translocation, while TBC1D1 had an inhibitory effect (26). Overexpression of TBC1D11 and TBC1D15 had no detectable effect on insulin-stimulated GLUT4 translocation while TBC1D13 caused marked inhibition (Fig. 1). All RabGAPs were expressed to similar levels and none affected surface GLUT4 in the basal state (data not shown). Knock down of TBC1D13 via siRNA had no significant effect on insulin-stimulated glucose uptake, which is a physiological measure of endogenous GLUT4 translocation to the PM in adipocytes (Fig. S1). However, this was likely due to the modest reduction in TBC1D13 levels observed, which could not be improved with a range of siRNAs targeting different regions of the transcript. This could be related to the fact that TBC1D13 is quite an abundant protein in adipocytes. Quantitative mass spectrometry of adipocyte proteins reveals that TBC1D13 is the third most abundant RabGAP in 3T3-L1 adipocytes being expressed at two times higher levels than AS160/TBC1D4, a known GLUT4 regulator in adipocytes (SJH and DEJ, unpublished data).
Figure 1.

TBC1D13 overexpression inhibits insulin-stimulated HA-GLUT4 translocation to the PM. HA-GLUT4 expressing adipocytes were electroporated with TagRFP as a control or FLAG-tagged TBC1D1, TBC1D4, TBC1D11, TBC1D13 or TBC1D15 as indicated. 3T3-L1 adipocytes were serum-starved in DMEM for 2 h and then incubated with or without 100nM insulin for 20 min. GLUT4 translocation to the PM was determined by surface HA staining. A) Representative images for adipocytes overexpressing TagRFP and TBC1D13 are shown. Arrowheads indicate transfected cells. Scale bar 10μm. B) Quantification of the number of 3T3-L1 adipocytes with surface HA-staining for TagRFP (n=3), TBC1D1 (n=3), TBC1D4 (n=3), TBC1D11 (n=2), TBC1D13 (n=5), and TBC1D15 (n=2). Statistical significance was determined by One-way ANOVA (*** = P < 0.001) and error bars represent one standard deviation (SD).
To rule out non-specific effects on vesicle transport (27–30) we examined the effects of TBC1D13 overexpression on ER to Golgi and endocytic trafficking. Overexpression of TBC1D13 in HeLa cells was without effect on the trafficking of the temperature sensitive tsVSV-G protein from the ER to the Golgi (Fig. S2), consistent with previous findings (31). Moreover, TBC1D13 overexpression had no detectable effect on endocytosis or recycling of the transferrin receptor in adipocytes, as examined by Alexa488-labelled transferrin uptake (Fig. S3A). To test whether the block in GLUT4 translocation with TBC1D13 overexpression was due to changes in HA-GLUT4 protein levels, HA-GLUT4 localisation or organelle morphology, 3T3-L1 adipocytes electroporated with Flag-tagged TBC1D13 were stained with Flag antibody, HA antibody or antibodies against organelle markers. Neither the localisation nor the protein expression level of HA-GLUT4 was affected by TBC1D13 overexpression. (Figure S3B,C). There was also no detectable morphological change in the Golgi and TGN in cells overexpressing TBC1D13 as determined by localisation of GM130 and Syntaxin16 (Figure S3D,E). Next we tested whether the block in insulin-stimulated GLUT4 translocation with TBC1D13 overexpression was due to defects in insulin signalling. Single cell analysis of pAkt levels (32) in TBC1D13-overexpressing and control cells. did not reveal any significant defect in insulin-dependent phosphorylation of Akt in cells overexpressing TBC1D13 (Fig. S4A,B). These data indicate that the TBC1D13 block is likely specific, involving one of the steps in GLUT4 trafficking to the PM.
TBC1D13 is expressed in adipocytes
To ascertain whether TBC1D13 has an endogenous role in adipocytes we generated an antibody to probe TBC1D13 expression. The TBC1D13 antibody immunolabelled a single band in 3T3-L1 adipocytes of the predicted molecular weight at ~ 45 kDa (Fig. 2A) and it specifically recognised the recombinant TBC1D13 protein (Fig. 2B). TBC1D13 was present in 3T3-L1 fibroblasts and it was up-regulated during adipocyte differentiation (Fig. 2C). Consistent with this, we identified by mass spectrometry that TBC1D13 is a highly abundant protein in 3T3-L1 adipocytes and the third most abundant RabGAP (SJH and DEJ, unpublished data). TBC1D13 was detected in several sub-cellular fractions obtained from adipocytes including HDM, LDM and cytosol and quantitative analysis of the relative amount in each fraction determined that 20 – 30% of TBC1D13 was associated with membranes with the remainder localised in the cytosol (Fig. 2D). The localisation of TBC1D13 was not significantly affected by insulin. IRAP, a constituent of GLUT4 vesicles, underwent insulin-stimulated translocation from LDM to the PM as previously shown (33). As GLUT4 and TBC1D13 are both present in HDM and LDM fractions we sought to determine whether these proteins colocalise in 3T3-L1 adipocytes (Fig. 2E). TBC1D13 and GLUT4 both localised to the perinuclear region and punctate structures within the cell periphery with limited colocalisation.
Figure 2.
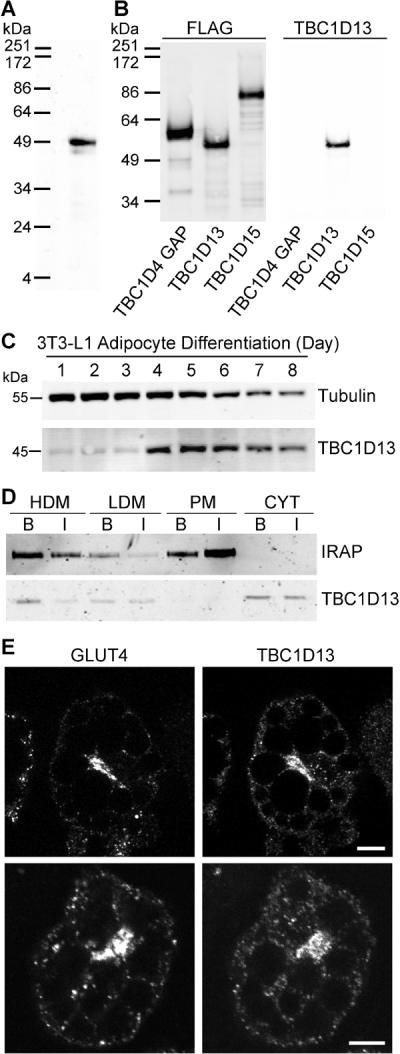
Characterisation of TBC1D13. A) TBC1D13 antibody recognises TBC1D13 in a 3T3-L1 adipocyte whole cell lysate. B) Purified FLAG-tagged TBC1D4 GAP domain, TBC1D13 and TBC1D15 were immunoblotted with FLAG and TBC1D13 antibody. C) TBC1D13 and Tubulin expression during a 3T3-L1 adipocyte differentiation time course. Tubulin is presented as a control. D) 3T3-L1 adipocytes were serum starved for 2 h and then incubated in the absence or presence of 100 nM insulin for 20 min. Cells were homogenised and cytosol, PM, HDM and LDM fractions were obtained by differential cell fractionation. 10 μg protein of each fraction was analysed by SDS-PAGE. Representative Western blots stained for TBC1D13 and IRAP are shown. E) TBC1D13 and GLUT4 localisation in 3T3-L1 adipocytes was determined by staining cells that were serum-starved for 2 h with TBC1D13 and GLUT4 antibody. Scale bar 10 μm.
TBC1D13-interacting proteins
To identify TBC1D13's substrate Rab GTPase(s) we employed a yeast two-hybrid (Y2H) strategy that has been used to identify substrate Rab GTPases for Gyp6p (34), RUTBC3 (27), TBC1D11 (28), and TBC1D20 (31). We constructed an Y2H library comprising 42 mammalian Rab GTPases representing all Rab sub-families. To increase the likelihood of a meaningful Rab/GAP interaction we used constitutively active Rab GTPases (Rab QL mutants) and the catalytically inactive TBC1D13 (TBC1D13 RA), as previously described (27). In this screen, TBC1D13 showed the strongest interaction with Rab10 and a weaker interaction with Rab1 (Figure 3A). A slight interaction was observed with Rab3a but subsequent studies showed this to be non-specific (data not shown). To determine if Rab1 and Rab10 bind selectively to TBC1D13 these two Rabs were screened against a panel of available RabGAPs (Figure 3B). In addition to TBC1D13, Rab1 interacted with TBC1D11/GAPCenA/ RabGAP1, while Rab10 interacted only with TBC1D13. No detectable interaction could be observed with TBC1D1 or TBC1D4 despite the fact that the TBC domains of these proteins display GAP activity toward Rab10 in vitro (12, 26).
Figure 3.

TBC1D13 interacts with Rab10. A) Catalytically inactive TBC1D13 was co-transformed with 46 constitutively active Rab GTPases into yeast strain AH109 and double transformants were serially diluted on –His/Leu/Trp selection plates. B) Constitutively active Rab1 or Rab10 were co-transformed with the indicated catalytically inactive RabGAPs. C) Catalytically inactive TBC1D13 was co-transformed with constitutively inactive (SN) or active (QL) Rab1, and constitutively inactive (TN), active (QL) or wildtype (WT) Rab10. D) Constitutively active Rab1 or Rab10 were co-transformed with the indicated full length and truncated catalytically inactive TBC1D13. Representative images of three independent experiments are shown.
To determine whether these interactions were nucleotide-dependent we examined the interaction of various Rab mutants with the catalytically inactive version of TBC1D13. We observed a significant interaction between constitutively active Rab1 QL and Rab10 QL with TBC1D13 RA (Fig. 3C). However, no interaction was observed using constitutively inactive Rabs (Rab1 SN; Rab10 TN). Immunoblotting of yeast lysates revealed that the Rab10 TN mutant was not expressed at the same level as other Rabs (data not shown). To rule out that the nucleotide-dependence of this interaction was due to reduced expression we next expressed Rab10 in HEK cells and examined the nucleotide-dependence of the interaction of this construct with recombinant TBC1D13 (35). GST-Rab10 was purified from HEK293 cells, loaded with GDP or GTPγS and incubated with lysate from FLAG-TBC1D13 WT or FLAG-TBC1D13 RA overexpressing HEK293 cells. We observed a specific interaction between both versions of TBC1D13 and GTPγS but not GDP loaded GST-Rab10 (Fig. 4). We used GST-Rab4 as a negative control in this assay and found a weak nucleotide-insensitive association that we interpret as non-specific binding.
Figure 4.

TBC1D13 binds to Rab10 in a GTP-dependent manner. The ability of Rab4 and Rab10 binding to TBC1D13 was determined using GST bead binding assay. GST-Rabs loaded with GDP or GTPγS were incubated with HEK293 lysate expressing either FLAG-TBC1D13WT or FLAG-TBC1D13RA. Rab interacting proteins were eluted by complexing free Mg2+. Starting material and eluates were immunoblotted with FLAG antibody. Representative image of three independent experiments is shown.
The TBC domain in TBC1D13 spans residues 32–370 and taking into the account the structure of the Gyp1 TBC domain and its domain prediction (36), the TBC1D13 TBC domain more likely spans form residue 32–400. We tested whether TBC1D13 binds to Rab10 via its 31 amino acid N-terminus or via the TBC domain (32–400) by using these truncation mutants in the Y2H system. Neither Rab10 nor Rab1 bound to either of these truncation mutants, indicating that Rab binding requires the full length TBC1D13 (Fig. 3D)
We next set out to determine if endogenous Rab10 and TBC1D13 interact in adipocytes. Endogenous TBC1D13 was immunoprecipitated from whole cell lysate using the TBC1D13 antibody and the interacting proteins were identified by LC-MS/MS. Table S1 shows proteins that were specifically enriched in the TBC1D13 immunoprecipitation (IP) but not identified in the IgG control IP.. Most notably, peptides from Rab10 were identified indicating that these proteins interact in vivo (Table S1).
We next wanted to test whether overexpression of constitutively active Rab 10, with a critical Gln mutated to Leu that reduces its intrinsic GTPase activity and renders it resistant towards RabGAP-mediated GTP hydrolysis (37), could change the localisation of TBC1D13 thus providing a further clue that these two proteins functionally interact. However, this was not possible since most transfected cells displayed high cytosolic Rab10 QL staining (data not shown).
TBC1D13 displays specific GAP activity toward Rab35
As TBC1D13 binds Rab1 and Rab10 in a nucleotide-dependent manner we next sought to investigate whether TBC1D13 accelerates GTP hydrolysis for these Rab GTPases in vitro. TBC1D13 was screened for GAP activity against a library of Rab GTPases where at least one member of each sub-family was accounted for (Figure 5A). Only one Rab GTPase was found to be a substrate for TBC1D13 in this assay while remaining Rabs did not show any significant activity with TBC1D13 addition. The GTP hydrolysis of Rab35 was accelerated by TBC1D13 with a specificity constant (kcat/Km) of 400 ±20 M−1s−1 (Figure 5B).
Figure 5.

TBC1D13 stimulates GTP hydrolysis on Rab35. A) Profile of the GAP activity of 6×His-TBC1D13 for 34 GST-Rab GTPases. Catalytic efficiency (kcat/Km) was determined from initial velocities as described in the methods. B) Kinetics of GST-Rab35 GTP hydrolysis in the presence and absence of 6×His-TBC1D13. Solid lines represent the model from a global fit with a Michaelis-Menten function as described in the methods. The mean and deviation for kinetic constants estimated from two independent experiments are as follows: kcat = 0.0024 ± 0.0004 s−1, Km = 6.3 ± 1.0 μM, kintr = 8 × 10−5 ± 6 × 10−5 s−1. The mean catalytic efficiency and deviation calculated from the ratio of kcat and Km is kcat/Km = 400 ± 20 M−1 s−1. Raw data were corrected by dividing by the emission intensity for the phosphate sensor alone (Iref). C) Rab effector pull-down from HEK293 cells co-expressing GFP-Rab35 and FLAG-TBC1D13 or empty vector control. Cells were serum-starved for 2 h and stimulated or not with 100 nM insulin for 2 min. GST-RBD35 pull-down and input were immunoblotted with antibodies against FLAG, GFP and GST as indicated. Quantifications of GFP-Rab35 normalised to input are indicated.
We next tested if TBC1D13 accelerates Rab35 GTP hydrolysis in vivo. We co-transfected GFP-Rab 35 with FLAG-TBC1D13 or empty vector into HEK293 cells and performed a Rab effector pulldown with the recently described Rab 35 binding domain (RBD35) of RUSC2 that specifically binds to GTP-loaded Rab35 (38). While insulin stimulation did not affect the GTP-loading of Rab35, overexpression of TBC1D13 reduced GTP-bound Rab35 by 60% (Fig. 5C), indicating that TBC1D13 accelerates Rab35 GTP hydrolysis in vivo.
Rab35 ameliorates the TBC1D13 block in insulin-stimulated GLUT4 translocation
TBC1D13 overexpression may inhibit GLUT4 translocation in adipocytes by inhibiting the action of its cognate Rab. We therefore overexpressed the catalytically inactive TBC1D13 RA in adipocytes. This still inhibited insulin-stimulated GLUT4 translocation but to a lesser extent (Fig. 5B). One possibility is that the GAP inactive TBC1D13 binds and traps the Rab substrate, thus sequestering it away and thereby affecting GLUT4 translocation to the PM. Another possibility is that TBC1D13 RA is still partially active as is the case for TBC1D14, which has just been reported to require mutations in two catalytic residues (Arg and Gln) to reverse a functional effect (39). The inhibitory effect on GLUT4 translocation due to overexpression of TBC1D13 might be reversed by co-expression of the constitutively active Rab. To examine this possibility we next determined if overexpression of either Rab35 or Rab10 could reverse the TBC1D13 block in GLUT4 translocation. Rab10 QL and Rab35 QL were previously shown to be functional as constitutively active Rabs (37, 40, 41). Overexpression of the Rab10 QL had no significant effect on insulin-stimulated GLUT4 translocation when expressed alone (41) (data not shown) nor did it reverse the TBC1D13 WT block in GLUT4 translocation (Fig. 6B). Overexpression of wild type Rab35 (data not shown) or Rab35 QL (Fig. 6B) had no detectable effect on GLUT4 translocation when expressed alone. Strikingly, the inhibition of insulin-stimulated GLUT4 translocation in cells overexpressing TBC1D13 WT was markedly reduced by co-expression of Rab35 QL (Fig. 6A,B). Furthermore, Rab35 QL also reversed the inhibition of TBC1D13 RA (Fig. 6B).
Figure 6.

TBC1D13 overexpression blocks insulin stimulated GLUT4 translocation in a Rab35-dependent manner. HA-GLUT4 expressing 3T3-L1 adipocytes were electroporated with wildtype (WT) or catalyically inactive (RA) FLAG-tagged TBC1D13 and with or without TagRFP-labelled constitutively active (QL) Rab35 and Rab10. Transfected cells were detected with TagRFP signal and/or the FLAG antibody. 3T3-L1 adipocytes were serum-starved in DMEM and then incubated with or without 100nM insulin for 20 min and GLUT4 translocation to the PM was determined by surface HA staining. A) Representative images of insulin-stimulated cells are shown. Arrowheads indicate transfected cells. Scale bar 10 μm. B) Surface HA fluorescence levels were quantified for transfected and non-transfected cells in A. The average HA-surface fluorescence of non-transfected cells was set to 100% for each analysed image. Quantification of independent experiments for Rab35QL (n=3), TBC1D13 WT (n=6), TBC1D13 WT plus Rab35QL (n=3), TBC1D13 RA (n=5), TBC1D13 RA plus Rab35QL (n=2), and TBC1D13 WT plus Rab10QL (n=2) are shown. Statistical significance was determined by One-way ANOVA (***=p<0.001, **=p<0.01, *=p<0.05) and error bars represent one SD.
Knockdown of Rab35 using siRNA in 3T3-L1 adipocytes did not result in any significant defect in insulin-stimulated glucose uptake (Fig. S5) although this may have been due to incomplete knock down or compensation by another related Rab. We were unable to analyse the effect of the dominant negative forms of the Rabs, because they did not express in adipocytes, as previously reported by others (41). These results indicate that TBC1D13 and Rab35 function co-operatively to regulate one of the steps involved in GLUT4 trafficking to the PM.
DISCUSSION
In this study, we have identified TBC1D13 as a Rab35 GAP and we provide a potential role for these proteins in insulin-stimulated GLUT4 trafficking in adipocytes. TBC1D13 specifically interacts with Rab10, a Rab protein previously implicated in GLUT4 trafficking (37, 41), without stimulating its GTP ase activity. Overexpression of TBC1D13 in adipocytes inhibited insulin-stimulated GLUT4 translocation and this effect was partially reversed by overexpression of constitutively active Rab35, whereas Rab10 was without effect. TBC1D13 was localised to the cytosol, perinuclear area, and punctate structures while Rab35 was found on the PM in adipocytes (42). This gives rise to a model where TBC1D13 may participate in a Rab cascade, involving Rab10 and Rab35, that regulates GLUT4 recycling in adipocytes.
TBC1D13 is expressed in a range of tissues, its expression is induced upon adipocyte differentiation and it is one of the most abundant RabGAPs in adipocytes. It is localised to the cytosol, perinuclear area, as well as to punctate structures scattered throughout the cell periphery. TBC1D13 is an unusual member of the TBC family. Whereas most TBC proteins are multi-domain proteins, TBC1D13 is comprised almost entirely of a GAP domain. The GAP domain of TBC1D13 is larger than that found in other TBC proteins due to the presence of several inserts, with one notable insert located between the catalytic motifs. Despite the presence of this insert, TBC1D13 displays significant and highly specific GAP activity toward Rab35 without showing significant GAP activity toward a range of other Rabs. This activity was striking since most TBC proteins do not display such restricted GAP activity. For example, TBC1D4 has GAP activity toward Rab2, Rab8, Rab10, and Rab14 (12). It should also be noted that the RabGAPs TBC1D10A-C also accelerate Rab35 GTP hydrolysis. However, TBC1D10A and TBC1D10B are much less abundant (3 and 18%, respectively) than TBC1D13 in 3T3-L1 adipocytes while TBC1D10C was not detected (SJH and DEJ, unpublished data) (30, 43). This indicates that GTP loading of Rab35 might be differentially regulated via the cell specific expression of its different cognate GAPs. Rab cascades have been described involving the co-ordinate regulation of two or more Rabs that function in series (15, 16). Here GTP loading of a downstream Rab in a vesicle transport reaction recruits a RabGAP to trigger the GTP hydrolysis and deactivation of the cognate upstream Rab. This ensures unidirectional flux along the pathway. For example, in yeast, the Rab Ypt32p recruits the RabGAP Gyp1p as an effector to regulate GTP loading of Ypt1p and disruption of this interaction caused a defect in membrane organisation. According to this model Rab10 would be downstream of Rab35 action and TBC1D13 might bind to Rab10 to deactivate Rab35. Rab35 is localised to the PM in adipocytes and in other cells (42, 44) and it has been reported to function in rapid recycling from early endosomes back to the PM (30, 44), a pathway likely traversed by GLUT4. Rab10 is localised to the Golgi and endosomes and plays a role in endosomal sorting and exocytosis (45–47). Rab10 has previously been implicated in insulin-stimulated GLUT4 translocation to the PM (20, 37, 41). The fact that TBC1D13 binds to Rab10 GTP in a non-catalytic manner but acts as a GAP for Rab35 poses the possibility that these two Rabs act in series and that TBC1D13 bridges this concerted action. One could envisage that in the absence of insulin TBC1D13 prevents rapid recycling to the PM possibly in a manner that is coupled to the regulation of the GTPase activity on Rab35, while with insulin the opposite occurs. TBC1D4 displays GAP activity toward Rab10 (12) and so this likely invokes a separate pair of Rabs downstream of Rab35 working in the same counter-current mechanism as described for Rab10 and Rab35 except in this instance it would involve Rab10 and its cognate downstream partner.
It is perhaps not surprising that we were unable to detect an interaction between TBC1D13 and Rab35 using Y2H because catalytic type interactions are likely to be more transient and less stable than effector type interactions. Although similar techniques have been used to identify pairings between RabGAPs and substrate Rab GTPases (27, 28) in our hands this methodology has yielded interactions that more closely resemble effector-like pairings. The functional relationship between Rab1, Rab10, Rab35 and TBC1D13 is intriguing in light of the sequence and structural similarities between these Rabs. Rab1a, Rab1b, and Rab35 are members of the same sub-family (1), although Rab35 localisation and function in the cell is different to Rab1a and Rab1b. Furthermore, the Rab1 and Rab8/10/13 sub-families are more closely related to each other than to other Rab sub-families (1). This indicates an evolutionary link between these molecules that could be related to their convergent interactions with TBC1D13.
It was somewhat disappointing that we were unable to observe any significant disruption of GLUT4 trafficking upon reduced expression of either Rab35 or TBC1D13 using siRNA expression in adipocytes. However, the largest knockdowns we have been able to achieve for TBC1D13 and Rab35 were 60% and 75%, respectively and these levels may not be sufficient to disrupt this point in the GLUT4 trafficking pathway, particularly in the case of TBC1D13, a highly abundant protein in adipocytes. Nevertheless, the observation that the block in insulin-stimulated GLUT4 trafficking observed with TBC1D13 overexpression could be overcome by co-expressing constitutively active Rab35 but not Rab10 and that other vesicular trafficking pathways were unaffected under these conditions, strongly supports the argument that this effect is quite specific.
The identification of Rab35 and TBC1D13 as two new players in the GLUT4 trafficking cascade provides new clues concerning this pathway. Previously a role for several other Rabs, including Rab4, Rab8, Rab10, Rab11, Rab13 and Rab14, in GLUT4 trafficking has been shown (11, 41, 48–50). These studies provide a picture of the complex trafficking itinerary undertaken by this molecule. Rab8 and Rab10 regulate endosomal sorting and exocytosis (45, 46) and it is known that GLUT4 follows this route. Rab11 and Rab4 regulate early to recycling endosome traffic (51–53) while Rab35 is predicted to control a separate trafficking pathway from early endosomes (44). A number of questions need to be answered to address these hypotheses. What is the location of the TBC1D13/Rab35 regulated site? Is the association between TBC1D13 and Rab35 regulated by insulin stimulation and what are the mechanisms that achieve this? What is the exact nature of the Rab35 step and what effectors does it recruit to achieve this? Understanding these functions of Rab35 and TBC1D13 will be critical in elaborating their role in insulin-stimulated GLUT4 translocation and how this and other regulatory nodes intersect to control glucose uptake into adipocytes.
MATERIALS AND METHODS
Reagents, Antibodies and Constructs
TBC1D13 was amplified by PCR from a 3T3-L1 adipocyte cDNA library provided by Dr. Alan Saltiel (54) and cloned into pGEMT-Easy (Promega). This construct was sequenced and identity confirmed as NM_146252. For RabGAPs TBC1D1, TBC1D11, and TBC1D15, cDNA was obtained from OpenBiosytems corresponding to NM_015173, NM_012197, NM_022771.4, respectively and cloned into pGEMT-Easy (Promega) by standard methodology. For all FLAG tagged constructs the corresponding cDNA was sub-cloned into the p3xFLAG-CMV10 vector (Sigma) to form an in frame C-terminal fusion. The eGFP-tsVSV-G construct was a gift from Dr. Jennifer Lippincott-Schwartz and the GST-RBD35 construct was kindly provided by Dr. Mitsunori Fukuda. Rab10 and Rab35 were cloned into TagRFP (Evrogen) and pDEST53 (Invitrogen) plasmids. Antibodies were purchased from Sigma (Mouse monoclonal anti-FLAG M2, Rabbit polyclonal anti-FLAG), Covance Research Products (HA antibody 16B12), BD Biosciences (GM130), Proteintech Group, Inc (Rab35), Roche Applied Science (mouse anti-GFP), Santa Cruz Biotechnology (rabbit anti-14-3-3) and Cell Signaling Technology (mouse anti-GST). Antibodies were kindly provided by Dr. Wanjin Hong (Syntaxin16, IMCB, Singapore) (55). The antibodies against GLUT4 and IRAP have previously been described (33, 56). TBC1D13 polyclonal rabbit antibody was raised against GST tagged full length TBC1D13 and affinity purified over a GST-TBC1D13 column. Rab35 and TBC1D13 siRNA were supplied as SMARTpools (Dharmacon) with four siRNA against each target. The Amaxa electroporation kit was purchased from Lonza AG. Matrigel was purchased from Becton Dickinson. Bicinchoninic acid (BCA) reagent and Supersignal West Pico chemiluminescent substrate were from Pierce. General chemicals were purchased from Sigma.
TBC1D13 immunoprecipitation
Serum starved 3T3-L1 adipocytes for 2 h prior to harvesting. For the whole cell lysate fractions the cells were washed twice with ice-cold PBS and scraped down in NP40 lysis buffer (1% NP40, 20 mM Tris-HCl pH 7.4, 137 mM NaCl, 10% v/v Glycerol, pH 7.4) supplemented with Complete protease inhibitors (Roche Applied Sciences) and phosphatase inhibitors (2 mM sodium orthovanadate, 1 mM pyrophosphate, 10 mM sodium fluoride). Samples were passed through a 25G needle ten times and solubilised on ice for 20 min. Lysates were cleared by centrifugation at 18,000 × g for 20 min at 4°C. The cleared supernatant was incubated with protein G sepharose beads (GE Healthcare Biosciences) and either TBC1D13 antibody or a rabbit IgG antibody as the negative control. Immunoprecipitations were incubated at 4°C for 2 h with rotation. After incubation, the beads were washed three times in NP40 Lysis buffer and two times in ice-cold PBS. The PBS was completely removed from the beads, which were then boiled in Laemmli sample buffer at 95°C for 5 min. The supernatant was collected and resolved by SDS-PAGE.
Mass Spectrometry analysis
Samples were resolved by SDS-PAGE and proteins were visualised with Sypro-Ruby staining (Molecular Probes) as per manufacturer's instructions. Proteins were visualised under ultra-violet light and excised from the gel. Gel slices were diced and washed with 50% acetonitrile in 250 mM NH4HCO3 at room temperature for 30 min, then incubated in 100% acetonitrile at room temperature for 10 min. The gel slice was incubated with TPCK treated Trypsin (Promega) at 12.5 ng/μl in 100 mM NH4HCO3 overnight at 37°C. 5% Formic acid was added to the gel and incubated for at least 1 hr at 37°C. An equal volume of 100% Acetonitrile was added to the slice and incubated at 37°C for at least 1 h after which an additional two times volume of 100% acetonitrile was added and incubated at room temperature for 10 min. The solution was retained and evaporated until dry using a SpeediVac. The sample was re-solubilised in 20 μl of 5% Formic acid. Samples were desalted and subjected to LC-MS/MS. MS/MS data analysis was performed using MaxQuant software (57) as previously described (42).
Yeast two-hybrid analysis
The Rab library was constructed as previously described (58). Briefly, the cDNA encoding the constitutively active version of each Rab GTPase was cloned by standard methodology into the pGBKT7 vector (Clontech) to form an in frame fusion with the DNA binding domain of the GAL4 protein. Wildtype and catalytically inactive mutants of each RabGAP and truncation mutants of TBC1D13 were sub-cloned into the pGADT7 vector (Clontech) to form an in frame fusion with the activation domain of the GAL4 protein. Inactive mutants were generated by site directed mutagenesis where the catalytic Arginine finger in each RabGAP was replaced with an Alanine residue. Plasmids for the Rab and RabGAP were co-transformed into chemically competent S.cerivisiae strain AH109 (Clontech) and selected for co-transformation by growth on –Leu/Trp plates. Co-transformants were selected for interaction by plating serial dilutions on –Leu/Trp/His plates.
Rab10 pulldown
Wildtype Rab10 was cloned into pDEST27 (Invitrogen) by Gateway cloning to form a C-terminal fusion with a Glutathione S-Transferase (GST) tag. The construct was transfected into HEK293 cells by lipofection (Invitrogen) and recombinant Rab10 was isolated using Glutathione Sepharose 4B (GE Healthcare Biosciences). Briefly, transfected HEK293 cells were incubated for 60 min in DMEM media with the addition of wortmannin 15 min before harvesting. Treated cells were washed twice with cold PBS and lysed in Rab NP40 Buffer (1% NP40, 10% Glycerol, 25 mM Tris pH 7.5, 137 mM NaCl, 5 mM MgCl2, 0.1 mM GDP, EDTA-free Complete protease inhibitors (Roche Applied Science), phosphatase inhibitors (as above). The lysate was incubated on ice for 20 min then centrifuged at 18,000 × g for 20 min at 4°C. Cleared lysate was incubated for 1–2 h with glutathione sepharose on a rotating wheel at 4°C. The beads were washed 3 times with Rab NP40 Buffer, followed by 2 times with PBS supplemented with 5 mM MgCl2, 0.1 mM GDP (Sigma) and inhibitors.
The immobilised GST-Rab10 was loaded with either GDP or GTPγS (Sigma) before incubation with HEK293 lysate (35). The Rab beads were washed once with Nucleotide Exchange (NE) Buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 10 mM EDTA, 5 mM MgCl2, 1 mM DTT) plus 10 μM GDP or 10 μM GTPγS, and then incubated for 20 min at 25°C in NE Buffer plus 1 mM GDP or 1 mM GTPγS. This wash and incubation step was repeated twice. Rab beads were then washed once with Nucleotide Stabilisation (NS) buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 5 mM MgCl2, 1 mM DTT) plus 10 μM GDP or 10 μM GTPγS and incubated 20 min at 25°C in NS buffer plus 1 mM GDP or 1 mM GTPγS.
HEK293 cells transfected with FLAG-tagged RabGAP were serum depleted for 2 h followed by incubation with wortmannin (100 nM) for 30 min and then lysed in NP40 Buffer (1% NP40, 10% Glycerol, 25 mM Tris pH 7.5, 137 mM NaCl, Complete protease inhibitors (Roche Applied Science) and phosphatase inhibitors). The lysate was incubated on ice for 1 h and centrifuged at 18,000 × g for 20 min at 4°C. Cleared lysate was supplemented with 5 mM MgCl2, and either 0.1 mM GDP or 0.1 mM GTPγS. The loaded Rab beads were then incubated with the respective lysates for 1–2 h at 4°C under rotation. The Rab beads were washed twice with NS buffer, then twice with NS buffer supplemented to 250 mM NaCl and then washed once with Wash Buffer (20 mM HEPES pH 7.5, 250 mM NaCl, 1 mM DTT). Proteins bound to the GST-Rab were then eluted by complexing Mg2+ in Elution Buffer (20 mM HEPES pH 7.5, 1.5 M NaCl, 20 mM EDTA, 1 mM DTT) plus 5 mM GDP or 1 mM GTPγS for 20 min at 25°C.
in vitro GAP assay
TBC1D13 was amplified using Vent polymerase and sub-cloned into a modified pET-15b vector, which incorporates a 6× His tag (MGHHHHHHGS). BL21(DE3)-RIPL cells transformed with the expression plasmid were cultured in 2×YT media and induced with 50 μM IPTG at an OD600 of 0.4 for 16 hrs at 22°C. Cells resuspended in 50 mM Tris, pH 8.0, 100 mM NaCl, 0.1% 2-mercaptoethanol, 1 mM PMSF, 0.2 mg/mL Lysozyme (Sigma), 0.01 mg/mL protease-free DNase I (Worthington) were lysed by sonication. The lysate was supplemented with 0.5% Triton X-100, centrifuged at 27000×g for 1 hr, and the clarified supernatant loaded onto a HisTrap HP column (GE Health). The column was washed with 50 mM Tris, pH 8.0, 500 mM NaCl, 0.1% 2-mercaptoethanol and eluted with a 20–300 mM Imidazole gradient. Further purification by anion exchange and gel filtration chromatography on HiTrap Q and Superdex-75 columns (GE Health Care) yielded a highly soluble protein with an estimated purity > 99% as judged by SDS-PAGE stained with Coomassie Blue. GST-Rab GTPases were expressed and purified as described (36).
The GAP activity of 6×His-TBC1D13 for 34 GST-Rab GTPases was profiled in 96 well half-area microplates (Corning Life Sciences) by monitoring the release of phosphate accompanying GTP hydrolysis. Released phosphate was detected as an increase in the fluorescence of a phosphate sensor consisting of the E. coli phosphate binding protein (PBP) labelled with N-[2-(1-maleimidyl) ethyl]-7-(diethylamino) coumarin-3-carboxamide (MDCC) labelled on a single cysteine residue near the phosphate binding site as described (59). GST-Rab GTPases were incubated for 1 hr at 25°C with a 25-fold molar excess of GTP in 20 mM Tris, pH 8.0, 150 mM NaCl, and 5 mM EDTA. Excess GTP was removed using a 10 ml D-Salt column (Pierce, Thermo Fisher Scientific) equilibrated with 20 mM Tris, pH 8.0, 150 mM NaCl. GAP-catalysed and intrinsic GTP hydrolysis reactions were initiated by mixing 50 μL of 4 μM GTP-loaded GST-Rab GTPases in 20 mM Tris, pH 8.0, 150 mM NaCl with an equivalent volume of 0–8 μM 6×His-TBC1D13 in 20 mM Tris, pH 8.0, 150 mM NaCl, 10 mM MgCl2, and 5 μM MDCC-PBP. The single turnover kinetics of intrinsic and 6×His-TBC1D13-accelerated GTP hydrolysis at 30°C were continuously monitored at excitation and emission wavelengths of 425 and 457 nm, respectively, using a Safire (Tecan) microplate spectrometer. For the initial profile of GAP activity, catalytic efficiencies (kcat/Km) were estimated by linear regression of the initial velocities as a function of five concentrations (0, 0.03125, 0.125, 0.5, 2, and 8 μM) of 6×His-TBC1D13. For more detailed kinetic characterization of the GAP activity for GST-Rab35, kinetic time courses were globally analysed by fitting with a pseudo-first order Michaelis–Menten model function I(t) = (I∞ − I0)(1 − exp(−kobst)) + I0, where the observed rate constant kobs = kintr + kcat [GAP] / (Km + [GAP]), the kinetic constants kcat, Km, and kintr were treated as global parameters, and the initial (I0) and final (I∞) emission intensities were treated as local parameters.
In vivo GAP assay using Rab effector pull-down
GST-RBD35 transformed BL21 cells were grown in LB media, induced with 0.5 mM IPTG at OD600 of 0.6 for 3 h. The culture was centrifuged at 5,000 × g for 10 min, lysed in PBS containing 1 mg/ml lysozyme and protease inhibitors (see above), incubated on ice for 30 min, followed by addition of 5 μg/ml DNase and 0.14% Triton X-100, incubation with rocking for 10 min at 4°C and centrifugation at 5,000 × g for 30 min. The supernatant was added to 200 μl glutathione sepharose beads and incubated over night, rotating at 4°C. The beads were washed with PBS, incubated with glutathione elution buffer (50 mM Tris pH 8.0, 10 mM reduced glutathione, 2 mM DTT) for 1 h at RT and the eluate was buffer exchanged into 50 mM Tris pH 8.0, 100 mM NaCl, 5 mM MgCl2 using 10 kD Amicon filters (Millipore). The protein concentration was determined using BCA assay.
The effector pull-down was performed as described in (37). HEK293 cells grown on 10-cm dishes were transiently transfected with GFP-tagged Rab35 (pDEST53) and FLAG-TBC1D13 WT or empty plasmid (3×FLAG-CMV10) at a 1:2 ratio to ensure cells expressing Rab35 also express TBC1D13 or empty plasmid. After 2 days cells were serum-starved in DMEM containing 0.2% BSA for 2 h and then stimulated or not with 100 nM insulin for 2 min. Cells were washed twice with ice-cold PBS on ice and lysed in 1 ml of lysis buffer (50 mM Tris pH 8.0, 100 mM NaCl, 5 mM MgCl2, 3% NP40) containing 100 μg/ml GST-RBD35 and protease and phosphatase inhibitors (as above). The lysate was passed 10 times through a 22G needle and then centrifuged at 16,000 × g for 10 min at 4°C. The supernatant was added to 40 μl of glutathione sepharose beads (GE Healthcare Biosciences) and incubated at 4°C for 1 h. The beads were washed 3 times with 1 ml of wash buffer (50 mM Tris pH 8.0, 100 mM NaCl, 5 mM MgCl2, 0.5% NP40) and then boiled in Laemmli sample buffer at 95°C for 5 min.
Cell Culture and Transfections
3T3-L1 fibroblasts were cultured and differentiated into adipocytes as described (21). HEK293 cells were cultured as per manufacturer's protocol. For transient transfection a total of 10 μg of DNA was used per 10 cm dish with 30 μl of Lipofectamine 2000 (Invitrogen) following standard protocols. 3T3-L1 fibroblasts were infected with a pBabepuro-HA-GLUT4 retrovirus as previously described (10). After 24 h, infected cells were selected with 2 μg/ml puromycin in DMEM/10% FCS (Invitrogen). Cells were grown and differentiated into adipocytes (10). Adipocytes at day 6–7 post differentiation were electroporated with indicated constructs as previously described (11) and used 48–72 h after electroporation.
Electroporation of siRNA was performed according to the manufacturer's protocol (60). Briefly, 2×106 3T3-L1 adipocytes were electroporated at day 6 post-differentiation with 2 μM siRNA. The adipocytes were re-seeded into matrigel-coated wells of 24-well plates and allowed to recover for 72 h before use.
Glucose Transport Assay
3T3-L1 adipocytes electroporated with the control siRNA or TBC1D13 siRNA pool were basalled for 2 h at 37°C in KRP buffer (12.5 mM Hepes [pH 7.4], 120 mM NaCl, 0.6 mM Na2HPO4, 6 mM KCl, 0.4 mM NaH2PO4, 1.2 mM MgSO4, 1mM CaCl2) with 0.2% BSA. Insulin stimulation was performed with 100 nM insulin for 20 min prior to glucose uptake. Glucose uptake was initiated by adding 0.25 μCi of 2-(1,2-3H[N])-deoxy-D-glucose (Perkin Elmer) in a final concentration of 50 μM 2-deoxy-D-glucose for 5 min at 37°C. Uptake was halted by three washes with ice-cold PBS. The adipocytes were solubilised in 0.4 ml of PBS with 1% Triton X-100 and glucose uptake was determined by scintillation counting.
Sub-cellular Fractionation of Adipocytes
Sub-cellular membrane fractions from 3T3-L1 adipocytes were prepared using a previously described differential centrifugation procedure (25, 61). Briefly, cells were homogenised in HES buffer (20 mM HEPES, 10 mM EDTA, 250 mM sucrose pH 7.4) containing protease and phosphatase inhibitors at 4°C. The lysates were centrifuged at 500 × g for 10 min to remove unbroken cells. The plasma membrane fraction was obtained after a 20 min centrifugation at 17,200 × g followed by centrifugation through sucrose (~2% of total protein). High-density microsomes (HDM) were obtained by centrifuging the 17,200 × g supernatant at 38,700 × g for 20 min (~15% of total protein) and low-density microsomes (LDM) were obtained by spinning the 38,700 × g supernatant at 150,000 × g, 75 min (~21% of total protein) with the supernatant collected as the cytosol fraction (~62% of total protein).
Trafficking assays and Immunofluorescence Microscopy
HA-GLUT4 translocation was performed as previously described (21). Briefly, 3T3-L1 adipocytes were cultured and electroporated and seeded on glass coverslips. Prior to incubation in the absence or presence of 100 nM insulin for 20 min, cells were serum starved for 2 h at 37°C. After washing in cold PBS, cells were fixed with 3% paraformaldehyde (ProSciTech) and quenched with 50 mM glycine in PBS and 2% bovine serum albumin. Cells were blocked in the same solution and either labelled for surface HA-GLUT4 and then permeabilised and labelled with the indicated antibodies. For each experiment 15–20 transfected cells were imaged for both qualitative HA-Rim count (Fig. 1) as well as surface HA fluorescence quantification (Fig. 6). The qualitative count was a simple score of the number of cells that exhibited surface HA staining (HA-Rim). For quantification of HA surface staining, images were collected with the same confocal settings and the HA-signal intensity was analysed using the Leica TCP SP confocal software (Leica Microsystems) as described (22). HA-GLUT4 expression levels (Fig. S3) and Akt phosphorylation (Fig. S4) were quantified in a similar manner. tsVSV-G trafficking was performed as previously described (62). HeLa cells were transfected with FLAG-TBC1D13 and/or GFP-tagged tsVSG-G by lipofection. Cells were reseeded on coverslips 48 h later and after 8 h were moved to incubation at 40°C. The cells were incubated at 40°C for 12 hand were either prepared for immunofluorescence detection or incubated at 32°C for 30 min before preparation for immunofluorescence. Transferrin recycling was performed as previously described (10). Briefly; 3T3-L1 adipocytes retrovirally expressing the Transferrin receptor (TfR) were electroporated with FLAG-TBC1D13 as described above. Cells were incubated for 2 h in serum free media and then in the presence of 50 μg/ml Tf-Alexa-488 (Invitrogen) for 2 h at 37°C. Primary antibodies were detected with anti-rabbit Cy2, Cy3; or anti-mouse Cy2, Cy3, Cy5; (Invitrogen) accordingly. Optical sections were obtained through separate scans for Cy2, Cy3 and Cy5 using the Leica TCP SP confocal laser-scanning microscope (Leica Microsystems).
Statistical Analysis
Data are expressed as means ± one standard deviation (SD). The p-values were calculated by a One-Way ANOVA with Bonferroni's Multiple Comparison post-test. Calculations were performed using GraphPad Prism (GraphPad Software).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Wanjin Hong for the Syntaxin 16 antibody, Dr. Alan Saltiel for the 3T3-L1 adipocyte cDNA library. We thank Dr. Matthew Prior and Dr. Mark Larance for technical advice with the mass spectrometry analysis. We thank Dr. Antony Cooper for critical reading of this manuscript. This work was supported by grants from the NHMRC of Australia and Diabetes Australia Research Trust (to D.E.J) and the National Institutes of Health (DK060564 to D.G.L.). D.E.J is a NHMRC Senior Principal Research Fellow.
REFERENCES
- 1.Pereira-Leal JB, Seabra MC. Evolution of the rab family of small GTP-binding proteins. Journal of Molecular Biology. 2001;313(4):889–901. doi: 10.1006/jmbi.2001.5072. [DOI] [PubMed] [Google Scholar]
- 2.Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2001;2(2):107–117. doi: 10.1038/35052055. [DOI] [PubMed] [Google Scholar]
- 3.Cai H, Reinisch K, Ferro-Novick S. Coats, Tethers, Rabs, and SNAREs Work Together to Mediate the Intracellular Destination of a Transport Vesicle. Developmental Cell. 2007;12(5):671–682. doi: 10.1016/j.devcel.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 4.Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10(8):513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- 5.Segev N. Ypt/Rab GTPases: Regulators of Protein Trafficking. Sci STKE. 2001;2001(100):re11. doi: 10.1126/stke.2001.100.re11. [DOI] [PubMed] [Google Scholar]
- 6.Neuwald AF. A shared domain between a spindle assembly checkpoint protein and Ypt/Rab-specific GTPase-activators. Trends in Biochemical Sciences. 1997;22(7):243–244. doi: 10.1016/s0968-0004(97)01073-6. [DOI] [PubMed] [Google Scholar]
- 7.Bryant NJ, Govers R, James DE. Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol. 2002;3(4):267–277. doi: 10.1038/nrm782. [DOI] [PubMed] [Google Scholar]
- 8.Huang S, Czech MP. The GLUT4 Glucose Transporter. Cell Metabolism. 2007;5(4):237–252. doi: 10.1016/j.cmet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 9.Slot JW, Geuze HJ, Gigengack S, Lienhard GE, James DE. Immuno-localization of the insulin regulatable glucose transporter in brown adipose tissue of the rat. The Journal of cell biology. 1991;113(1):123–135. doi: 10.1083/jcb.113.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shewan AM, van Dam EM, Martin S, Luen TB, Hong W, Bryant NJ, James DE. GLUT4 Recycles via a trans-Golgi Network (TGN) Subdomain Enriched in Syntaxins 6 and 16 But Not TGN38: Involvement of an Acidic Targeting Motif. Mol Biol Cell. 2003;14(3):973–986. doi: 10.1091/mbc.E02-06-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larance M, Ramm G, Stöckli J, van Dam EM, Winata S, Wasinger V, Simpson F, Graham M, Junutula JR, Guilhaus M, James DE. Characterization of the Role of the Rab GTPase-activating Protein AS160 in Insulin-regulated GLUT4 Trafficking. J Biol Chem. 2005;280(45):37803–37813. doi: 10.1074/jbc.M503897200. [DOI] [PubMed] [Google Scholar]
- 12.Miinea CP, Sano H, Kane S, Sano E, Fukuda M, Peranen J, Lane WS, Lienhard GE. AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem J. 2005;391(1):87–93. doi: 10.1042/BJ20050887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jedrychowski MP, Gartner CA, Gygi SP, Zhou L, Herz J, Kandror KV, Pilch PF. Proteomic analysis of GLUT4 storage vesicles reveals LRP1 to be an important vesicle component and target of insulin signaling. J Biol Chem. 2010;285(1):104–114. doi: 10.1074/jbc.M109.040428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ortiz D, Medkova M, Walch-Solimena C, Novick P. Ypt32 recruits the Sec4p guanine nucleotide exchange factor, Sec2p, to secretory vesicles; evidence for a Rab cascade in yeast. J Cell Biol. 2002;157(6):1005–1015. doi: 10.1083/jcb.200201003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rink J, Ghigo E, Kalaidzidis Y, Zerial M. Rab conversion as a mechanism of progression from early to late endosomes. Cell. 2005;122(5):735–749. doi: 10.1016/j.cell.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 16.Rivera-Molina FE, Novick PJ. A Rab GAP cascade defines the boundary between two Rab GTPases on the secretory pathway. Proceedings of the National Academy of Sciences. 2009;106(34):14408–14413. doi: 10.1073/pnas.0906536106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kane S, Sano H, Liu SCH, Asara JM, Lane WS, Garner CC, Lienhard GE. A Method to Identify Serine Kinase Substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase activating protein (GAP) domain. J Biol Chem. 2002;277(25):22115–22118. doi: 10.1074/jbc.C200198200. [DOI] [PubMed] [Google Scholar]
- 18.Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS, Garner CW, Lienhard GE. Insulin-stimulated Phosphorylation of a Rab GTPase-activating Protein Regulates GLUT4 Translocation. J Biol Chem. 2003;278(17):14599–14602. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- 19.Eguez L, Lee A, Chavez JA, Miinea CP, Kane S, Lienhard GE, McGraw TE. Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metabolism. 2005;2(4):263–272. doi: 10.1016/j.cmet.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 20.Sano H, Peck GR, Kettenbach AN, Gerber SA, Lienhard GE. Insulin-stimulated GLUT4 protein translocation in adipocytes requires the Rab10 guanine nucleotide exchange factor Dennd4C. J Biol Chem. 2011;286(19):16541–16545. doi: 10.1074/jbc.C111.228908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramm G, Larance M, Guilhaus M, James DE. A Role for 14-3-3 in Insulin-stimulated GLUT4 Translocation through Its Interaction with the RabGAP AS160. J Biol Chem. 2006;281(39):29174–29180. doi: 10.1074/jbc.M603274200. [DOI] [PubMed] [Google Scholar]
- 22.Stöckli J, Davey JR, Hohnen-Behrens C, Xu A, James DE, Ramm G. Regulation of Glucose Transporter 4 Translocation by the Rab Guanosine Triphosphatase-Activating Protein AS160/TBC1D4: Role of Phosphorylation and Membrane Association. Mol Endocrinol. 2008;22(12):2703–2715. doi: 10.1210/me.2008-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zeigerer A, McBrayer MK, McGraw TE. Insulin stimulation of GLUT4 exocytosis, but not its inhibition of endocytosis, is dependent on RabGAP AS160. Mol Biol Cell. 2004;15(10):4406–4415. doi: 10.1091/mbc.E04-04-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quon MJ, Guerre-Millo M, Zarnowski MJ, Butte AJ, Em M, Cushman SW, Taylor SI. Tyrosine kinase-deficient mutant human insulin receptors (Met1153-->Ile) overexpressed in transfected rat adipose cells fail to mediate translocation of epitope-tagged GLUT4. Proc Natl Acad Sci U S A. 1994;91(12):5587–5591. doi: 10.1073/pnas.91.12.5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Piper RC, Hess LJ, James DE. Differential sorting of two glucose transporters expressed in insulin-sensitive cells. Am J Physiol Cell Physiol. 1991;260(3):C570–580. doi: 10.1152/ajpcell.1991.260.3.C570. [DOI] [PubMed] [Google Scholar]
- 26.Roach WG, Chavez JA, Miinea CP, Lienhard GE. Substrate specificity and effect on GLUT4 translocation of the Rab GTPase-activating protein TBC1D1. Biochem J. 2007;403(2):353–358. doi: 10.1042/BJ20061798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haas AK, Fuchs E, Kopajtich R, Barr FA. A GTPase-activating protein controls Rab5 function in endocytic trafficking. Nat Cell Biol. 2005;7(9):887–893. doi: 10.1038/ncb1290. [DOI] [PubMed] [Google Scholar]
- 28.Fuchs E, Haas AK, Spooner RA, Yoshimura S, Lord JM, Barr FA. Specific Rab GTPase-activating proteins define the Shiga toxin and epidermal growth factor uptake pathways. J Cell Biol. 2007;177(6):1133–1143. doi: 10.1083/jcb.200612068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sklan EH, Serrano RL, Einav S, Pfeffer SR, Lambright DG, Glenn JS. TBC1D20 Is a Rab1 GTPase-activating Protein That Mediates Hepatitis C Virus Replication. J Biol Chem. 2007;282(50):36354–36361. doi: 10.1074/jbc.M705221200. [DOI] [PubMed] [Google Scholar]
- 30.Patino-Lopez G, Dong X, Ben-Aissa K, Bernot KM, Itoh T, Fukuda M, Kruhlak MJ, Samelson LE, Shaw S. Rab35 and Its GAP EPI64C in T Cells Regulate Receptor Recycling and Immunological Synapse Formation. J Biol Chem. 2008;283(26):18323–18330. doi: 10.1074/jbc.M800056200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haas AK, Yoshimura S, Stephens DJ, Preisinger C, Fuchs E, Barr FA. Analysis of GTPase-activating proteins: Rab1 and Rab43 are key Rabs required to maintain a functional Golgi complex in human cells. J Cell Sci. 2007;120(17):2997–3010. doi: 10.1242/jcs.014225. [DOI] [PubMed] [Google Scholar]
- 32.Ng Y, Ramm G, Burchfield JG, Coster AC, Stöckli J, James DE. Cluster analysis of insulin action in adipocytes reveals a key role for Akt at the plasma membrane. J Biol Chem. 2010;285(4):2245–2257. doi: 10.1074/jbc.M109.060236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin S, Rice JE, Gould GW, Keller SR, Slot JW, James DE. The glucose transporter GLUT4 and the aminopeptidase vp165 colocalise in tubulo-vesicular elements in adipocytes and cardiomyocytes. J Cell Sci. 1997;110(Pt 18):2281–2291. doi: 10.1242/jcs.110.18.2281. [DOI] [PubMed] [Google Scholar]
- 34.Will E, Gallwitz D. Biochemical characterization of Gyp6p, a Ypt/Rab-specific GTPase-activating protein from yeast. J Biol Chem. 2001;276(15):12135–12139. doi: 10.1074/jbc.M011451200. [DOI] [PubMed] [Google Scholar]
- 35.Christoforidis S, Zerial M. Purification and Identification of Novel Rab Effectors Using Affinity Chromatography. Methods. 2000;20(4):403–410. doi: 10.1006/meth.2000.0953. [DOI] [PubMed] [Google Scholar]
- 36.Pan X, Eathiraj S, Munson M, Lambright DG. TBC-domain GAPs for Rab GTPases accelerate GTP hydrolysis by a dual-finger mechanism. Nature. 2006;442(7100):303–306. doi: 10.1038/nature04847. [DOI] [PubMed] [Google Scholar]
- 37.Sano H, Roach WG, Peck GR, Fukuda M, Lienhard GE. Rab10 in insulin-stimulated GLUT4 translocation. Biochem J. 2008;411(1):89–95. doi: 10.1042/BJ20071318. [DOI] [PubMed] [Google Scholar]
- 38.Fukuda M, Kobayashi H, Ishibashi K, Ohbayashi N. Genome-wide investigation of the Rab binding activity of RUN domains: development of a novel tool that specifically traps GTP-Rab35. Cell Struct Funct. 2011;36(2):155–170. doi: 10.1247/csf.11001. [DOI] [PubMed] [Google Scholar]
- 39.Longatti A, Lamb CA, Razi M, Yoshimura S, Barr FA, Tooze SA. TBC1D14 regulates autophagosome formation via Rab11- and ULK1-positive recycling endosomes. J Cell Biol. 2012 doi: 10.1083/jcb.201111079. doi: 10.1083/jcb.201111079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marat AL, Ioannou MS, McPherson PS. Connecdenn 3/DENND1C binds actin linking Rab35 activation to the actin cytoskeleton. Mol Biol Cell. 2012;23(1):163–175. doi: 10.1091/mbc.E11-05-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sano H, Eguez L, Teruel MN, Fukuda M, Chuang TD, Chavez JA, Lienhard GE, McGraw TE. Rab10, a Target of the AS160 Rab GAP, Is Required for Insulin-Stimulated Translocation of GLUT4 to the Adipocyte Plasma Membrane. Cell Metabolism. 2007;5(4):293–303. doi: 10.1016/j.cmet.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 42.Prior MJ, Larance M, Lawrence RT, Soul J, Humphrey S, Burchfield J, Kistler C, Davey JR, La-Borde PJ, Buckley M, Kanazawa H, Parton RG, Guilhaus M, James DE. Quantitative proteomic analysis of the adipocyte plasma membrane. J Proteome Res. 2011;10(11):4970–4982. doi: 10.1021/pr200446r. [DOI] [PubMed] [Google Scholar]
- 43.Hsu C, Morohashi Y, Yoshimura S, Manrique-Hoyos N, Jung S, Lauterbach MA, Bakhti M, Gronborg M, Mobius W, Rhee J, Barr FA, Simons M. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A-C. J Cell Biol. 2010;189(2):223–232. doi: 10.1083/jcb.200911018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kouranti I, Sachse M, Arouche N, Goud B, Echard A. Rab35 regulates an endocytic recycling pathway essential for the terminal steps of cytokinesis. Curr Biol. 2006;16(17):1719–1725. doi: 10.1016/j.cub.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 45.Babbey CM, Ahktar N, Wang E, Chen CC-H, Grant BD, Dunn KW. Rab10 Regulates Membrane Transport through Early Endosomes of Polarized Madin-Darby Canine Kidney Cells. Mol Biol Cell. 2006;17(7):3156–3175. doi: 10.1091/mbc.E05-08-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schuck S, Gerl MJ, Ang A, Manninen A, Keller P, Mellman I, Simons K. Rab10 is Involved in Basolateral Transport in Polarized Madin-Darby Canine Kidney Cells. Traffic. 2007;8(1):47–60. doi: 10.1111/j.1600-0854.2006.00506.x. [DOI] [PubMed] [Google Scholar]
- 47.Michaux G, Dyer CE, Nightingale TD, Gallaud E, Nurrish S, Cutler DF. A role for Rab10 in von Willebrand factor release discovered by an AP-1 interactor screen in C. elegans. J Thromb Haemost. 2011;9(2):392–401. doi: 10.1111/j.1538-7836.2010.04138.x. [DOI] [PubMed] [Google Scholar]
- 48.Cormont M, Bortoluzzi MN, Gautier N, Mari M, van Obberghen E, Le Marchand-Brustel Y. Potential role of Rab4 in the regulation of subcellular localization of Glut4 in adipocytes. Mol Cell Biol. 1996;16(12):6879–6886. doi: 10.1128/mcb.16.12.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kessler A, Tomas E, Immler D, Meyer HE, Zorzano A, Eckel J. Rab11 is associated with GLUT4-containing vesicles and redistributes in response to insulin. Diabetologia. 2000;43(12):1518–1527. doi: 10.1007/s001250051563. [DOI] [PubMed] [Google Scholar]
- 50.Sun Y, Bilan PJ, Liu Z, Klip A. Rab8A and Rab13 are activated by insulin and regulate GLUT4 translocation in muscle cells. Proc Natl Acad Sci U S A. 2010;107(46):19909–19914. doi: 10.1073/pnas.1009523107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van der Sluijs P, Hull M, Webster P, Male P, Goud B, Mellman I. The small GTP-binding protein rab4 controls an early sorting event on the endocytic pathway. Cell. 1992;70(5):729–740. doi: 10.1016/0092-8674(92)90307-x. [DOI] [PubMed] [Google Scholar]
- 52.Ullrich O, Reinsch S, Urbe S, Zerial M, Parton RG. Rab11 regulates recycling through the pericentriolar recycling endosome. J Cell Biol. 1996;135(4):913–924. doi: 10.1083/jcb.135.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sonnichsen B, De Renzis S, Nielsen E, Rietdorf J, Zerial M. Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J Cell Biol. 2000;149(4):901–914. doi: 10.1083/jcb.149.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ribon V, Printen JA, Hoffman NG, Kay BK, Saltiel AR. A Novel, Multifunctional c-Cbl Binding Protein in Insulin Receptor Signaling in 3T3-L1 Adipocytes. Mol Cell Biol. 1998;18(2):872–879. doi: 10.1128/mcb.18.2.872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mallard F, Tang BL, Galli T, Tenza D, Saint-Pol A, Yue X, Antony C, Hong W, Goud B, Johannes L. Early/recycling endosomes-to-TGN transport involves two SNARE complexes and a Rab6 isoform. J Cell Biol. 2002;156(4):653–664. doi: 10.1083/jcb.200110081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.James DE, Brown R, Navarro J, Pilch PF. Insulin-regulatable tissues express a unique insulin-sensitive glucose transport protein. Nature. 1988;333:183–185. doi: 10.1038/333183a0. [DOI] [PubMed] [Google Scholar]
- 57.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 58.Westlake CJ, Junutula JR, Simon GC, Pilli M, Prekeris R, Scheller RH, Jackson PK, Eldridge AG. Identification of Rab11 as a small GTPase binding protein for the Evi5 oncogene. Proceedings of the National Academy of Sciences. 2007;104(4):1236–1241. doi: 10.1073/pnas.0610500104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brune M, Hunter JL, Corrie JE, Webb MR. Direct, real-time measurement of rapid inorganic phosphate release using a novel fluorescent probe and its application to actomyosin subfragment 1 ATPase. Biochemistry. 1994;33(27):8262–8271. doi: 10.1021/bi00193a013. [DOI] [PubMed] [Google Scholar]
- 60.Jiang ZY, Zhou QL, Coleman KA, Chouinard M, Boese Q, Czech MP. Insulin signaling through Akt/protein kinase B analyzed by small interfering RNA-mediated gene silencing. Proc Natl Acad Sci U S A. 2003;100(13):7569–7574. doi: 10.1073/pnas.1332633100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marsh B, Alm R, McIntosh S, James D. Molecular regulation of GLUT-4 targeting in 3T3-L1 adipocytes. J Cell Biol. 1995;130(5):1081–1091. doi: 10.1083/jcb.130.5.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJM, Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature. 1997;389(6646):81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.