

Abstract
AIM: To identify the prevalence of pre-S2 start codon mutations and to assess their association with liver disease progression.
METHODS: The mutations were identified by direct sequencing from 73 asymptomatic carriers, 66 chronic hepatitis (CH), 66 liver cirrhosis (LC) and 63 hepatocellular carcinoma (HCC) patients. Statistical significances were determined using Fisher’s exact test, χ2 test, and t-test analyses whenever appropriate. Pre-S mutation as a risk factor for advanced liver disease was estimated by unconditional logistic regression model adjusted with age, sex, and hepatitis B e antigen (HBeAg). P < 0.05 was considered significant.
RESULTS: Mutation of the hepatitis B virus (HBV) pre-S2 start codon was found in 59 samples from 268 subjects (22.0%), with higher prevalence in patients with cirrhosis 27/66 (40.9%) followed by HCC 18/63 (28.6%), chronic hepatitis 12/66 (18.2%) and asymptomatic carriers 2/73 (2.7%) (P < 0.001). Logistic regression analysis showed that pre-S2 start codon mutation was an independent factor for progressive liver disease. Other mutations, at T130, Q132, and A138, were also associated with LC and HCC, although this was not statistically significant when adjusted for age, sex, and HBeAg. The prevalence of pre-S2 start codon mutation was higher in HBV/B than in HBV/C (23.0% vs 19.1%), whilst the prevalence of T130, Q132, and A138 mutation was higher in HBV/C than in HBV/B. The prevalence of pre-S2 start codon mutation was higher in LC (38.9%) and HCC (40.0%) than CH (5.6%) in HBeAg(+) group, but it was similar between CH, LC and HCC in HBeAg(-) group.
CONCLUSION: Pre-S2 start codon mutation was higher in Indonesian patients compared to other Asian countries, and its prevalence was associated with advanced liver disease, particularly in HBeAg(+) patients.
Keywords: Hepatitis B virus, Pre-S2 start codon, Liver disease, Hepatitis B e antigen seroconversion, Indonesia
INTRODUCTION
Hepatitis B virus (HBV) infection is one of the most important infectious diseases worldwide. It is the major cause of chronic hepatitis (CH), liver cirrhosis (LC), and hepatocellular carcinoma (HCC). About 40% of the world’s population has had contact with, or are carriers of, HBV and it is estimated that more than 350 million people world wide have chronic liver infection[1]. Indonesia has a moderate to high endemnicity of HBV infection[2], perhaps due to the lack of proper health facilities, poor economical status, less public awareness, or incomplete vaccination.
HBV is a relaxed circular, partially double stranded 3.2 kb DNA virus[3]. It has four overlapping reading frames that encode Polymerase, Core, X and Envelope proteins[4]. The Envelope proteins are produced from a single open reading frame with three different translation sites, pre-S1, pre-S2, and S. It produces three forms of HBV surface proteins (HBs), which are the large (L), middle (M) and small (S) HBs[5]. The major component of the Envelope protein is S protein which consists of 226 amino acids and drives particle budding. The M protein is composed of the S protein with an additional of 55 amino acids termed pre-S2 attached to the N-terminus. The L protein is the M protein with an additional 108 or 119 genotype-dependent amino acids attached to the N-terminus[6].
The pre-S1 and pre-S2 region is the region with the most variability in the HBV genome[7]. This variability is in the form of deletions, insertions, or synonymous or non-synonymous nucleotide substitutions. The pre-S1 and pre-S2 regions encode the T- and B-cell epitopes which play roles in allowing neutralizing antibodies to bind, and consequent immune protection[8,9]. Therefore, HBV pre-S mutant variants may emerge as a result of selective immune pressure.
The high prevalence of HBV pre-S mutations in Asia, including pre-S deletion and pre-S2 start codon mutations, has been reported, and it has been demonstrated that these mutations were associated with progressive liver disease[10]. In addition, amino acid substitution from Phenylalanine to Lysine at codon 141 in pre-S2 region (F141L) is also associated with HCC in patients infected with HBV genotype C[11]. Furthermore, study of hepatitis B e antigen (HBeAg) (-) patients in China revealed that pre-S deletions alone, or in combination with mutations in precore and basal core promoter (BCP) of HBV, are also associated with advanced liver disease[12].
Studies on the prevalence of HBV genotypes, BCP and precore mutations, and their association with severity of liver disease in Indonesia have previously been reported[13-15]. We have recently published a molecular epidemiological study on the prevalence of pre-S deletion mutation in Indonesian subjects[16], but this paper did not describe the incidence of synonymous and non-synonymous mutations in the pre-S region. Therefore, we extended this study to investigate the prevalence of pre-S2 start codon mutation and its association with severe liver disease in Indonesian patients. In addition, other mutations in the pre-S2 regions were analyzed.
MATERIALS AND METHODS
Samples
Blood samples were obtained from 270 HBV carriers. Two subjects with genotype D were excluded due to its relatively low prevalence in the collective samples and also in Asian population in general. A total of 268 subjects were included in the study: 73 asymptomatic carriers (AC) and 195 liver disease patients which composed of 66 CH, 66 LC, and 63 HCC. Asymptomatic carrier samples were taken from donors with positive HBsAg from the Blood Transfusion Unit (Red Cross Makassar, South Sulawesi, Indonesia), between February and August 2007. Chronic hepatitis samples were from patients positive for HBsAg for more than six months, and who had more than twice the normal alanine aminotransferase (ALT) level. Liver cirrhosis was diagnosed by liver function tests and ultrasonography, whilst the diagnosis of HCC was either on the basis of ultrasonography as well as an elevated serum-fetoprotein (AFP) level (≥ 200 ng/mL), or by needle aspiration liver biopsy for samples in which the AFP level was low. Samples from CH, LC, and HCC patients were collected from Cipto Mangunkusumo Hospital, Gatot Soebroto Hospital, Klinik Hati, Jakarta, Siloam Hospital Lippo Karawaci, Tangerang, Mataram General Hospital, Mataram, Wahidin Sudirohusodo Hospital, Makassar, and M. Djamil Hospital, Padang, from May 2006 to February 2010. None of the samples had coinfection with hepatitis C virus. The HBsAg test for all samples was performed using a commercially available enzyme-linked immunosorbent assay, kit (Abbott Laboratories, Chicago, IL, United States). The HBeAg status of samples was tested by using rapid test (Intec, Xiamen, China) as described previously[16]. Blood samples were separated into plasma and stored at -70 °C until use. The study was approved by the Committee on Health Research Ethics of the Mochtar Riady Institute for Nanotechnology and informed consent was obtained from each subject.
Viral DNA extraction, polymerase chain reaction and sequencing
HBV DNA was extracted from 200 μL plasma using the QIAamp DNA blood mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions, and 80 μL of eluted DNA was stored at -70 °C until use. Pre-S region was amplified by nested polymerase chain reaction (PCR) using PCR Core System (Promega, Madison, WI, United States) and two sets of primers as previously described[16]. The first round was performed with primers PS1 (5’-GGGTCACCTTATTCTTGGGA-3’, position 2814-2833) and PS2 (5’-CCCCGCCTGTAACACGAGCA-3’, position 208-189). For the second round, primers PS3 (5’-TTGGGAACAAGATCTACAGC-3’, position 2828-2847) and PS4 (5’-GTCCTGATGCGATGTTCTCC-3’, position 176-157) were used. First and second round PCR were performed with the same profile for 36 cycles of 94 °C for 1 min, 58 °C for 30 s and 72 °C for 1 min in a thermal cycler. The PCR products were purified with Wizard® SV Gel and PCR Clean-Up System (Promega, Madison, WI, United States), directly sequenced employing an ABI 3130xl Genetic Analyzer (Applied Biosystems, Inc., Foster City, CA, United States) with the Big Dye Terminator V3.1 Cycle Sequencing kit (Applied Biosystems, Inc.) using primers PS3 and PS4[13-16].
Hepatitis B virus genotyping and pre-S mutations analysis
The HBV genotype was determined by phylogenetic analysis of the pre-S sequences. The sequences were compared to 74 HBV reference strains of eight HBV genotypes (A-H) from GenBank. Alignment and phylogenetic trees were constructed using molecular evolutionary genetics analysis 4 software (Center for Evolutionary Functional Genomics, Tempe, AZ, United States)[17]. Pre-S amino acid sequences were aligned and compared between each group to detect the mutations.
Statistical analysis
All statistical analyses were performed using SPSS 17.0 software for Windows (SPSS Inc., Chicago, IL, United States). Statistical significances were determined using Fisher’s exact test, χ2 test, and t-test analyses whenever appropriate. The odds ratio and 95% CI were estimated by unconditional logistic regression model to evaluate the association of Pre-S mutation as a risk factor for advanced liver disease adjusted with age, sex and HBeAg. P < 0.05 was considered to be statistically significant.
RESULTS
Demographic data of subjects
Demographic data of subjects are shown in Table 1. A total of 268 subjects comprising 73 AC, 66 CH, 66 LC and 63 HCC were included in the study. The male/female ratio was significantly different among the groups (P = 0.002). The mean of ages for AC, CH, LC, and HCC was 32.3 ± 9.1, 41.9 ± 13.3, 50.1 ± 10.9 and 48.6 ± 11.6 years, respectively, and it was significantly different among the group (P < 0.001). Ratio of HBV/B to HBV/C showed no significant difference among the groups (P = 0.267). Of 268 subjects, 235 subjects were available for HBeAg rapid test analysis. Of 235 subjects, 82 (34.9%) had HBeAg(+) and 153 (65.1%) had HBeAg(-) and the distribution was significantly different among the group (P < 0.001).
Table 1.
Demographic data and the prevalence of pre-S mutations associated with progressive liver disease
| AC | CH | LC | HCC | All | P value (all) | P value (AC-CH vs LC) | P value (AC-CH vs HCC) | |
| n (%) | 73 (27.2) | 66 (24.6) | 66 (24.6) | 63 (23.5) | 268 (100.0) | - | - | - |
| Gender, n (%) | ||||||||
| Male | 69 (94.5) | 49 (74.2) | 46 (69.7) | 49 (77.7) | 213 (79.5) | 0.002 | 0.027 | 0.314 |
| Female | 4 (5.5) | 17 (25.8) | 20 (30.3) | 14 (22.2) | 55 (20.5) | |||
| Age (yr, mean ± SD) | 32.3 ± 9.1 | 41.9 ± 13.3 | 50.1 ± 10.9 | 48.6 ± 11.6 | 42.9 ± 13.3 | < 0.001 | < 0.001 | < 0.001 |
| Genotype, n (%) | ||||||||
| B | 56 (76.7) | 46 (69.7) | 46 (69.7) | 52 (82.5) | 200 (74.6) | 0.267 | 0.453 | 0.393 |
| C | 17 (23.3) | 20 (30.3) | 20 (30.3) | 11 (17.5) | 68 (25.4) | |||
| HBeAg1 | ||||||||
| HBeAg(+) | 13 (22.0) | 36 (56.3) | 18 (30.5) | 15 (28.3) | 82 (34.9) | < 0.001 | 0.222 | 0.144 |
| HBeAg(-) | 46 (78.0) | 28 (43.8) | 41 (69.5) | 38 (71.7) | 153 (65.1) | |||
| Mutation | ||||||||
| M120 | 2 (2.7) | 12 (18.2) | 27 (40.9) | 18 (28.6) | 59 (22.0) | < 0.001 | < 0.001 | 0.002 |
| T130 | 8 (11.0) | 19 (28.8) | 18 (27.3) | 25 (39.7) | 70 (26.1) | 0.0018 | 0.311 | 0.054 |
| Q132 | 11 (15.1) | 19 (28.8) | 21 (31.8) | 26 (41.3) | 77 (28.7) | 0.008 | 0.111 | 0.04 |
| A138 | 10 (13.7) | 21 (31.8) | 22 (33.3) | 26 (41.3) | 79 (29.5) | 0.003 | 0.089 | 0.094 |
Number of samples for HBeAg. n (all) = 235, n (AC:CH:LC:HCC) = 59:64:59:53. AC: Asymptomatic carrier; CH: Chronic hepatitis; LC: Liver cirrhosis; HCC: Hepatocellular carcinoma; HBeAg: Hepatitis B e antigen.
Prevalence of pre-S2 start codon mutation and its association with the risk of advanced liver disease
Pre-S2 start codon mutations were found in 22.0% (59/268) of the total samples (Table 1). The mutation (M120) was either an amino acid substitution from Methionine to other amino acids or a deletion mutation (Figure 1). The prevalence of this mutation was significantly different among the groups (P < 0.001), and was increasingly common as disease progressed from AC (2.7%) to CH (18.2%) and LC (40.9%), but was less common in those with HCC (28.6%) than with LC but this difference was not statistically significant (Figure 2A). Multivariate regression analysis (based on 235 subjects with complete HBeAg) adjusted with age, gender and HBeAg demonstrated that M120 mutation was an independent factor in the development of progressive liver disease [OR 3.996 (1.830-8.729), P = 0.0005] (Table 2). These results implied that this mutation was associated with progressive liver disease and more particularly with LC than HCC.
Figure 1.

Amino acid alignment of 59 hepatitis B virus sequences harboring pre-S2 start codon mutation with wild type reference M54923 (genotype B). Positions of mutations are shown in black shades (M120, T130, Q132, and A138). Dots represent identical amino acids to the consensus sequence. Dashes represent deletion mutation. The names of the samples are indicated with each respective genotype in parentheses. AC: Asymptomatic carrier; CH: Chronic hepatitis; LC: Liver Cirrhosis; HCC: Hepatocellular carcinoma.
Figure 2.
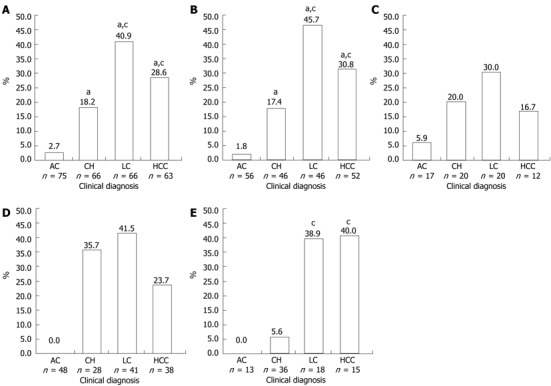
Prevalence of pre-S2 start codon mutation in all samples and in patients infected with hepatitis B virus/B, hepatitis B virus/C, hepatitis B e antigen (+), hepatitis B e antigen (-). A: Prevalence of pre-S2 start codon mutation in all samples; B: Prevalence of pre-S2 start codon mutation in patients infected with hepatitis B virus (HBV)/B; C: Prevalence of pre-S2 start codon mutation in patients infected with HBV/C; D: Prevalence of pre-S2 start codon mutation in patients with hepatitis B e antigen (HBeAg) (-); E: Prevalence of pre-S2 start codon mutation in patients with HBeAg(+). aP < 0.05 vs AC; cP < 0.05 vs CH. AC: Asymptomatic carrier; CH: Chronic hepatitis; LC: Liver cirrhosis; HCC: Hepatocellular carcinoma.
Table 2.
Pre-S mutation as risk factor for progressive liver disease adjusted with age, sex, and hepatitis B e antigen (n = 235)
| P-value | OR (95% CI) | P-value | OR adjusted (95% CI) | |
| AC-CH vs LC-HCC | ||||
| Age, mean ± SD | 1.99E-11 | 1.099 (1.069-1.130) | ||
| Gender (male) | 0.049 | 0.531 (0.281-1.002) | ||
| HBeAg(-) | 0.096 | 1.585 (0.921-2.730) | ||
| Mutation | ||||
| M120 | 3.23E-06 | 4.942 (2.427-10.064) | 0.0005 | 3.996 (1.830-8.729) |
| T130 | 0.078 | 1.684 (0.940-3.016) | 0.531 | 1.246 (0.626-2.483) |
| Q132 | 0.029 | 1.885 (1.064-3.339) | 0.352 | 1.379 (0.701-2.712) |
| A138 | 0.041 | 1.801 (1.020-3.178) | 0.580 | 1.209 (0.617-2.370) |
| AC-CH vs LC | ||||
| Age, mean ± SD | 8.86E-09 | 1.096 (1.063-1.131) | ||
| Gender (male) | 0.027 | 0.442 (0.213-0.920) | ||
| HBeAg(-) | 0.222 | 1.508 (0.778-2.922) | ||
| Mutation | ||||
| M120 | 9.50E-07 | 6.343 (2.878-13.980) | 4.00E-05 | 6.406 (2.640-15.540) |
| T130 | 0.311 | 1.440 (0.710-2.918) | 0.672 | 1.198 (0.518-2.770) |
| Q132 | 0.111 | 1.740 (0.878-3.449) | 0.364 | 1.457 (0.646-3.287) |
| A138 | 0.089 | 1.791 (0.911-3.523) | 0.485 | 1.330 (0.597-2.964) |
| AC-CH vs HCC | ||||
| Age, mean ± SD | 2.13E-07 | 1.084 (1.052-1.118) | ||
| Gender (male) | 0.314 | 0.663 (0.298-1.479) | ||
| HBeAg(-) | 0.144 | 1.677 (0.835-3.372) | ||
| Mutation | ||||
| M120 | 0.002 | 3.651 (1.570-8.490) | 0.052 | 2.538 (0.992-6.494) |
| T130 | 0.054 | 1.987 (0.981-4.022) | 0.402 | 1.413 (0.629-3.174) |
| Q132 | 0.041 | 2.056 (1.024-4.129) | 0.498 | 1.321 (0.590-2.953) |
| A138 | 0.094 | 1.811 (0.900-3.644) | 0.744 | 1.144 (0.509-2.571) |
AC: Asymptomatic carrier; CH: Chronic hepatitis; LC: Liver cirrhosis; HCC: Hepatocellular carcinoma; CI: Confidence interval; OR: Odds ratio; HBeAg: Hepatitis B e antigen.
Prevalence of other mutations in the pre-S2 region
Beside the pre-S2 start codon (M120) mutation, other mutations in the pre-S region were also analyzed. It was found that the prevalence of three amino acid changes in the pre-S2 region showed significant differences among the groups (Table 1). Amino acid substitution from Threonine to other amino acids or deletion at codon 130 (T130) was detected in 26.1% of the samples, and the percentage increased in more severe liver disease (P = 0.002). Changes from Glutamine to other amino acids, or deletion at codon 132 (Q132), was found in 28.7% of the samples, and the prevalence was also increased with the severity of liver disease (P = 0.008). Similarly, amino acid substitution or deletion at Alanine 138 (A138) was found in 29.5% of samples and was highly prevalent in advanced liver disease (P = 0.003). Multivariate regression analysis showed that none of the mutations had significant association with progressive liver disease (Table 2).
Comparison of pre-S mutations in hepatitis B virus genotypes B and C
The samples were grouped into HBV/B and HBV/C and there were no significant differences in male/female ratios, mean age, or pattern of clinical diagnoses between the two groups (data not shown). Pre-S2 start codon mutation (M120) was more prevalent in HBV/B (23.0%) than in HBV/C (19.1%) (Table 3). In HBV/B, M120 mutation was associated with severity of liver disease (P < 0.001), but no association was found in HBV/C. The highest prevalence of M120 mutation in HBV/B was found in the LC group (45.7%), which was higher than that in the HCC group (30.8%), although not statistically significant (Figure 2B). Two other mutations in pre-S2 region (T130 and A138) showed significant association with the progress of liver disease in HBV/B (P = 0.017 and P = 0.042, respectively). In HBV/C, there was no association of the M120 mutation or the other mutations with progressive liver disease (Table 3, Figure 2C).
Table 3.
Prevalence of pre-S mutations in hepatitis B virus genotype B and C associated with progressive liver disease
| Mutations |
Genotype B |
Genotype C |
||||||||||
| AC | CH | LC | HCC | All | P-value | AC | CH | LC | HCC | All | P-value | |
| n = 56 | n = 46 | n = 46 | n = 52 | n = 200 | n = 17 | n = 20 | n = 20 | n = 12 | n = 68 | |||
| M120 | 1 (1.8) | 8 (17.4) | 21 (45.7) | 16 (30.8) | 46 (23.0) | < 0.001 | 1 (5.9) | 4 (20.0) | 6 (30.0) | 2 (16.7) | 13 (19.1) | 0.324 |
| T130 | 6 (10.7) | 11 (23.9) | 10 (21.7) | 19 (36.5) | 46 (23.0) | 0.017 | 2 (11.8) | 8 (40.0) | 8 (40.0) | 6 (50.0) | 24 (35.3) | 0.098 |
| Q132 | 9 (16.1) | 11 (23.9) | 12 (26.1) | 20 (38.5) | 52 (26.0) | 0.067 | 2 (11.8) | 8 (40.0) | 9 (45.0) | 6 (50.0) | 25 (36.8) | 0.081 |
| A138 | 8 (14.3) | 12 (26.1) | 13 (28.3) | 20 (38.5) | 53 (26.5) | 0.042 | 2 (11.8) | 9 (45.0) | 9 (45.0) | 6 (50.0) | 26 (38.2) | 0.070 |
AC: Asymptomatic carrier; CH: Chronic hepatitis; LC: Liver cirrhosis; HCC: Hepatocellular carcinoma.
Comparison of pre-S mutations in HBeAg(-) and in HBeAg(+) patients
As shown in Table 4, the HBeAg(-) and HBeAg(+) groups differed significantly in the ratio of males to females, mean age, and pattern of clinical diagnoses. More male subjects were found in the HBeAg(-) group than in the HBeAg(+) group (83.0% vs 70.7%, P = 0.035). The mean age of subjects in the HBeAg(-) was greater than in HBeAg(+) group (44.7 ± 13.9 years vs 39.2 ± 11.9 years, P = 0.003). The prevalence of M120 mutations was higher in the HBeAg(-) group than in the HBeAg(+) group (23.5% vs 18.3%) (Table 5). The prevalence of this mutation was significantly different among the samples with different clinical diagnoses both in HBeAg(-) and HBeAg(+) patients. In the HBeAg(-) group, the highest prevalence of M120 mutation was found in LC (41.5%), followed by CH (35.7%) and HCC (23.7%), and no mutations were found in the AC group. However, no statistically significant differences were found between CH, LC and HCC (Figure 2D). Among HBeAg(+) patients, the highest frequency of M120 mutation was detected in those with HCC (40.0%), followed by those with LC (38.9%), and CH (5.6%), and no mutations were found in the AC group. Significant differences were found between those with CH and LC and CH and HCC, but not between those with LC and HCC (Figure 2E). On the other hand, no significant correlation of T130, Q132 and A138 mutations with severity of liver disease was found in either the HBeAg(+) or HBeAg(-) groups (Table 5).
Table 4.
Demographic data of samples with hepatitis B e antigen (+) and hepatitis B e antigen (-) n (%)
| HBeAg(+) | HBeAg(-) | P-value | |
| 82 (34.9) | 153 (65.1) | ||
| Gender, | |||
| Male | 58 (70.7) | 127 (83.0) | 0.028 |
| Female | 24 (29.3) | 26 (17.0) | |
| Age, mean ± SD | 39.2 ± 11.9 | 44.7 ± 13.9 | 0.003 |
| Genotype, | |||
| B | 56 (68.3) | 116 (75.8) | 0.215 |
| C | 26 (31.7) | 37 (24.2) | |
| Clinical diagnosis, | |||
| AC | 13 (15.9) | 46 (30.1) | < 0.001 |
| CH | 36 (43.9) | 28 (18.3) | |
| LC | 18 (22.0) | 41 (26.8) | |
| HCC | 15 (18.3) | 38 (24.8) |
AC: Asymptomatic carrier; CH: Chronic hepatitis; LC: Liver cirrhosis; HCC: Hepatocellular carcinoma; HBeAg: Hepatitis B e antigen.
Table 5.
Prevalence of pre-S mutations in hepatitis B e antigen (-) and hepatitis B e antigen (+) associated with progressive liver disease
| Mutation |
HBeAg(-) |
HBeAg(+) |
||||||||||
| AC | CH | LC | HCC | All | P-value | AC | CH | LC | HCC | All | P-value | |
| n = 46 | n = 28 | n = 41 | n = 38 | n = 155 | n = 13 | n = 36 | n = 18 | n = 15 | n = 82 | |||
| M12 | 0 (0.0) | 10 (35.7) | 17 (41.5) | 9 (23.7) | 36 (23.5) | < 0.001 | 0 (0.0) | 2 (5.6) | 7 (38.9) | 6 (40.0) | 15 (18.3) | < 0.001 |
| T130 | 6 (13.0) | 10 (35.7) | 11 (26.8) | 13 (34.2) | 40 (26.1) | 0.082 | 2 (15.4) | 9 (25.0) | 6 (33.3) | 6 (40.0) | 23 (28.0) | 0.474 |
| Q13 | 6 (13.0) | 10 (35.7) | 12 (29.3) | 14 (36.8) | 42 (27.5) | 0.057 | 3 (23.1) | 9 (25.0) | 8 (44.4) | 6 (40.0) | 26 (31.7) | 0.389 |
| A13 | 6 (13.0) | 10 (35.7) | 13 (31.7) | 13 (34.2) | 42 (27.5) | 0.072 | 2 (15.4) | 11 (30.6) | 8 (44.4) | 6 (40.0) | 27 (32.9) | 0.344 |
AC: Asymptomatic carrier; CH: Chronic hepatitis; LC: Liver cirrhosis; HCC: Hepatocellular carcinoma; HBeAg: Hepatitis B e antigen.
DISCUSSION
Many studies have been reported on the association of pre-S mutations with severity of liver disease. Studies from Taiwan and China demonstrated that the pre-S deletion mutation is associated with HCC[18-20]. In addition, the pre-S2 deletion mutation was also specifically associated with increased risk of HCC in Asian children[21]. Other studies also reported that pre-S deletion and pre-S2 start codon mutations was found to be significantly associated with HCC[22,23]. A meta analysis study including 43 reports found that mutations at the promoter sites of pre-S1 and pre-S2 are significantly associated with an increased risk of HCC[24]. However, the diagnostic or predictive value of those mutations for HCC is limited because the frequencies of these mutations in the patients with HCC were low; pre-S1 and pre-S2 promoter mutations were 19.7% and 15.3%, respectively[24].
Recently, we reported on the prevalence of pre-S deletion mutation in Indonesian HBV carriers. The prevalence of pre-S deletion was 12.1% which is considered low compared to previous reports from other populations[16]. In the present study, which extends the analysis of this group of patients, pre-S2 start codon mutations were found in 22.0% of the samples. In Indonesia, it therefore appears that the pre-S2 start codon mutation is a more common type of mutation compared to the pre-S deletion mutation. The prevalence of pre-S2 start codon mutations was higher in the cirrhosis group than the HCC group, which is in accordance with the previous report[22]. Both pre-S2 deletion and start codon mutations cause alterations or loss of the M-protein, which has the T- and B-cell epitopes. These altered M-proteins can stimulate hepatocyte proliferation, and cause the formation of type II ground glass hepatocytes[25,26]. In this study, it was found that this mutation is more prevalent in the liver cirrhosis group, which was older than the other groups, indicating prolonged infection by HBV in this group. It has been suggested that pre-S2 deletion mutations, which affect the T-cell epitope and hence the ability of the virus to be neutralized, represent mutants that are selected under immune pressure, and are thus able to persist during chronic HBV infection[26]. Pre-S2 start codon mutations may have the same effect, since the mutation prevents production of M-protein. Furthermore, it has been reported that the loss of M-protein does not affect virus infectivity, but instead it may function as a spacer that supports conformational changes of L protein, or as a subdomain of the L protein in virus entry[6]. In this way it might allow mutants that lack M protein to emerge without affecting the virus life cycle.
Other mutations in the pre-S2 region were shown to be significantly associated with progressive liver disease. However regression analysis showed these amino acid changes or deletion at three codon sites, T130, Q132, and A138 were not statistically significantly associated with the increased risk of cirrhosis and HCC. The prevalence of these three mutations was considerably higher in the HCC group (38.5%-40.0%). Few studies on amino acid substitutions in the pre-S2 region and their association with progressive liver disease have been reported. An amino acid change from Glycine to Arginine at pre-S2 at codon 149 (G149R) was found in 52.9% of chronic carriers with spontaneous clearance of HBV surface antigen[27]. More recently it was found that a mutation at position 141 (F141L mutation) was associated with increased risk of HCC in patients with HBV/C infections[11]. However the mutation found in this study did not correlate with coexistence of HBsAg and anti-HBs, but may alter the B- and T-cell epitopes of the S protein, and thereby increase the risk of cirrhosis and HCC by decreasing immune recognition.
In the present study, the prevalence of pre-S2 start codon mutation was higher in HBV/B than HBV/C, although the difference was not statistically significant. In addition, a significant association of pre-S2 start codon mutation with severe liver disease was found in HBV/B, while there was no significant difference in the frequency of this mutation in HBV/C. A study in Thailand revealed a higher prevalence of pre-S2 deletions and preS2 start codon mutation in HBV/C than in HBV/B[28]. Another recent report showed a higher prevalence of pre-S2 deletions in HBV/C and this was associated with HCC in children[29]. The differences between previous studies and the present study are probably due to the difference in percentage of HBV/C samples in subjects included in the studies (the number of HBV/C infections was lower in the present study). On the other hand, in the present study, the prevalence of the three other mutations in the pre-S2 region (T130, Q132, and A138) was higher in those with HBV/C than those with HBV/B. In those with HBV/B, the incidence of T130 and A138 mutations was significantly different between the disease groups, and was increased with advanced liver disease (Table 3). However, the prevalence of these mutations was not significantly different between disease groups with HBV/C, possibly due to the low number of HBV/C samples included in this study.
The prevalence of pre-S2 start codon mutations was higher in HBeAg(-) samples than in HBeAg(+) samples (23.5% vs 18.3%), but interestingly in the HCC group the prevalence of this mutation was higher in HBeAg(+) samples than in HBeAg(-) samples (40.0% vs 23.7%) (Table 5). No mutations were found in asymptomatic carriers of either group. The prevalence of mutations in the HBeAg(+) group showed significant association between chronic hepatitis and cirrhosis as well as HCC, while in the HBeAg(-) group there were no differences found among CH, LC and HCC. Previous studies have described the roles of pre-S deletions on the progression of liver disease in HBeAg(-) patients in longitudinal study, and found that pre-S deletions were significantly associated with the development of liver cirrhosis[12]. In another longitudinal study observing the natural course of HBV there was no clear relationship between HBeAg seroconversion and pre-S deletions[30]. The present study was a cross-sectional study which is limited by uncertainty whether these mutations preceded the development of advanced liver disease. However, it does suggest that pre-S2 start codon mutations can be used as a predictor for development of advanced liver disease in HBeAg(+) patients (Figure 2E), but not in HBeAg(-) patients (Figure 2D). Interestingly, the frequency of T130, Q132, and A138 mutations was similar between the two groups (Table 5), indicating that the mutations in this region are not related to HBeAg seroconversion. In summary, our study demonstrated that the pre-S2 start codon mutation has high prevalence in Indonesia. This mutation may serve as biomarker for prediction of development of advanced liver disease in HBeAg(+) patients who are infected with HBV genotype B.
ACKNOWLEDGMENTS
We thank Mardiana Radjuni, Griskalia Christine and Shinta Soraya for sample collection and Dr. David Vaux (The Walter and Eliza Hall Institute, Australia) for critical reading of the manuscript.
COMMENTS
Background
Hepatitis B virus (HBV) infection is a serious worldwide health problem. Indonesia is one of the countries with high endemicity of hepatitis B related disease caused by HBV. Particular mutations of the HBV genome have been associated with severe liver disease. Studies on HBV molecular epidemiology associated with the development of progressive liver disease in Indonesia are still very scarce. This study is to characterize the prevalence of pre-S2 mutation in HBV associated with advanced liver disease in Indonesia.
Research frontiers
Pre-S mutations have been reported to be associated with advanced liver disease. Pre-S deletion and pre-S2 start codon mutations were the most common form of mutations at the pre-S region of HBV. Previous study had shown the low prevalence of pre-S deletion in Indonesian patients; however the prevalence of the pre-S2 start codon mutation has not been fully investigated. In this study, the authors demonstrated that the pre-S2 start codon mutation could serve as a biomarker for liver disease progression.
Innovations and breakthroughs
Previous studies have reported the high prevalence of HBV pre-S deletions and its association with advanced liver disease. But contrary to reports from studies of other populations, previous and current studies showed that pre-S2 start codon mutations and not pre-S deletion may serve as potential biomarkers for progressive liver disease in Indonesia.
Applications
By characterizing the prevalence of the HBV pre-S2 start codon mutation among different clinical diagnoses of different HBV genotype and hepatitis B e antigen (HBeAg) presence, the pre-S2 start codon mutation may serve as a potential biomarker for prediction of development of advanced liver disease in HBeAg(+) patients who are infected with HBV genotype B.
Terminology
HBV surface (HBs) gene has three different translation sites, pre-S1, pre-S2, and S, which produce large-, middle-, and small-HBs protein, respectively. Pre-S2 start codon mutation is a substitution or deletion of Methionine at the pre-S2 translation site which abolished the M HBs protein.
Peer review
The manuscript is interesting because the authors suggest a way to predict liver disease progression according to the appearance of a mutation at pre-S2 start codon in a subset of HBV infected patients.
Footnotes
Supported by MRIN Funding, Budget, No. cc041/2010
Peer reviewers: Dr. Juan Ramón Larrubia, Gastroenterology Unit and Liver Research Unit, Guadalajara University Hospital, University of Alcalá, Donante de Sangre s/n, 19002 Guadalajara, Spain; Dr. Eric WC Tse, Department of Medicine, Queen Mary Hospital, Hong Kong, China
S- Editor Gou SX L- Editor O’Neill M E- Editor Zhang DN
References
- 1.Goldstein ST, Zhou F, Hadler SC, Bell BP, Mast EE, Margolis HS. A mathematical model to estimate global hepatitis B disease burden and vaccination impact. Int J Epidemiol. 2005;34:1329–1339. doi: 10.1093/ije/dyi206. [DOI] [PubMed] [Google Scholar]
- 2.Sastrosoewignjo RI, Sandjaja B, Okamoto H. Molecular epidemiology of hepatitis B virus in Indonesia. J Gastroenterol Hepatol. 1991;6:491–498. doi: 10.1111/j.1440-1746.1991.tb00894.x. [DOI] [PubMed] [Google Scholar]
- 3.Delius H, Gough NM, Cameron CH, Murray K. Structure of the hepatitis B virus genome. J Virol. 1983;47:337–343. doi: 10.1128/jvi.47.2.337-343.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Locarnini S. Molecular virology of hepatitis B virus. Semin Liver Dis. 2004;24 Suppl 1:3–10. doi: 10.1055/s-2004-828672. [DOI] [PubMed] [Google Scholar]
- 5.Schmitt S, Glebe D, Alving K, Tolle TK, Linder M, Geyer H, Linder D, Peter-Katalinic J, Gerlich WH, Geyer R. Analysis of the pre-S2 N- and O-linked glycans of the M surface protein from human hepatitis B virus. J Biol Chem. 1999;274:11945–11957. doi: 10.1074/jbc.274.17.11945. [DOI] [PubMed] [Google Scholar]
- 6.Ni Y, Sonnabend J, Seitz S, Urban S. The pre-s2 domain of the hepatitis B virus is dispensable for infectivity but serves a spacer function for L-protein-connected virus assembly. J Virol. 2010;84:3879–3888. doi: 10.1128/JVI.02528-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lauder IJ, Lin HJ, Lau JY, Siu TS, Lai CL. The variability of the hepatitis B virus genome: statistical analysis and biological implications. Mol Biol Evol. 1993;10:457–470. doi: 10.1093/oxfordjournals.molbev.a040013. [DOI] [PubMed] [Google Scholar]
- 8.Milich DR, McLachlan A, Chisari FV, Nakamura T, Thornton GB. Two distinct but overlapping antibody binding sites in the pre-S(2) region of HBsAg localized within 11 continuous residues. J Immunol. 1986;137:2703–2710. [PubMed] [Google Scholar]
- 9.Milich DR, Hughes JL, McLachlan A, Langley KE, Thornton GB, Jones JE. Importance of subtype in the immune response to the pre-S(2) region of the hepatitis B surface antigen. I. T cell fine specificity. J Immunol. 1990;144:3535–3543. [PubMed] [Google Scholar]
- 10.Huy TT, Ushijima H, Win KM, Luengrojanakul P, Shrestha PK, Zhong ZH, Smirnov AV, Taltavull TC, Sata T, Abe K. High prevalence of hepatitis B virus pre-s mutant in countries where it is endemic and its relationship with genotype and chronicity. J Clin Microbiol. 2003;41:5449–5455. doi: 10.1128/JCM.41.12.5449-5455.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mun HS, Lee SA, Kim H, Hwang ES, Kook YH, Kim BJ. Novel F141L pre-S2 mutation in hepatitis B virus increases the risk of hepatocellular carcinoma in patients with chronic genotype C infections. J Virol. 2011;85:123–132. doi: 10.1128/JVI.01524-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen CH, Hung CH, Lee CM, Hu TH, Wang JH, Wang JC, Lu SN, Changchien CS. Pre-S deletion and complex mutations of hepatitis B virus related to advanced liver disease in HBeAg-negative patients. Gastroenterology. 2007;133:1466–1474. doi: 10.1053/j.gastro.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 13.Utama A, Purwantomo S, Siburian MD, Dhenni R, Gani RA, Hasan I, Sanityoso A, Miskad UA, Akil F, Yusuf I, et al. Hepatitis B virus subgenotypes and basal core promoter mutations in Indonesia. World J Gastroenterol. 2009;15:4028–4036. doi: 10.3748/wjg.15.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Utama A, Octavia TI, Dhenni R, Miskad UA, Yusuf I, Tai S. Hepatitis B virus genotypes/subgenotypes in voluntary blood donors in Makassar, South Sulawesi, Indonesia. Virol J. 2009;6:128. doi: 10.1186/1743-422X-6-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Utama A, Siburian MD, Purwantomo S, Intan MD, Kurniasih TS, Gani RA, Achwan WA, Arnelis , Lukito B, Harmono T, et al. Association of core promoter mutations of hepatitis B virus and viral load is different in HBeAg(+) and HBeAg(-) patients. World J Gastroenterol. 2011;17:708–716. doi: 10.3748/wjg.v17.i6.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Utama A, Siburian MD, Fanany I, Intan MD, Dhenni R, Kurniasih TS, Lelosutan SA, Achwan WA, Arnelis , Yusuf I, et al. Low prevalence of hepatitis B virus pre-S deletion mutation in Indonesia. J Med Virol. 2011;83:1717–1726. doi: 10.1002/jmv.22172. [DOI] [PubMed] [Google Scholar]
- 17.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 18.Lin CL, Liu CH, Chen W, Huang WL, Chen PJ, Lai MY, Chen DS, Kao JH. Association of pre-S deletion mutant of hepatitis B virus with risk of hepatocellular carcinoma. J Gastroenterol Hepatol. 2007;22:1098–1103. doi: 10.1111/j.1440-1746.2006.04515.x. [DOI] [PubMed] [Google Scholar]
- 19.Fang ZL, Sabin CA, Dong BQ, Wei SC, Chen QY, Fang KX, Yang JY, Huang J, Wang XY, Harrison TJ. Hepatitis B virus pre-S deletion mutations are a risk factor for hepatocellular carcinoma: a matched nested case-control study. J Gen Virol. 2008;89:2882–2890. doi: 10.1099/vir.0.2008/002824-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yeung P, Wong DK, Lai CL, Fung J, Seto WK, Yuen MF. Association of hepatitis B virus pre-S deletions with the development of hepatocellular carcinoma in chronic hepatitis B. J Infect Dis. 2011;203:646–654. doi: 10.1093/infdis/jiq096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abe K, Thung SN, Wu HC, Tran TT, Le Hoang P, Truong KD, Inui A, Jang JJ, Su IJ. Pre-S2 deletion mutants of hepatitis B virus could have an important role in hepatocarcinogenesis in Asian children. Cancer Sci. 2009;100:2249–2254. doi: 10.1111/j.1349-7006.2009.01309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi MS, Kim DY, Lee DH, Lee JH, Koh KC, Paik SW, Rhee JC, Yoo BC. Clinical significance of pre-S mutations in patients with genotype C hepatitis B virus infection. J Viral Hepat. 2007;14:161–168. doi: 10.1111/j.1365-2893.2006.00784.x. [DOI] [PubMed] [Google Scholar]
- 23.Cao Z, Bai X, Guo X, Jin Y, Qian G, Tu H. High prevalence of hepatitis B virus pre-S mutation and its association with hepatocellular carcinoma in Qidong, China. Arch Virol. 2008;153:1807–1812. doi: 10.1007/s00705-008-0176-9. [DOI] [PubMed] [Google Scholar]
- 24.Liu S, Zhang H, Gu C, Yin J, He Y, Xie J, Cao G. Associations between hepatitis B virus mutations and the risk of hepatocellular carcinoma: a meta-analysis. J Natl Cancer Inst. 2009;101:1066–1082. doi: 10.1093/jnci/djp180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan YF, Lu CC, Chen WC, Yao WJ, Wang HC, Chang TT, Lei HY, Shiau AL, Su IJ. Prevalence and significance of hepatitis B virus (HBV) pre-S mutants in serum and liver at different replicative stages of chronic HBV infection. Hepatology. 2001;33:277–286. doi: 10.1053/jhep.2001.21163. [DOI] [PubMed] [Google Scholar]
- 26.Su IJ, Wang HC, Wu HC, Huang WY. Ground glass hepatocytes contain pre-S mutants and represent preneoplastic lesions in chronic hepatitis B virus infection. J Gastroenterol Hepatol. 2008;23:1169–1174. doi: 10.1111/j.1440-1746.2008.05348.x. [DOI] [PubMed] [Google Scholar]
- 27.Yeh CT, Chang MH, Lai HY, Chang ML, Chu CM, Liaw YF. Identification of a novel pre-S2 mutation in a subgroup of chronic carriers with spontaneous clearance of hepatitis B virus surface antigen. J Gastroenterol Hepatol. 2003;18:1129–1138. doi: 10.1046/j.1440-1746.2003.03146.x. [DOI] [PubMed] [Google Scholar]
- 28.Suwannakarn K, Tangkijvanich P, Thawornsuk N, Theamboonlers A, Tharmaphornpilas P, Yoocharoen P, Chongsrisawat V, Poovorawan Y. Molecular epidemiological study of hepatitis B virus in Thailand based on the analysis of pre-S and S genes. Hepatol Res. 2008;38:244–251. doi: 10.1111/j.1872-034X.2007.00254.x. [DOI] [PubMed] [Google Scholar]
- 29.Huang HP, Hsu HY, Chen CL, Ni YH, Wang HY, Tsuei DJ, Chiang CL, Tsai YC, Chen HL, Chang MH. Pre-S2 deletions of hepatitis B virus and hepatocellular carcinoma in children. Pediatr Res. 2010;67:90–94. doi: 10.1203/PDR.0b013e3181c1b0b7. [DOI] [PubMed] [Google Scholar]
- 30.Yeung P, Wong DK, Lai CL, Fung J, Seto WK, Yuen MF. Profile of pre-S deletions in the natural history of chronic hepatitis B infection. J Med Virol. 2010;82:1843–1849. doi: 10.1002/jmv.21901. [DOI] [PubMed] [Google Scholar]