

Abstract
As a leading cause of childhood mortality worldwide, selection pressure by Plasmodium falciparum continues to shape the human genome. Severe disturbances within the microcirculation result from the adhesion of infected erythrocytes to host receptors on monocytes, platelets, and endothelium. In this prospective study, we compared expression of all major host cytoadhesion receptors among Ugandan children presenting with uncomplicated malaria (n = 1078) versus children with severe malaria (n = 855), including cerebral malaria (n = 174), severe anaemia (n = 522), and lactic acidosis (n = 154). We report a significant survival advantage attributed to blood group O and increased monocyte expression of CD36 and ICAM1 (CD54). The high case fatality rate syndromes of cerebral malaria and lactic acidosis were associated with high platelet CD36 expression and thrombocytopenia, and severe malaria anaemia was characterized by low ICAM1 expression. In a logistic regression model of disease severity, odds ratios for the mitigating effects of blood group O, CD36, and ICAM1 phenotypes were greater than that of sickle haemoglobin. Host genetic adaptations to Plasmodium falciparum suggest new potential malaria treatment strategies.
Keywords: Malaria, ABO, Anaemia, Platelet, CD36, ICAM1
Although the human genome has been remarkably altered by selective pressure from malaria (Weatherall, 2008), infection with Plasmodium falciparum continues to kill large numbers of children born within malaria endemic regions characterized by limited health resources (World Health Organization [WHO], 2011). Prior to developing adaptive immunity and gaining access to therapy, malaria-infected children depend upon host attributes that optimize parasite clearance or mitigate lethal pathophysiology (Stevenson & Riley, 2004). The best appreciated adaptations to P. falciparum, such as sickle haemoglobin and thalassaemia, appear to be concerned not with invasion resistance, but rather with earlier clearance of infected erythrocytes (iRBC) within the spleen as a result of modified red cell fitness (Modiano, 1998). Furthermore, the most lethal malaria syndromes appear to associate with enhanced iRBC adhesion (Miller et al, 2002; Rowe et al, 2009a), wherein parasite traffic to the spleen is reduced by systemic (“extrasplenic”) sequestration of iRBC. Although favourable to survival of the parasite, adhesion and sequestration of iRBC result in microvascular obstruction and tissue hypoperfusion in the host (van der Heyde et al, 2006). Whether human hosts show any evidence of reduced or differential expression of adhesion receptors (Cserti-Gazdewich et al, 2011) as a counter-adaptation to iRBC cytoadhesion has not been fully investigated and is the focus of this study.
The most appealing candidates for possible co-evolution are host receptors of adhesion with P. falciparum Erythrocyte Membrane Protein (PfEMP-1) (Cooke et al, 2004). In laboratory studies, erythrocytes expressing blood group A1, the primordial human ABO type (Calafell et al, 2008), exhibit strong rosette formation through binding of the lectin-like domain of PfEMP-1 (Carlson & Wahlgren, 1992; Vigan-Womas et al, 2012), while the evolutionarily more recent group O erythrocytes rosette least (Rowe et al, 2007). PfEMP-1 contains alternative domains that bind the host receptors ICAM1 (CD54) and CD36. It has been proposed that their expression on monocytes may benefit the host by mediating immune clearance of iRBCs (McGilvray et al, 2000; Baratin et al, 2007). However, ICAM1 and CD36 are also expressed on endothelial cells, promote adhesion of iRBC to the vasculature (Yipp et al, 2000), and therefore may be detrimental to the host. The biology of sequestration via CD36 is further complicated by its expression on platelets (as platelet glycoprotein IV). Platelets expressing CD36 may mediate microvascular occlusion either by facilitating clumping of iRBC (Pain et al, 2001) or by bridging adhesion of iRBC to endothelial cells (Bridges et al, 2010).
Consistent with human-parasite co-evolution, prior studies have observed a higher ratio of blood group O-to-A (Cserti & Dzik, 2007) and a higher prevalence of ICAM1 (Fernandez-Reyes et al, 1997) and CD36 (Gelhaus et al, 2001; Fry et al, 2009) mutations among individuals who reside in areas endemic for P. falciparum. However, prior clinical studies have either examined cytoadhesion receptors individually, not accounted for the effect of other clinical risk factors, or have had small sample sizes. As a result, the relative contribution of the major host cytoadhesion receptors to malaria pathogenesis has remained incompletely characterized (Rowe et al, 2009b; Ochola et al, 2011).
To test the hypothesis that host blood cell phenotypes are associated with malaria severity, we conducted the Cytoadherence in Paediatric Malaria (CPM) Study in a cohort of two thousand children in Uganda. We compared expression of the major host cytoadhesion receptors across a spectrum of malaria severity, quantified their relative effect size compared with other recognized factors that influence severity (such as sickle trait), and examined their distributions in three severe malaria syndromes.
Methods
Study population
This prospective case-control study enrolled children, age 6 months to 12 years, presenting with either initially severe or uncomplicated P falciparum malaria to Mulago Hospital's Acute Care Unit in meso-endemic Kampala, Uganda between October 15, 2007 and October 30, 2009. Children with severe malnutrition were ineligible for enrolment. The malaria diagnosis was established first with clinical symptoms and a positive screening thick smear. Two blinded reviewers of thin and thick smears at a reference parasitology laboratory independently confirmed the diagnosis and parasite density using leucocyte counts (Figs 2 in supporting information).
Fig. 2.
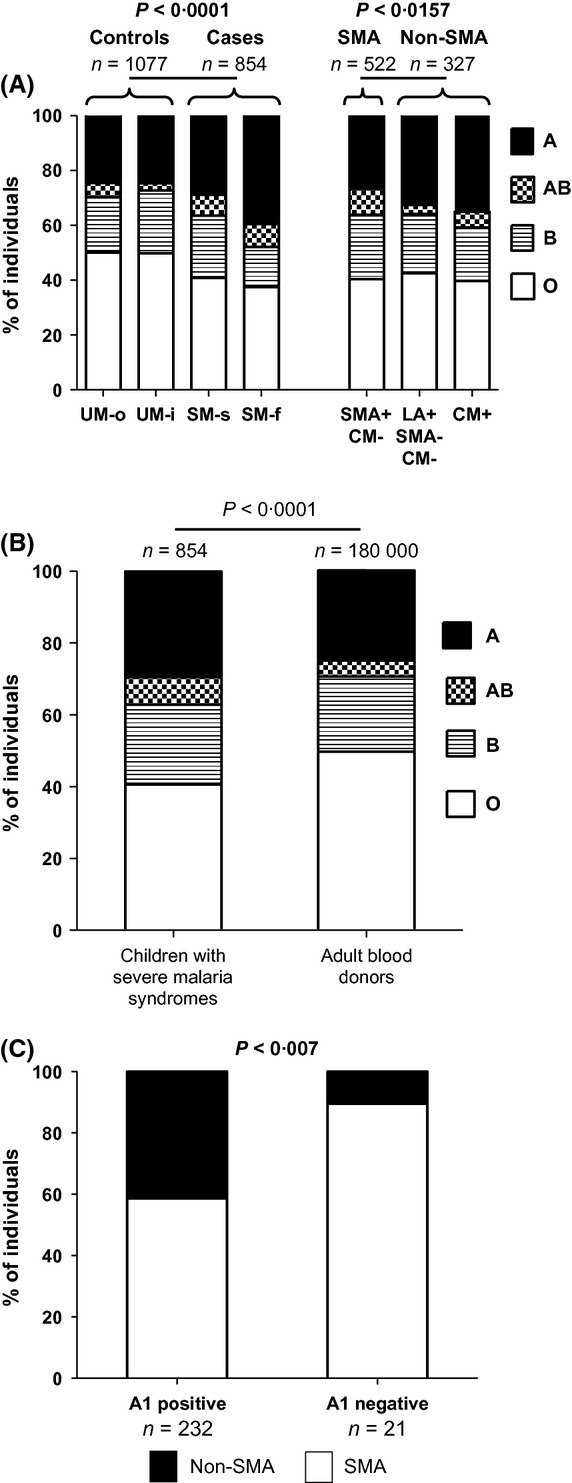
ABO blood groups in patients with malaria. (A) ABO groups according to the spectrum of disease severity. Controls have a higher prevalence of group O, and cases have a higher prevalence of non-group O. (B) ABO distributions among the cases (severe or fatal malaria) are significantly different from that observed in healthy adult Ugandan blood donors. (C) Among group A or AB cases, A1 positive cases are more likely to have CM or LA than those negative for A1. UM-o, uncomplicated malaria treated as an outpatient; UM-i, uncomplicated malaria treated as an inpatient. SM-s, severe malaria, survivor; SM-f, severe malaria, fatal; SMA, severe malaria anaemia; LA, lactic acidosis; CM, cerebral malaria.
Oversight
Approval for the study was granted by Makerere University Faculty of Medicine Research Ethics Committee, the Toronto Academic Health Science Network Research Ethics Board, and the Uganda National Council for Science and Technology. The study was registered as NCT 00707200 at http://www.clinicaltrials.gov.
Consent form
The consent form was available in both English and Luganda and was approved by research ethicists in Toronto, Canada and Kampala, Uganda, in accordance with Good Clinical Practice Guidelines. The research officers, fluent in both languages, were trained in the administration and documentation of informed consent from the parents or legal guardians of paediatric subjects. Parents/guardians retained a copy of consent forms with supplementary study summary pamphlets and investigator contact information.
Data collection
Each enrolled patient was evaluated and treated by physicians experienced in malaria care, using all available clinical resources to assess the presence of other conditions. Data were recorded on a hard-copy Case Report Form (CRF), available at http://www.cd36malaria.org. After each subject's discharge, data were transferred to a digital CRF. Regular fortnightly conference calls (n = 55) reviewed logistics, tabulated accrual statistics, and monitored study quality control. The accuracy of data transfer to the digital CRF was audited on each 25th CRF quarterly. The digital CRF (prepared in FileMaker Pro 9·0 v 1, Santa Clara, CA, USA) mirrored the paper CRF with multiple features to prevent transcription errors. Paper and digital CRFs were kept in a secure, locked environment during enrollment and transferred at the conclusion of enrollment to study headquarters for use in resolution of any discrepancies in the database.
The electronic study database, prepared from exports of the digital CRF, underwent extensive testing for data integrity, consistency, and accuracy. Scrutiny included range value testing, missing data testing, logical tests on data consistency across fields, and multiple comparisons with the original paper CRF record. Strict version control was used. All data analyses were done on a single version of a finalized corrections dataset.
Clinical procedures and categorization
Patients were observed until discharge or death by two study physicians. Prior to analysis, each patient was categorized at final disposition as a case or control, and if failing to meet pre-specified definitions, was excluded. Controls were those with uncomplicated malaria (UM), defined as the absence of any impairment of consciousness or hypoxia, with peripheral blood lactate <5 mmol/l and haemoglobin (Hb) >70 g/l without transfusion. Cases were those with severe malaria (SM) including severe malaria anaemia (SMA), defined as Hb < 50 g/l (or <60 g/l after transfusion); lactic acidosis (LA), defined as blood lactate >5 mmol/l; hypoxia, defined as oxygen saturation <90% while breathing ambient air; or cerebral malaria (CM), as defined below.
Definition of cerebral malaria (CM)
WHO guidelines provide general criteria to characterize cerebral malaria, i.e., Blantyre Coma Scale (BCS) ≤2 with depressed levels or loss of consciousness (LOC) persisting >1 h after any possible prior convulsions (Idro et al, 2005). Due to recent evidence for the non-specificity of WHO criteria for CM when compared with autopsy confirmation or expert retinoscopy (Taylor et al, 2004), and because the neurological literature on LOC has established that prolonged post-ictal states and/or anticonvulsants may themselves affect level of consciousness, we opted for a stricter screening definition of CM that accepted coma under the following conditions:
Coma was present for more than 6 h, particularly if following convulsions, and
Coma was not attributable to high fevers, hypoglycaemia, meningitis, non-malaria-related pre-existing neurological abnormalities, or drugs, such as anticonvulsants or other agents with sedative/hypnotic effects (WHO, 1986).
To increase the specificity of our categorization of CM cases, we used objective clinical criteria that included both WHO features of severe disease, and a case report form-derived scoring system. A case was designated as probable to definite CM (n = 174) if the patient had coma plus >3 of the following 10 WHO malaria severity criteria: >2 seizures in 24 h, respiratory distress, jaundice, haemoglobinuria, spontaneous bleeding, hypoglycaemia (glucose < 2·2 mmol/l), lactic acidosis (lactate >5 mmol/l), normocytic severe anaemia, hyperparasitaemia >5%, or new acute renal failure; or if the patient had a cumulative score of ≥3 points (n = 169) using the following scale:
-
Coma:
1 point: survivor with timing of coma to seizures not specified.
2 points: fatal case with timing of coma to seizures not specified.
3 points: coma present >6 h after last seizure.
-
Blantyre Score <3:
1 point: survivor with timing of Blantyre score to seizures not specified.
2 points: fatal case with timing of Blantyre score to seizures not specified.
3 points: score <3 and performed >6 h after last seizure.
-
Seizures witnessed in hospital:
1 point: survivor with ≤3 observed seizures (or number not recorded) in 24 h.
2 points: fatality with ≤3 observed seizures (or number not reported) in 24 h.
6 points: >3 observed seizures in 24 h.
-
Blood sugar:
0 points: blood sugar not tested at time of coma or Blantyre assessment.
0 points: blood sugar ≥3 mmol/l at time of coma or Blantyre score assessment.
−1 point: blood sugar <3 mmol/l at time of coma or Blantyre score assessment.
-
Lumbar puncture (performed in n = 56):
0 points: not performed.
1 point: performed and did not show signs of meningitis.
-
Retinoscopy (performed in n = 12):
0 points: if negative or not performed.
2 points: if positive.
Laboratory procedures
Patients were excluded if human immunodeficiency virus (HIV) positive. 740 patients were tested for HIV in Kampala using a screening assay (Clearview® STAT-PAK®, Inverness Medical, Louisville CO, USA). Negative screening tests were re-tested with a second assay (Uni-Gold™ Recombigen® HIV, Trinity Biotech PLC, Wicklow, Ireland), and positive screening tests confirmed with a second enzyme-linked assay (Murex HIV Ag/Ab Combination; Abbott Diagnostics Division, Dartford, UK; or Vironistika® HIV Uni-Form II Ag/Ab; bioMérieux, Lyon, France). Archived plasma aliquots from the remaining 1331 who were not tested in Kampala (or not already excluded) were tested with a high-sensitivity commercial assay for antigen and antibody at Mount Sinai Hospital's Microbiology Laboratory in Toronto, Canada (Architect System HIV Ag/Ab Combo; Abbott Diagnostics Division, Weisbaden, Germany). 45 HIV+ subjects were excluded from analysis. Of the 733 subjects identified as HIV-negative by laboratory testing in Kampala, 0·5% of samples were randomly re-assessed in Toronto to validate the negative result.
ABO blood groups were determined locally on peripheral blood collected in EDTA- anticoagulated tubes. ABO Blood Group Reagents (Anti-A BioClone®, Anti-B BioClone®, Anti-D for Slide and Modified Tube Tests, ORTHO® Anti-A1 Lectin; Ortho-Clinical Diagnostics, Inc, Raritan, NJ, USA) were applied to whole blood for direct haemagglutination typing according to manufacturer's directions.
Monocyte CD54, monocyte CD36, and platelet CD36 were determined in a blinded fashion by flow cytometry (Joint Clinical Research Centre, Kampala) according to previously published methods established for malaria patients (Cserti-Gazdewich et al, 2009, 2010; Dzik et al, 2010) measuring median fluorescence intensity (MFI) (Fig 3 in supporting information). Values for monocyte CD36 MFI were corrected for the effect of platelet count prior to analysis (Dzik et al, 2010). Complete blood counts were determined by Coulter AC*T8 (Coulter Corp, Miami, FL, USA). Peripheral blood lactate was measured with a point-of-care device (Lactate-Pro, Arkray Inc, Kyoto, Japan). Oxygen saturation was measured by finger oximetry (Nonin Corp, Plymouth, MN, USA). The presence of haemoglobin S (HbS) was detected by solubility (SickleDex®, Streck Laboratories, Omaha, NB, USA).
Fig. 3.
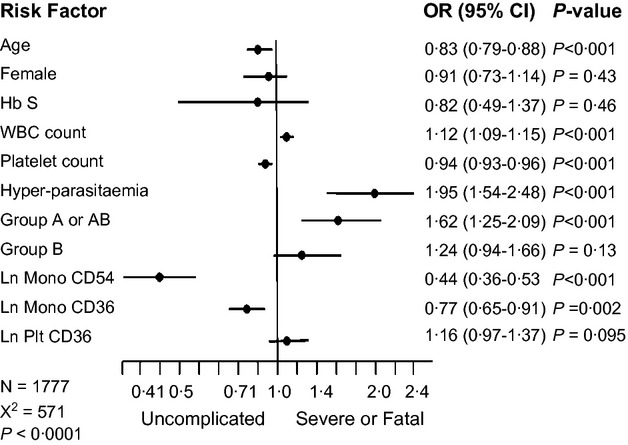
Factors associated with uncomplicated or severe malaria. Results of logistic regression model showing the odds ratios (OR, black circles) and 95% confidence intervals (95% CI, horizontal lines) for uncomplicated versus severe or fatal malaria according to input risk factors. Odds ratios for age are scaled per year and for white blood cell count (WBC) and platelet count (Plt) are scaled in increments of 109/l and 10 × 109/l, respectively. Mono, monocyte; Hb S, haemoglobin S.
Statistical analysis
Continuous data are reported as a median with inter-quartile ranges, and compared using a Wilcoxon test with α = 0·05. Categorical data were compared using the chi-square test with α = 0·05. All comparisons were two tailed.
Logistic regression was used to measure factors associated with severe malaria. Input variables in the model were age, sex, presence of haemoglobin S, leucocyte count, platelet count, presence of hyperparasitaemia, ABO group, monocyte CD54, monocyte CD36 and platelet CD36 (Table I in supporting information). Because group A and AB individuals strongly express the A antigen, they were entered together in the model. Because CD36 and CD54 MFI approximate a log-normal distribution (Fig 4 in supporting information), log-transformed values were entered in the model. Odds ratios (OR) and significance were computed using commercial software (Stata® ver 11.1; Stata Corp, College Station, TX, USA).
Fig. 4.
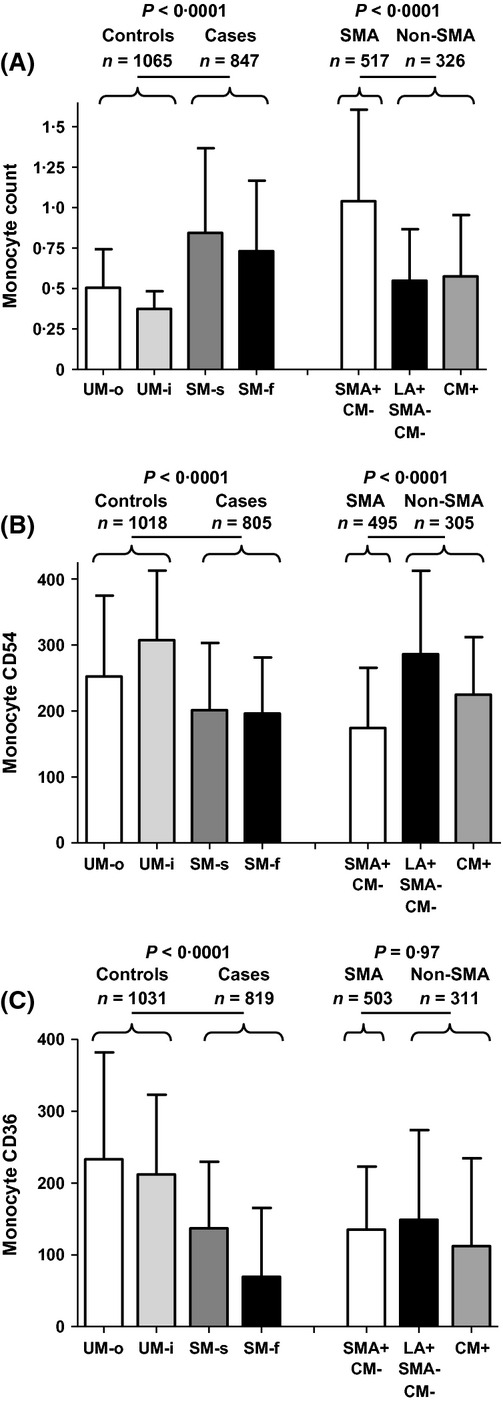
Monocyte CD54 and CD36 expression in patients with malaria. (A) The median and inter-quartile range of monocyte counts is shown. (B) The median and inter-quartile range of CD54 (ICAM1) expression on monocytes (measured as median fluorescence intensity using flow cytometry). Low monocyte CD54 expression is seen among cases with SMA. (C) The median and inter-quartile range of CD36 expression on monocytes (measured as median fluorescence intensity using flow cytometry). A progressive decline in monocyte CD36 expression is associated with categories of increasing disease severity. UM-o, uncomplicated malaria treated as an outpatient; UM-i, uncomplicated malaria treated as an inpatient; SM-s, severe malaria; survivor. SM-f, severe malaria, fatal; SMA, severe malaria anaemia; LA, lactic acidosis; CM, cerebral malaria.
The study was powered to detect a difference in the proportion of cases with blood group A or AB compared with the proportion of controls with group A or AB. Assuming a baseline prevalence of group A or AB ranging from 0·25 to 0·4, our study, with approximately 1000 cases and 1000 controls, could detect a 6% or greater difference in the proportion of group A or AB among cases versus controls with at least 80% power and a type I error probability of 0·05.
Results
Patients
A total of 2092 patients were enrolled, with 81 exclusions (45 HIV+, 35 falsely positive for P falciparum, 1 not of predefined age-range). Of the 2011, 78 were excluded from analysis because at final disposition, they did not meet pre-study definitions of uncomplicated malaria (UM) or severe malaria (SM). Controls included UM outpatients (UM-o, n = 316, 29·1%) and UM inpatients (UM-i, n = 762, 39·4%). Cases comprised SM survivors (SM-s n = 807, 41·7%) and fatalities (SM-f, n = 48, 5·6%) (Fig 1A). Median hospital length of stay was 2, 3 and 1 d among UM-i, SM-s and SM-f, respectively.
Fig. 1.
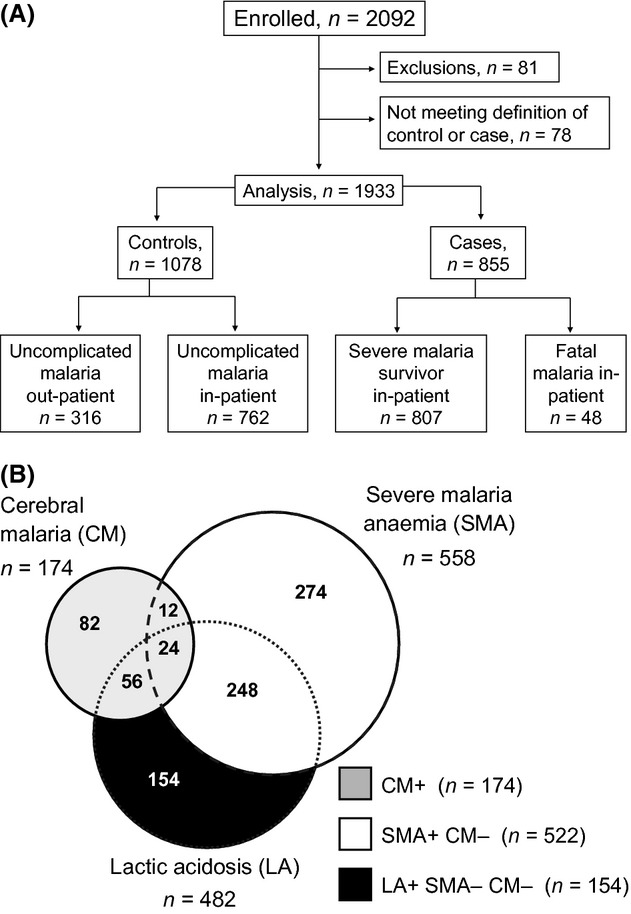
Study design and patient categories. (A) Pre-specified categories of patients enrolled. The analysis of severity of disease compares controls (uncomplicated malaria, UM, n = 1078) with cases (severe malaria, SM, n = 855). (B) Distribution of the three principal severe malaria syndromes among the cases. For analysis, cases are classified into three non-overlapping categories: all cerebral malaria (CM) (grey); severe malaria anaemia (SMA) without CM (white); and isolated lactic acidosis (LA) not attributable to SMA or CM (black).
Illness in the great majority was attributable exclusively to malaria rather than to other infections. None of the 56 lumbar punctures in suspected cases of cerebral malaria showed meningitis. In four chest radiographs of suspected pneumonia, only one showed infiltrates. Parenteral antibiotics were empirically offered in one case of unconfirmed meningitis and 38 cases of unconfirmed pneumonia.
Table I compares baseline patient features between UM and SM. Children with UM and SM had similar sex ratios and prevalence of haemoglobin S (HbS). Cases were significantly younger, had a lower body mass index, had been unwell for longer before hospital presentation, and had a greater extent of palpable splenomegaly. Although median parasitaemia was similar in cases and controls, hyperparasitaemia (>5% of erythrocytes parasitized) was present twice as often among cases (48% vs. 25%). Median (interquartile range) parasitaemia in children positive for HbS was half that of children negative for HbS [44 200 (16 800–141 000) vs. 88 400 (27 400–216 000), P = 0·0031]. As shown in Table I, SM was also characterized by lower platelet counts, a higher prevalence of severe thrombocytopenia, and higher leucocyte and monocyte counts.
Table I.
Clinical and laboratory findings in children with Plasmodium falciparum malaria
| Comparison by severity (n = 1933) | Comparison by severe syndrome type (n = 850) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Controls n = 1078 uncomplicated malaria (UM) | Cases n = 855 severe malaria (SM) | P value UM vs. SM | Severe malaria anaemia (SMA)* n = 522 | Isolated lactic acidosis (LA)† n = 154 | Cerebral malaria (CM)‡ n = 174 | |||||||
| n | n | n | n | n | P value SMA vs. non-SMA ¶ | |||||||
| Patient demographic factors: median, [IQR] or n (%) | ||||||||||||
| Age (years) | 2·9 [1·6–5·1] | 1078 | 1·8 [1·1–3·1] | 855 | <0·0001 | 1·6 [1·0–2·6] | 522 | 2·4 [1·2–3·9] | 154 | 2·51 [1·5–3·9] | 174 | <0·0001 |
| Body mass index | 15·4 [14·0–17·0] | 797 | 14·8 [13·6–16·5] | 655 | <0·0001 | 14·8 [13·6–16·2] | 414 | 15·2 [14·1–17·7] | 115 | 14·7 [13·3–16·7] | 122 | 0·086 |
| Sex (female: male) | 521:557 (48:52) | 1078 | 402:453 (47:53) | 855 | 0·60 | 242:280 (46:54) | 522 | 69:85 (45:55) | 154 | 90:84 (52:48) | 174 | 0·60 |
| Presence of HbS | 57 (6%) | 1045 | 43 (5%) | 826 | 0·89 | 30 (6%) | 505 | 7 (5%) | 149 | 6 (4%) | 167 | 0·33 |
| Malaria presentation features: median, [IQR] or n (%) | ||||||||||||
| Days unwell prior to hospitalization | 3 [2–4] | 1078 | 3 [3–5] | 855 | <0·0001 | 4 [3–5] | 522 | 3 [2–4] | 154 | 3 [3–4] | 174 | <0·0001 |
| Palpable spleen below costal margin (cm) | 0 | 674 | 2 [0–4] | 504 | <0·0001 | 3 [0–4] | 307 | 0·75 [0–3] | 108 | 2 [0–3] | 87 | 0·0002 |
| Haemoglobin (g/l) | 93 [82–104] | 1078 | 45 [36–63] | 855 | n/a | 38 [32–44] | 522 | 72 [59–87] | 154 | 69 [52–82] | 174 | n/a |
| SMA (%) | 0 | 1078 | 558 (65%) | 855 | n/a | 522 (100%) | 522 | 0 (0%) | 154 | 36 (21%) | 174 | n/a |
| MCV (fl) | 84 [78–89] | 1077 | 84 [78–90] | 855 | 0·61 | 85 [79–93] | 522 | 83 [78–88] | 154 | 84 [79–89] | 174 | 0·039 |
| Parasitized RBCs/μl | 83 000 [29 000–190 000] | 1063 | 91 100 [21 600-263 200] | 831 | 0·13 | 67 600 [14 800–191 800] | 503 | 187 000 [61 900–462 600] | 152 | 124 000 [25 600– 386 800] | 171 | <0·0001 |
| % RBCs parasitized | 2·2 [0·8–5·0] | 1062 | 4·6 [1·2–12·9] | 831 | <0·0001 | 4·2 [1·0–11·4] | 503 | 6·6 [2·4–14·3] | 152 | 4·6 [1·1–16·2] | 171 | 0·039 |
| Hyper-parasitaemia (>5% iRBC), (%) | 264 (25%) | 1063 | 402 (48%) | 831 | <0·0001 | 232 (46%) | 503 | 83 (54%) | 152 | 85 (50%) | 171 | 0·11 |
| Platelet count (×109/l) | 136 [81–217] | 1078 | 103 [60–170] | 854 | <0·0001 | 123 [79–181] | 522 | 78 [42–125] | 154 | 73 [43–130] | 174 | <0·0001 |
| Severe (<50 × 109/l) thrombocytopenia | 104 (10%) | 1078 | 166 (19%) | 854 | <0·0001 | 60 (11%) | 522 | 48 (31%) | 154 | 57 (33%) | 174 | <0·0001 |
| Leucocyte count (×109/l) | 7·8 [5·9–10·3] | 1072 | 11·1 [7·7–16·7] | 853 | <0·0001 | 12·6 [8·6–18·7] | 520 | 9·1 [6·6–11·5] | 154 | 9·4 [7·1–14·3] | 174 | <0·0001 |
| Absolute monocyte count (×109/l) | 0·5 [0·3–0·8] | 1065 | 0·8 [0·5–1·4] | 847 | <0·0001 | 1·0 [0·6–1·6] | 517 | 0·5 [0·3–0·9] | 154 | 0·6 [0·3–0·9] | 174 | <0·0001 |
| Lactate (mM) | 2·2 [1·6–3·0] | 1052 | 5·6 [3·1–8·3] | 851 | n/a | 4·8 [2·9–8·8] | 521 | 6·4 [5·7–8·0] | 154 | 4·3 [2·7–8·1] | 171 | 0·0045 |
| Patients with lactic acidosis (>5 mmol/l) (%) | 0 (0%) | 1052 | 482 (56%) | 851 | n/a | 248 (48%) | 521 | 154 (100%) | 154 | 80 (47%) | 171 | <0·0001 |
| Oximetry saturation (%) | 99 [97–100] | 1052 | 97 [94–99] | 849 | n/a | 97 [95–99] | 519 | 98 [96–99] | 154 | 96 [94–98] | 171 | 0·069 |
| Patients with hypoxia (SpO2 <90%) (%) | 0 (0%) | 1052 | 43 (5%) | 849 | n/a | 22 (4%) | 519 | 6 (4%) | 154 | 10 (6%) | 171 | 0·77 |
| Patients with respiratory distress (%) | 74 (7%) | 1078 | 518 (61%) | 855 | <0·0001 | 310 (59%) | 522 | 78 (51%) | 154 | 125 (72%) | 174 | 0·51 |
| Cerebral malaria (%) | 0 (0%) | 1078 | 174 (20%) | 855 | n/a | 0 (0%) | 522 | 0 (0%) | 154 | 174 (100%) | 174 | n/a |
| Deaths (%) | 0 (0%) | 1078 | 48 (4·5%) | 855 | n/a | 10 (1·9%) | 522 | 5 (3·3%) | 154 | 33 (19·0%) | 174 | <0·0001 |
| Host phenotypes for receptors of parasite cytoadhesion ligands: median, [IQR] or n (%) | ||||||||||||
| Group A | 266 (24·7%) | 1077 | 252 (29·5%) | 854 | <0·0001 | 140 (26·8%) | 522 | 50 (32·7%) | 153 | 61 (35·1%) | 174 | 0·016 |
| Group AB | 36 (3·3%) | 65 (7·6%) | 49 (9·4%) | 5 (3·3%) | 10 (5·7%) | |||||||
| Group B | 238 (22·1%) | 190 (22·2%) | 122 (23·4%) | 33 (21·6%) | 34 (19·5%) | |||||||
| Group O | 537 (49·9%) | 347 (40·6%) | 211 (40·4%) | 65 (42·5%) | 69 (39·7%) | |||||||
| A1 positive (A1 or A1B) | 226 (90·0%) | 251 | 227 (92·3%) | 246 | 0·47 | 132 (88·6%) | 149 | 43 (100%) | 43 | 50 (96·2%) | 52 | 0·007 |
| A1 negative (non-A1 and non-A1B) | 25 (10·0%) | 19 (7·7%) | 17 (11·4%) | 0 (0%) | 2 (3·8%) | |||||||
| Monocyte CD54 MFI | 290 [189–404] | 1018 | 201 [118–298] | 805 | <0·0001 | 174 [98–264] | 495 | 286 [188–406] | 143 | 225 [135–311] | 162 | <0·0001 |
| Monocyte CD36 MFI | 216 [121–341] | 1031 | 134 [69–229] | 819 | <0·0001 | 135 [72–222] | 503 | 149 [72–271] | 147 | 112 [58–231] | 164 | 0·97 |
| Platelet CD36 MFI | 140 [82–215] | 1046 | 141 [80–208] | 828 | 0·79 | 129 [77–192] | 507 | 166 [100–243] | 149 | 146 [86–230] | 167 | 0·0009 |
UM, uncomplicated malaria; SM, severe malaria; IQR, interquartile range; MCV, mean corpuscular volume; RBC, red blood cell; iRBC, infected red blood cell; MFI, median fluorescence intensity; n/a Not applicable for comparison.
Severe Malaria Anaemia (SMA) with or without lactic acidosis but excluding cerebral malaria (CM).
Isolated Lactic Acidosis (LA) excludes SMA or CM.
Cerebral Malaria (CM) includes all cases of CM with or without LA or SMA.
non-SMA refers to isolated LA combined with CM, as defined above.
SM cases reflected severe malaria anaemia (SMA, n = 558), lactic acidosis (LA, n = 482), and cerebral malaria (CM, n = 174) with some expected overlap (Fig 1B). Hypoxia was present in 43 children and was the only SM manifestation in five. For the analysis of SM syndromes in Table I and Fig 5, SM cases were classified into three non-overlapping groups as follows: all CM (n = 174); SMA with or without LA (SMA, n = 522); and isolated LA without either SMA or CM (isolated LA, n = 154), Table I, Fig 1B. Because we hypothesize that SMA is a syndrome characterized by less receptor-dependent microvascular occlusion than the syndromes of CM and isolated LA, these severe syndromes were compared accordingly. SMA patients were significantly younger, had evidence of greater splenomegaly, and a lower burden of parasitaemia (Table I). In contrast, children with CM or isolated LA had lower platelet, leucocyte and monocyte counts, and higher blood lactate levels. The case fatality rate (CFR) was nearly ten-fold higher for those with CM compared with SMA (19% vs. 2%, P < 0·0001).
Fig. 5.
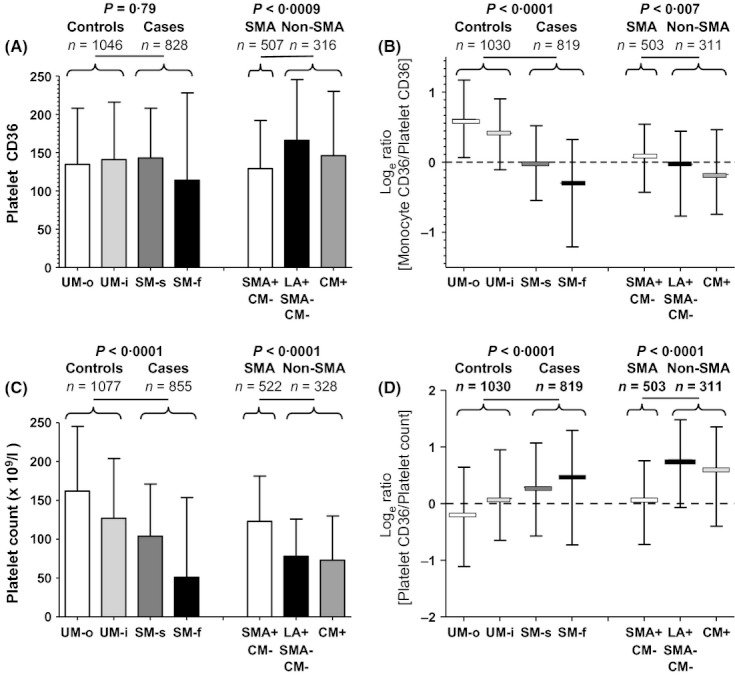
CD36 Expression and platelet count in patients with malaria. (A) The median and inter-quartile range of platelet CD36 expression (measured as median fluorescence intensity through flow cytometry) is shown. (B) A progressive decline in the median and inter-quartile range of the loge of the ratio of monocyte-to-platelet CD36 expression according to disease severity is shown. (C) Increasing disease severity and cases of CM or LA are associated with progressive thrombocytopenia. (D) A progressive rise in the median and inter-quartile range of the loge of the ratio of platelet CD36 expression to platelet count with disease severity and with CM and LA is shown. UM-o, uncomplicated malaria treated as an outpatient; UM-i, uncomplicated malaria treated as an inpatient; SM-s, severe malaria, survivor; SM-f, severe malaria, fatal; SMA, severe malaria anaemia; LA, lactic acidosis; CM, cerebral malaria.
ABO blood group
The distribution of ABO types varied substantially between UM vs. SM, χ2 = 29·57, P < 0·0001. Group A or AB prevalence was 9·1% higher in SM (37·1%) compared with UM (28·0%), P < 0·0001; while group O prevalence was 9·3% higher in UM (49·9%) compared with SM (40·6%), P < 0·0001. This was further reflected in a consistent gradient of changing ABO proportions across severity subgroups (UM-o, UM-i, SM-s, SM-f) (Fig 2A). Group A or AB was more likely than group O to have a fatal versus uncomplicated outcome [OR = 2·27, 95% confidence interval (CI) 1·21–4·28, P = 0·047]. In logistic regression analysis, group A or AB remained significantly associated with SM (OR = 1·50, 95% CI 1·2–2·1, P = 0·0008) after controlling for the effect of age, gender, HbS, platelet count, leucocyte count, hyperparasitaemia, monocyte CD54, monocyte CD36 and platelet CD36 (Fig 3). The ABO distribution was also vastly different between enrolled children with SM (Table I) and the 180 000 healthy adults who donated blood in 2008 to the nationalized Uganda Blood Transfusion Service (A: 24·9%, B: 20·9%, AB: 4·5%, O: 49·7%; personal communication, Dr Dorothy Kyeyune, Nakasero Blood Transfusion Service) (Fig 2B).
The distribution of ABO groups remained significantly different in SM for SMA versus non-SMA cases (χ2 = 10·36, P < 0·0157) (Fig 2A). Although the proportion of group O patients did not vary within the three SM subgroups, (40%, 42%, 40%), the non-O distributions consisted of more group A patients among those with CM or isolated LA versus SMA (33·9% vs. 26·8%, P = 0·033), and correspondingly more group B (B or AB) among those with SMA [32·8% (23·4% + 9·4%) vs. 25·1% (20·5% + 4·6%), P = 0·021]. When group A or AB cases were further sub-classified according to the presence or absence of the A1 antigen, a larger proportion of the patients with CM or isolated LA were represented among those positive for A1 (P = 0·016, Fig 2C). The A1 antigen was present in 97·9% of non-SMA subjects, while A1-negative variants were present in a 5-fold higher proportion (11·4%) in SMA (OR 5·99, 95% CI 1·35–26·56) (Table II in supporting information).
Monocyte CD54 (ICAM1)
Monocyte levels were significantly higher in SMA compared with the other severe malaria syndromes, P < 0·0001 (Fig 4A). Median monocyte CD54 (ICAM1) expression per cell was significantly lower in SMA compared with other SM syndromes (P < 0·0001) or in cases compared with controls (P < 0·0001) (Fig 4B).
Monocyte CD36 and platelet CD36
Monocyte CD36 levels were significantly higher in UM versus SM (P < 0·0001) (Fig 4C). Among cases, however, median monocyte CD36 values did not differ by syndromes. Platelet CD36 expression, in contrast, did not differ between SM and UM, but was significantly higher among those with CM or isolated LA compared with SMA (P = 0·0009), (Fig 5A). Given higher relative monocyte CD36 in UM and higher relative platelet CD36 in non-SMA, the monocyte-to-platelet CD36 ratio best accounted for the differential tissue expression effects of CD36 (Fig 5B). This ratio decreased in proportion with increasing disease severity (P < 0·0001), and with CM and isolated LA.
Thrombocytopenia is recognized by the WHO as a manifestation of severe malaria (WHO, 2000). The present study confirmed this association (Table I) and further found that platelet counts were significantly lower in syndromes associated with cytoadhesion (CM and isolated LA) compared with SMA (Fig 5C). We found that the relationship between platelet CD36 expression and platelet count, reflecting CD36 expression per platelet, varied with both severity and syndrome type. UM-o patients had the highest platelet counts with lowest observed platelet CD36 expression; whereas CM and isolated LA had lowest platelet counts with highest platelet CD36 expression (Fig 5D).
Host receptor phenotypes in fatal and uncomplicated malaria
Table II compares host receptor phenotypes between children with fatal or uncomplicated malaria outcomes. The prevalence of group A or AB was significantly higher (47·9% vs. 28·0%, P = 0·005) in SM-f versus UM, while monocyte ICAM1 and CD36 expression were significantly higher in UM compared with SM-f (P < 0·0001).
Table II.
Host receptor phenotypes in fatal and uncomplicated malaria
| Prevalence in (or value in) fatal malaria | Prevalence in (or value in) uncomplicated malaria | P-value | |
|---|---|---|---|
| Group A or AB | 23/48 47·9% | 302/1077 28·0% | 0·005 |
| Monocyte CD54: median MFI | 196 (95–281) N = 44 | 290 (189–404) N = 1018 | <0·0001 |
| Monocyte CD36: median MFI | 69 (32–160) N = 44 | 215 (121–340) N = 1031 | <0·0001 |
| Platelet CD36: median MFI | 114 (50–222) N = 47 | 140 (82–215) N = 1046 | 0·148 |
| Log e (monoCD36/pltCD36): median | −0·29 (−1·20 to +0·30) N = 44 | +0·48 (−0·07 to +0·95) N = 1030 | <0·0001 |
MFI, median fluorescence intensity; monoCD36, monocyte CD36; pltCD36, platelet CD36.
Discussion
This prospective study of P. falciparum infection demonstrated a relationship between the expression of host receptors for ligands of cytoadhesion and disease severity. The malarial syndromes marked most by hypoperfusion, namely CM and LA, are of particular significance owing to their unrivalled case fatality rates (Marsh et al, 1995). Cytoadhesion results in sequestration and rosetting of iRBCs (Ho et al, 1991; Pongponratn et al, 1991; Silamut et al, 1999; Doumbo et al, 2009), impaired microvascular perfusion, and endothelial damage (van der Heyde et al, 2006). Despite parasiticides, cytoadhesion continues until iRBC structures are cleared (Hughes et al, 2010). We studied children because of their higher relative risk of fatal outcomes, their comparatively less-developed adaptive immunity, and their least-selected distribution of severity-mitigating traits on initial presentation. Our hypothesis was focused not on the absence or presence of malaria (in the matter of its invasion), but on host factors associated with clinical severity.
Factors associated with mortality prior to reproductive age are those most likely to shape the human genome. While previous studies (Table III in supporting information) (Rowe et al, 2007; Fry et al, 2008) have suggested an association between ABO and severity, we document for the first time an increased odds ratio of death associated with A or AB types, providing clinical evidence to substantiate malaria as an evolutionary driver of human ABO blood group distributions (Cserti & Dzik, 2007). We found the relative risk of fatal malaria to UM was 2·3-fold higher for group A or AB versus group O. Among SM cases, the ratio of A to B types was 1·5-fold higher in the syndromes of CM and isolated LA when compared with SMA. We also identified for the first time that group A1 (the wild-type with the highest A antigen expression (Berneman et al, 1991)) was over-represented in the severe malaria syndromes of CM and isolated LA. We propose that genetic selection against the primordial A1 antigen results in enrichment for either group O in association with UM, or for weaker non-A1 types or group B in association with syndromes such as SMA.
This study is novel for the simultaneous analysis of the major malaria cytoadhesion receptors found on erythrocytes, leucocytes, and platelets together with established clinical factors across a spectrum of disease severity. In a logistic regression model, the odds ratio for the harm of blood group A or AB or the benefit of group O remained significant even after controlling for numerous clinical factors including HbS (Fig 3). Consistent with prior reports on its protective effect (Aidoo et al, 2002; Williams et al, 2005; Jallow et al, 2009; Bougouma et al, 2012; Gong et al, 2012), we observed significantly lower levels of parasitaemia in those with HbS. However, perhaps due to the low overall prevalence of HbS in our study population, the proportion of HbS-positive patients was not significantly different in UM vs. SM. In addition, our study design focused on malaria severity, and excluded uninfected controls or asymptomatic parasitaemia. Prior reports have highlighted the role of HbS during these earlier stages of infection (Gong et al, 2012), which our study was not designed to assess. For example, Crompton et al (2008) concluded that the presence of HbS was associated with delayed onset of disease, and Verra et al (2007) proposed that the protective effect of HbS was related to enhanced innate immunity, while Gong et al (2012) inferred both innate and acquired immunity played age-dependent protective roles. Other studies have discovered that the protective effects of HbS may also result from mechanisms including, but not limited to, cytoadhesion (Cholera et al, 2008; Ferreira et al, 2011).
That malaria is likely to be changing global distributions of ABO types (Cserti & Dzik, 2007; Cserti-Gazdewich et al, 2011) is supported not only by our severity or survival associations, but also by the large differences noted between our SM cases and healthy Ugandan adult blood donors. Because the burden of malaria surged over the last several decades despite control efforts, nearly all Ugandan blood donors have successfully overcome prior and frequent exposures to P. falciparum, especially in rural areas with higher endemicity than present in our study. Thus, malaria selection may be a plausible explanation for the shift in ABO distributions that we observed within a single nation and generation.
ICAM1 is expressed on the endothelium and may serve as a sequestration receptor for iRBC. The level of expression of ICAM1 on monocytes has had an uncertain overall role in malaria. Monocyte ICAM1 may indirectly enhance iRBC immune clearance but may conversely co-localize with iRBC in CM (Stevenson & Riley, 2004; Baratin et al, 2007). We found a surprising and significant association between lower monocyte ICAM1 expression in SMA compared with other severe syndromes. This low expression may reflect down-regulation (or inhibition of upregulation) (Skorokhod et al, 2004) of ICAM1 by haemozoin (Schwarzer et al, 1998), which has been linked to the pathogenesis of SMA (Perkins et al, 2011). Alternatively, enhanced clearance of monocytes with high ICAM1 expression may have occurred. Congruent with data from Kenya (Novelli et al, 2010), we found that the clinical factors associated with SMA were younger age, more palpable splenomegaly, and higher monocyte counts.
The effects of CD36 in malaria have likewise been particularly controversial. On monocytes, CD36 supports phagocytosis of iRBC (McGilvray et al, 2000); while on integumentary tissues (such as the fat or skin), it detains iRBC and may enhance vector re-transmission. Cytoadhesion in CM has been proposed to result from bridging iRBCs to endothelial cells via CD36 expression on platelets (Pain et al, 2001; Bridges et al, 2010). For example, Wassmer et al (2004) demonstrated in-vitro that binding of iRBCs to brain endothelial cells was mediated by platelets and was CD36-dependent. Because genetic studies (Fry et al, 2009; Cserti-Gazdewich et al, 2011) have provided conflicting conclusions, we measured CD36 expression phenotypically rather than inferring expression from genotypes. We demonstrated that monocyte CD36 expression was significantly higher in UM versus SM, and that platelet CD36 expression was significantly higher in severe cytoadhesive syndromes. We suggest that the most clinically informative measure of CD36 is the ratio of its monocyte (beneficial) to platelet (harmful) expression, with ratio values inversely proportional to severity and with low ratios associated with CM and isolated LA. This is in keeping with our previously proposed model of the clinical significance of CD36 resulting from differential tissue expression (Cserti-Gazdewich et al, 2011). Genetic alterations assumed to diminish CD36 expression across tissues may in fact have inhomogeneous effects, as shown by zygosity studies where heterozygous deficiency resulted in substantially decreased expression on platelets compared with monocytes (Imai et al, 2002), thus potentially maximizing the benefit of a “carrier” state for CD36 deficiency (Cserti-Gazdewich et al, 2011). As with HbS in malaria, CD36 polymorphisms may exhibit a heterozygous advantage.
Thrombocytopenia is a hallmark of Plasmodial infections, with several mechanisms unique in P. falciparum, including platelet binding to iRBCs and endothelial cells (de Mast, 2011; de Mast et al, 2010). We confirm an association between thrombocytopenia and SM, as well as CM and isolated LA, syndromes speculated to result in part from cytoadhesion.
The strengths of our study include its mechanism-driven design of case evaluation, large-volume accrual, completeness of case reporting, laboratory blinding, and use of validated assays for host receptors of adhesion ligands. In this largest prospective study of malaria outcomes according to host adhesion receptors, limitations nevertheless included: not characterizing parasite-encoded adhesins or performing adhesion assays with patient-derived iRBC isolates (Ochola et al, 2011); inability to directly measure endothelial ligand expression; and not having quantified ICAM1 and CD36 expression through illness and recovery. We do not yet know which factors control expression of ICAM1 and CD36, nor how well their expression on blood cells correlates with endothelial cells. We therefore cannot conclude whether the observed relative expression of host receptors of adhesion were the causes or associated effects of outcomes. Despite attempts to match UM and SM for age prospectively, while not assessing for pre-existing humoral malaria immunity, we saw the expected enrichment of older (and possibly adaptively protected) children among those with UM. Nevertheless, our results remained significant after adjusting for age. Finally, the study's single site location, despite serving as a national referral centre, may have emphasized adaptations that are nevertheless regional and not necessarily generalizable to other malaria-endemic areas.
In summary, we have demonstrated that host phenotypes for receptors of parasite cytoadhesion ligands associate with the clinical severity of malaria, as well as with high mortality manifestations. Specifically, SMA exhibited low ICAM1 expression, whereas CM or isolated LA demonstrated a high prevalence of group A1 and an elevated ratio of platelet-to-monocyte CD36. Informed by the molecular interactions of parasite and human, the measurable “panel” of ABO type, monocyte ICAM1 and CD36 expression, and platelet CD36 expression translates clinically. The determination of ABO type, an unchanging attribute and a categorical measure of the host, is of particular appeal because of its ease of testing in resource-limited settings, and its significance at the level of malaria mortality. The observation that ABO is an independent risk factor for survival among children with malaria provides the most compelling explanation to date for evolution away from the A1 wild-type and towards the weaker subgroups of A, group B or AB, and most importantly group O. Finally, these results invite approaches to malaria treatment that transcend the challenge of developing new cost-prohibitive drug therapies to which the parasite may also gain resistance. Re-directing our focus on co-evolutionary interactions between host and parasite suggests new lines of research. For example, the use of “universal” group O RBCs rather than type-specific RBCs may prove especially advantageous for group A1 patients. Because in vitro studies document that soluble blood group substances can block the formation of iRBC rosettes (Barragan et al, 2000), competitive inhibition of cytoadhesion using plasma-sourced soluble A/B substances or other targeted inhibitors may merit clinical investigation. Medicine may thus strive to recapitulate the advantages already gained by natural selection.
Acknowledgments
Grant support was provided by the International Society of Blood Transfusion Foundation (ISBT), the National Blood Foundation (NBF), the University of Toronto Dean's Fund for New Faculty, and the Robert and Evelyn Luick Fund. We are grateful to all the paediatric patients and their families who agreed to participate in this study, the staff of the MU-UCSF MoLab, the staff of the JCRC Flow Cytometry Lab, the director of the Uganda National Blood Transfusion Service (Dr Dorothy Kyeyune), biostatistical support (Dr Brian Healy, Massachusetts General Hospital), and our web design and database support (Eileen Selogie). Support for reagents and equipment was provided by Beckman Coulter Corporation, Becton Dickinson [BD Biosciences], Masimo Corporation, Whatman Corporation, Bayer, Nonin Medical, and Heart to Heart International.
Authorship contributions
CMC-G, AD, CM, DN-S, AM, and WD conceived and designed the study, and all authors participated in the administrative/technical operations of the project and the idea development, review, preparation, and final approval of this paper. AD and CM enrolled and followed up patients. AD, CM, IS, and CMC-G acquired data and performed data entry. Data analysis and interpretation were done mainly by CMC-G and WD. The text was drafted by CMC-G and WD with contributions from all other authors.
Disclosure of conflicts of interest
None.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Data S1.
{kind=link}
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Aidoo M, Terlouw DJ, Kolczak MS, McElroy PD, ter Kuile FO, Kariuki S, Nahlen BL, Lal AA, Udhayakumar V. Protective effects of the sickle cell gene against malaria morbidity and mortality. Lancet. 2002;359:1311–1312. doi: 10.1016/S0140-6736(02)08273-9. [DOI] [PubMed] [Google Scholar]
- Baratin M, Roetynck S, Pouvelle B, Lemmers C, Viebig NK, Johansson S, Bierling P, Scherf A, Gysin J, Vivier E, Ugolini S. Dissection of the role of PfEMP1 and ICAM-1 in the sensing of Plasmodium-falciparum-infected erythrocytes by natural killer cells. PLoS ONE. 2007;2:e228. doi: 10.1371/journal.pone.0000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barragan A, Kremsner PG, Wahlgren M, Carlson J. Blood group A antigen is a coreceptor in Plasmodium falciparum rosetting. Infection & Immunity. 2000;68:2971–2975. doi: 10.1128/iai.68.5.2971-2975.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berneman ZN, van Bockstaele DR, Uyttenbroeck WM, Van Zaelen C, Cole-Dergent J, Muylle L, Peetermans ME. Flow-cytometric analysis of erythrocytic blood group A antigen density profile. Vox Sanguinis. 1991;61:265–274. doi: 10.1111/j.1423-0410.1991.tb00958.x. [DOI] [PubMed] [Google Scholar]
- Bougouma EC, Tiono AB, Ouedraogo A, Soulama I, Diarra A, Yaro JB, Ouedaogo E, Sanon S, Konate AT, Nebie I, Watson N, Sanza M, Dube TJ, Sirima SB. Haemoglobin variants and Plasmodium falciparum malaria in children under five years of age living in a high and seasonal malaria transmission area of Burkina Faso. Malaria Journal. 2012;11:154. doi: 10.1186/1475-2875-11-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges DJ, Bunn J, van Mourik JA, Grau G, Preston RJ, Molyneux M, Combes V, O'Donnell JS, de Laat B, Craig A. Rapid activation of endothelial cells enables Plasmodium falciparum adhesion to platelet-decorated von Willebrand factor strings. Blood. 2010;115:1472–1474. doi: 10.1182/blood-2009-07-235150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calafell F, Roubinet F, Ramirez-Soriano A, Saitou N, Bertranpetit J, Blancher A. Evolutionary dynamics of the human ABO gene. Human Genetics. 2008;124:123–135. doi: 10.1007/s00439-008-0530-8. [DOI] [PubMed] [Google Scholar]
- Carlson J, Wahlgren M. Plasmodium falciparum erythrocyte rosetting is mediated by promiscuous lectin-like interactions. Journal of Experimental Medicine. 1992;176:1311–1317. doi: 10.1084/jem.176.5.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholera R, Brittain NJ, Gillrie MR, Lopera-Mesa TM, Diakite SA, Arie T, Krause MA, Guindo A, Tubman A, Fujioka H, Diallo DA, Doumbo OK, Ho M, Wellems TE, Fairhurst RM. Impaired cytoadherence of Plasmodium falciparum-infected erythrocytes containing sickle hemoglobin. Proceedings of the National Academy of Sciences USA. 2008;105:991–996. doi: 10.1073/pnas.0711401105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke BM, Mohandas N, Coppel RL. Malaria and the red blood cell membrane. Seminars in Hematology. 2004;41:173–188. doi: 10.1053/j.seminhematol.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Crompton PD, Traore B, Kayentao K, Doumbo S, Ongoiba A, Diakite SA, Krause MA, Doumtabe D, Kone Y, Weiss G, Huang CY, Doumbia S, Guindo A, Fairhurst RM, Miller LH, Pierce SK, Doumbo OK. Sickle cell trait is associated with a delayed onset of malaria: implications for time-to-event analysis in clinical studies of malaria. Journal of Infectious Diseases. 2008;198:1265–1275. doi: 10.1086/592224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cserti CM, Dzik WH. The ABO blood group system and Plasmodium falciparum malaria. Blood. 2007;110:2250–2258. doi: 10.1182/blood-2007-03-077602. [DOI] [PubMed] [Google Scholar]
- Cserti-Gazdewich CM, Dzik WH, Dorn ME, Quagliaroli RO, Xu S, Ssewanyana I, Nayyar R, Preffer FI. Quantitation of CD36 (platelet glycoprotein IV) expression on platelets and monocytes by flow cytometry: application to the study of Plasmodium falciparum malaria. Cytometry Part B Clinical Cytometry. 2009;76:127–134. doi: 10.1002/cyto.b.20443. [DOI] [PubMed] [Google Scholar]
- Cserti-Gazdewich CM, Dzik WH, Erdman L, Ssewanyana I, Dhabangi A, Musoke C, Kain KC. Combined measurement of soluble and cellular ICAM-1 among children with Plasmodium falciparum malaria in Uganda. Malaria Journal. 2010;9:233. doi: 10.1186/1475-2875-9-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cserti-Gazdewich CM, Mayr WR, Dzik WH. Plasmodium falciparum malaria and the immunogenetics of ABO, HLA, and CD36 (platelet glycoprotein IV) Vox Sanguinis. 2011;100:99–111. doi: 10.1111/j.1423-0410.2010.01429.x. [DOI] [PubMed] [Google Scholar]
- Doumbo OK, Thera MA, Kone AK, Raza A, Tempest LJ, Lyke KE, Plowe CV, Rowe JA. High levels of Plasmodium falciparum rosetting in all clinical forms of severe malaria in African children. American Journal of Tropical Medicine and Hygiene. 2009;81:987–993. doi: 10.4269/ajtmh.2009.09-0406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzik WH, Cserti-Gazdewich CM, Ssewanyana I, Delelys M, Preffer FI. When monocytes and platelets compete: the effect of platelet count on the flow cytometric measurement of monocyte CD36. Cytometry Part B Clinical Cytometry. 2010;78:81–87. doi: 10.1002/cyto.b.20504. [DOI] [PubMed] [Google Scholar]
- Fernandez-Reyes D, Craig AG, Kyes SA, Peshu N, Snow RW, Berendt AR, Marsh K, Newbold CI. A high frequency African coding polymorphism in the N-terminal domain of ICAM-1 predisposing to cerebral malaria in Kenya. Human Molecular Genetics. 1997;6:1357–1360. doi: 10.1093/hmg/6.8.1357. [DOI] [PubMed] [Google Scholar]
- Ferreira A, Marguti I, Bechmann I, Jeney V, Chora A, Palha NR, Rebelo S, Henri A, Beuzard Y, Soares MP. Sickle hemoglobin confers tolerance to Plasmodium infection. Cell. 2011;145:398–409. doi: 10.1016/j.cell.2011.03.049. [DOI] [PubMed] [Google Scholar]
- Fry AE, Griffiths MJ, Auburn S, Diakite M, Forton JT, Green A, Richardson A, Wilson J, Jallow M, Sisay-Joof F, Pinder M, Peshu N, Williams TN, Marsh K, Molyneux ME, Taylor TE, Rockett KA, Kwiatkowski DP. Common variation in the ABO glycosyltransferase is associated with susceptibility to severe Plasmodium falciparum malaria. Human Molecular Genetics. 2008;17:567–576. doi: 10.1093/hmg/ddm331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry AE, Ghansa A, Small KS, Palma A, Auburn S, Diakite M, Green A, Campino S, Teo YY, Clark TG, Jeffreys AE, Wilson J, Jallow M, Sisay-Joof F, Pinder M, Griffiths MJ, Peshu N, Williams TN, Newton CR, Marsh K, Molyneux ME, Taylor TE, Koram KA, Oduro AR, Rogers WO, Rockett KA, Sabeti PC, Kwiatkowski DP. Positive selection of a CD36 nonsense variant in sub-Saharan Africa, but no association with severe malaria phenotypes. Human Molecular Genetics. 2009;18:2683–2692. doi: 10.1093/hmg/ddp192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelhaus A, Scheding A, Browne E, Burchard GD, Horstmann RD. Variability of the CD36 gene in West Africa. Human Mutations. 2001;18:444–450. doi: 10.1002/humu.1215. [DOI] [PubMed] [Google Scholar]
- Gong L, Maiteki-Sebuguzi C, Rosenthal PJ, Hubbard AE, Drakeley CJ, Dorsey G, Greenhouse B. Evidence for both innate and acquired mechanisms of protection from Plasmodium falciparum in children with sickle cell trait. Blood. 2012;119:3808–3814. doi: 10.1182/blood-2011-08-371062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Heyde HC, Nolan J, Combes V, Gramaglia I, Grau GE. A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends in Parasitology. 2006;22:503–508. doi: 10.1016/j.pt.2006.09.002. [DOI] [PubMed] [Google Scholar]
- Ho M, Singh B, Looareesuwan S, Davis TM, Bunnag D, White NJ. Clinical correlates of in vitro Plasmodium falciparum cytoadherence. Infection and Immunity. 1991;59:873–878. doi: 10.1128/iai.59.3.873-878.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes KR, Biagini GA, Craig AG. Continued cytoadherence of Plasmodium falciparum infected red blood cells after antimalarial treatment. Molecular and Biochemical Parasitology. 2010;169:71–78. doi: 10.1016/j.molbiopara.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idro R, Jenkins NE, Newton CR. Pathogenesis, clinical features, and neurological outcome of cerebral malaria. Lancet Neurology. 2005;4:827–840. doi: 10.1016/S1474-4422(05)70247-7. [DOI] [PubMed] [Google Scholar]
- Imai M, Tanaka T, Kintaka T, Ikemoto T, Shimizu A, Kitaura Y. Genomic heterogeneity of type II CD36 deficiency. Clinica Chimica Acta. 2002;321:97–106. doi: 10.1016/s0009-8981(02)00102-x. [DOI] [PubMed] [Google Scholar]
- Jallow M, Teo YY, Small KS, Rockett KA, Deloukas P, Clark TG, Kivinen K, Bojang KA, Conway DJ, Pinder M, Sirugo G, Sisay-Joof F, Usen S, Auburn S, Bumpstead SJ, Campino S, Coffey A, Dunham A, Fry AE, Green A, Gwilliam R, Hunt SE, Inouye M, Jeffreys AE, Mendy A, Palotie A, Potter S, Ragoussis J, Rogers J, Rowlands K, Somaskantharajah E, Whittaker P, Widden C, Donnelly P, Howie B, Marchini J, Morris A, Sanjoaquin M, Achidi EA, Agbenyega T, Allen A, Amodu O, Corran P, Djimde A, Dolo A, Doumbo OK, Drakeley C, Dunstan S, Evans J, Farrar J, Fernando D, Hien TT, Horstmann RD, Ibrahim M, Karunaweera N, Kokwaro G, Koram KA, Lemnge M, Makani J, Marsh K, Michon P, Modiano D, Molyneux ME, Mueller I, Parker M, Peshu N, Plowe CV, Puijalon O, Reeder J, Reyburn H, Riley EM, Sakuntabhai A, Singhasivanon P, Sirima S, Tall A, Taylor TE, Thera M, Troye-Blomberg M, Williams TN, Wilson M, Kwiatkowski DP. Genome-wide and fine-resolution association analysis of malaria in West Africa. Nature Genetics. 2009;41:657–665. doi: 10.1038/ng.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh K, Forster D, Waruiru C, Mwangi I, Winstanley M, Marsh V, Newton C, Winstanley P, Warn P, Peshu N, Pasvol G, Snow R. Indicators of life-threatening malaria in African children. New England Journal of Medicine. 1995;332:1399–1404. doi: 10.1056/NEJM199505253322102. [DOI] [PubMed] [Google Scholar]
- de Mast Q. Hematological Alterations in Malaria. Nijmegen: Radboud University Nijmegen Medical Center; 2011. PhD Thesis. [Google Scholar]
- de Mast Q, de Groot PG, van Heerde WL, Roestenberg M, van Velzen JF, Verbruggen B, Roest M, McCall M, Nieman AE, Westendorp J, Syafruddin D, Fijnheer R, van Dongen-Lases EC, Sauerwein RW, van der Ven AJ. Thrombocytopenia in early malaria is associated with GP1b shedding in absence of systemic platelet activation and consumptive coagulopathy. British Journal of Haematology. 2010;151:495–503. doi: 10.1111/j.1365-2141.2010.08399.x. [DOI] [PubMed] [Google Scholar]
- McGilvray ID, Serghides L, Kapus A, Rotstein OD, Kain KC. Nonopsonic monocyte/macrophage phagocytosis of Plasmodium falciparum-parasitized erythrocytes: a role for CD36 in malarial clearance. Blood. 2000;96:3231–3240. [PubMed] [Google Scholar]
- Miller LH, Baruch DI, Marsh K, Doumbo OK. The pathogenic basis of malaria. Nature. 2002;415:673–679. doi: 10.1038/415673a. [DOI] [PubMed] [Google Scholar]
- Modiano G. An appraisal of the best known erythrocytic genetic adaptations of man to malaria and its implications. International Journal of Anthropology. 1998;13:295–314. [Google Scholar]
- Novelli EM, Hittner JB, Davenport GC, Ouma C, Were T, Obaro S, Kaplan S, Ong'echa JM, Perkins DJ. Clinical predictors of severe malarial anaemia in a holoendemic Plasmodium falciparum transmission area. British Journal of Haematology. 2010;149:711–721. doi: 10.1111/j.1365-2141.2010.08147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochola LB, Siddondo BR, Ocholla H, Nkya S, Kimani EN, Williams TN, Makale JO, Liljander A, Urban BC, Bull PC, Szestak T, Marsh K, Craig AG. Specific receptor usage in Plasmodium falciparum cytoadherence is associated with disease outcome. PLoS One. 2011;6:e14741. doi: 10.1371/journal.pone.0014741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pain A, Ferguson DJ, Kai O, Urban BC, Lowe B, Marsh K, Roberts DJ. Platelet-mediated clumping of Plasmodium falciparum-infected erythrocytes is a common adhesive phenotype and is associated with severe malaria. Proceedings of the National Academy of Sciences USA. 2001;98:1805–1810. doi: 10.1073/pnas.98.4.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins DJ, Were T, Davenport GC, Kempaiah P, Hittner JB, Ong'echa JM. Severe malarial anemia: innate immunity and pathogenesis. International Journal of Biological Sciences. 2011;7:1427–1442. doi: 10.7150/ijbs.7.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pongponratn E, Riganti M, Punpoowong B, Aikawa M. Microvascular sequestration of parasitized erythrocytes in human falciparum malaria: a pathological study. American Journal of Tropical Medicine and Hygiene. 1991;44:168–175. doi: 10.4269/ajtmh.1991.44.168. [DOI] [PubMed] [Google Scholar]
- Rowe JA, Handel IG, Thera MA, Deans AM, Lyke KE, Kone A, Diallo DA, Raza A, Kai O, Marsh K, Plowe CV, Doumbo OK, Moulds JM. Blood group O protects against severe Plasmodium falciparum malaria through the mechanism of reduced rosetting. Proceedings of the National Academy of Sciences USA. 2007;104:17471–17476. doi: 10.1073/pnas.0705390104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe JA, Claessens A, Corrigan RA, Arman M. Adhesion of Plasmodium falciparum-infected erythrocytes to human cells: molecular mechanisms and therapeutic implications. Expert Reviews in Molecular Medicine. 2009a;11:e16. doi: 10.1017/S1462399409001082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe JA, Opi DH, Williams TN. Blood groups and malaria: fresh insights into pathogenesis and identification of targets for intervention. Current Opinion in Hematology. 2009b;16:480–487. doi: 10.1097/MOH.0b013e3283313de0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer E, Alessio M, Ulliers D, Arese P. Phagocytosis of the malarial pigment, hemozoin, impairs expression of major histocompatibility complex class II antigen, CD54, and CD11c in human monocytes. Infection and Immunity. 1998;66:1601–1606. doi: 10.1128/iai.66.4.1601-1606.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silamut K, Phu NH, Whitty C, Turner GD, Louwrier K, Mai NT, Simpson JA, Hien TT, White NJ. A quantitative analysis of the microvascular sequestration of malaria parasites in the human brain. American Journal of Pathology. 1999;155:395–410. doi: 10.1016/S0002-9440(10)65136-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorokhod OA, Alessio M, Mordmuller B, Arese P, Schwarzer E. Hemozoin (malarial pigment) inhibits differentiation and maturation of human monocyte-derived dendritic cells: a peroxisome proliferator-activated receptor-gamma-mediated effect. Journal of Immunology. 2004;173:4066–4074. doi: 10.4049/jimmunol.173.6.4066. [DOI] [PubMed] [Google Scholar]
- Stevenson MM, Riley EM. Innate immunity to malaria. Nature Reviews in Immunology. 2004;4:169–180. doi: 10.1038/nri1311. [DOI] [PubMed] [Google Scholar]
- Taylor TE, Fu WJ, Carr RA, Whitten RO, Mueller JS, Fosiko NG, Lewallen S, Liomba NG, Molyneux ME. Differentiating the pathologies of cerebral malaria by postmortem parasite counts. Nature Medicine. 2004;10:143–145. doi: 10.1038/nm986. [DOI] [PubMed] [Google Scholar]
- Verra F, Simpore J, Warimwe GM, Tetteh KK, Howard T, Osier FH, Bancone G, Avellino P, Blot I, Fegan G, Bull PC, Williams TN, Conway DJ, Marsh K, Modiano D. Haemoglobin C and S role in acquired immunity against Plasmodium falciparum malaria. PLoS One. 2007;2:e978. doi: 10.1371/journal.pone.0000978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigan-Womas I, Guillotte M, Juillerat A, Hessel A, Raynal B, England P, Cohen JH, Bertrand O, Peyrarad T, Bentley GA, Lewit-Bentley A, Mercereau-Puijalon O. Structural basis for the ABO blood-group dependence of Plasmodium falciparum rosetting. PLoS Pathogens. 2012;8:e1002781. doi: 10.1371/journal.ppat.1002781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassmer SC, Lepolard C, Traore B, Pouvelle B, Gysin J, Grau GE. Platelets reorient Plasmodium falciparum-infected erythrocyte cytoadhesion to activated endothelial cells. Journal of Infectious Diseases. 2004;189:180–189. doi: 10.1086/380761. [DOI] [PubMed] [Google Scholar]
- Weatherall DJ. Genetic variation and susceptibility to infection: the red cell and malaria. British Journal of Haematology. 2008;141:276–286. doi: 10.1111/j.1365-2141.2008.07085.x. [DOI] [PubMed] [Google Scholar]
- WHO. Severe and complicated malaria. World Health Organization Malaria Action Programme. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1986;80(Suppl):3–50. [PubMed] [Google Scholar]
- WHO. Severe falciparum malaria. World Health Organization, Communicable Diseases Cluster. Transactions of the Royal Society of Tropical Medicine and Hygiene. 2000;94(Suppl. 1):S1–S90. [PubMed] [Google Scholar]
- WHO. World Malaria Report. Geneva, Switzerland: World Health Organization; 2011. [Google Scholar]
- Williams TN, Mwangi TW, Wambua S, Alexander ND, Kortok M, Snow RW, Marsh K. Sickle cell trait and the risk of Plasmodium falciparum malaria and other childhood diseases. Journal of Infectious Diseases. 2005;192:178–186. doi: 10.1086/430744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yipp BG, Anand S, Schollaardt T, Patel KD, Looareesuwan S, Ho M. Synergism of multiple adhesion molecules in mediating cytoadherence of Plasmodium falciparum-infected erythrocytes to microvascular endothelial cells under flow. Blood. 2000;96:2292–2298. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.