

Abstract
The IκB kinase (IKKα, β and the regulatory subunit IKKγ) complex regulates nuclear factor of κB (NF-κB) transcriptional activity, which is upregulated in many chronic inflammatory diseases. NF-κB signaling promotes inflammation and limits muscle regeneration in Duchenne muscular dystrophy (DMD), resulting in fibrotic and fatty tissue replacement of muscle that exacerbates the wasting process in dystrophic muscles. Here we examined whether dominant-negative forms of IKKα (IKKα-dn) and IKKβ (IKKβ-dn) delivered by adeno-associated viral (AAV) vectors to the gastrocnemius (GAS) and tibialis anterior (TA) muscles of 1, 2, and 11 month-old mdx mice, a murine DMD model, block NF-κB activation and increase muscle regeneration. At one month post-treatment, the levels of nuclear NF-κB in locally treated muscle were decreased by gene transfer with either AAV-CMV-IKKα-dn or AAV-CMV-IKKβ-dn, but not by IKK wild-type controls (IKKα and β) or PBS. Although treatment with AAV-IKKα-dn or AAV-IKKβ-dn vectors had no significant effect on muscle regeneration in young mdx mice treated at 1 and 2 months of age and collected 1 month later, treatment of old (11 month) mdx with AAV-CMV-IKKα-dn or AAV-CMV-IKKβ-dn significantly increased levels of muscle regeneration. In addition, there was a significant decrease in myofiber necrosis in the AAV-IKKα-dn and AAV-IKKβ-dn treated mdx muscle in both young and old mice. These results demonstrate that inhibition of IKKα or IKKβ in dystrophic muscle reduces the adverse effects of NF-κB signaling, resulting in a therapeutic effect. Moreover, these results clearly demonstrate the therapeutic benefits of inhibiting NF-κB activation by AAV gene transfer in dystrophic muscle to promote regeneration, particularly in older mdx mice, and block necrosis.
Keywords: AAV, NF-kappa B, DMD, and muscle regeneration
Introduction
Duchenne muscular dystrophy (DMD) is the most common muscular dystrophy and is caused by recessive mutations in the dystrophin gene.1 Progressive muscle weakness and degeneration usually lead to loss of independent ambulation by the middle of the second decade and a fatal outcome due to cardiac or respiratory failure by the third decade. 2 In DMD patients, loss of sarcolemmal dystrophin and the dystrophin-associated glycoprotein (DAG) complex promotes muscle fiber damage during muscle contraction.3–7 This process results in efflux of creatine kinase (CK), influx of calcium ions, and recruitment of T cells, macrophages, and mast cells to the damaged muscle, causing progressive myofiber necrosis.2 Muscle regeneration is the initial response to damage, but muscle progenitor cells are exhausted with continued cycles of necrosis over time, thus giving way to accumulated fibrosis and fatty deposits that exacerbate the wasting process in DMD.8 Currently, there are no effective treatments available for DMD. Adeno-associated viral (AAV) vector-mediated gene transfer of mini-dystrophin has shown great promise in mouse models, but did not completely prevent fibrosis and necrosis in dystrophic muscles or reverse the muscle wasting process in the dystrophin/utrophin double knockout mouse (dKO), a DMD model with genetic loss of both dystrophin and utrophin.9–11 The efficacy of AAV-mediated mini-dystrophin gene transfer to ameliorate the muscle wasting process is limited, in part, by the inflammatory micro-environment. Therefore, the ability to modulate chronic inflammation in dystrophic muscle is important for optimizing therapeutic strategies to treat muscle wasting and DMD.
NF-κB is a ubiquitously expressed transcription factor in mammals. In the classical activation pathway, NF-κB signaling occurs primarily by way of a p50/p65 heterodimer.12 In most cells the majority of NF-κB is localized in the cytoplasmic compartment, maintained in an inactive state by binding to the inhibitor of NF-κB (IκB).13 The classical pathway of NF-κB activation occurs through IκB kinase (IKK), a large 700–900 kDa complex containing two catalytic subunits, IKKα and IKKβ, and a regulatory subunit IKKγ.14 In response to a multitude of factors, including lipopolysaccharide (LPS), tumor necrosis factor alpha (TNFα), interleukin-1β(IL-1β) and IL6, IKK is activated and phosphorylates IκB, leading to its ubiquitination and subsequent degradation by the 26S proteasome.15 IκB degradation allows the translocation of the NF-κB heterodimer to the nucleus where it binds to its cognate DNA site and interacts with the basal transcription factors and co-activators to induce gene expression.16 NF-κB plays a key role promoting the inflammatory response in muscular dystrophy.17,18 Elevated levels of NF-κB have been detected in more than one type of muscular dystrophy, including DMD19 and limb-girdle muscular dystrophy (LGMD).20 A recent study demonstrated that chronic activation of NF-κB signaling, often mediated by IKKβ, is required for DMD pathology by acting both on immune cells and damaged skeletal muscles to promote inflammation and inhibit myogenic differentiation of muscle precursors in mdx mice. 19 Although IKKα regulates the muscle mitochondrial content and function,21 it is unclear whether IKKα is involved in chronic inflammation in dystrophic muscle. The p65 subunit of NF-κB was implicated in regulating muscle pathology in mdx mice by a genetic experiment in which dystrophin-deficient mdx mice were bred with mice that carry a heterozygous deletion of the p65. The mdx-heterozygous p65 cross resulted in improved dystrophic pathology characterized by enhanced muscle regeneration.22 Also, targeted ablation of IKKβ was found to improve the morphological and functional phenotypes of mdx muscle. 2222 Collectively, these studies demonstrate the critical role of IKK/NF-κB signaling in the progression of DMD and implicate the NF-κB signaling pathway as a potential therapeutic target for this disease.
More recently, modulation of NF-κB by systemic treatment with a synthetic antioxidant such as pyrrolidine dithiocarbamate (PDTC)17 or with a peptide inhibitor of IKK β,22 reduced inflammation and necrosis while enhancing regeneration. However, these approaches require repeated treatments and have the potential for systemic side effects. AAV-based gene therapy is a promising therapeutic approach for the treatment of DMD, as demonstrated by preclinical studies in mdx or dKO mouse9,23,24 and canine models.25 We, therefore, tested the therapeutic effects of treatment with AAV vectors carrying either a dominant negative IKKα (IKKα-dn) or IKKβ (IKKβ-dn). The efficacy of AAV vector-mediated IKKα-dn and IKKβ-dn gene transfer was assessed in locally treated skeletal muscle of young and old mdx mice.
Results
Dominant negative IKK (IKKβ-dn) blocks the NF-κB pathway in mouse macrophages
We generated AAV-based vectors carrying the cDNAs for dominant negative mutants (IKKα-dn and IKKβ-dn) and wild-type IKKα and IKKβ(IKK α-wt and IKKβ-wt). To confirm that the vectors expressed functional proteins, the ability of AAV-IKKβ-dn and AAV-IKKβ-wt to regulate activation of NF-κB was examined by measuring TNFα and IL-10 expression following transduction of mouse macrophages, called RAW cells. Following treatment of the macrophages with lipopolysaccharide (LPS) stimulation, there was a reduction in the levels of TNF-α and IL-10 in the culture medium of the IKKβ-dn treated cells compared to either IKKβ-wt + LPS (* p<0.05) or LPS groups (# p<0.05). Moreover, we found that the IKKβ-wt treatment resulted in increased TNF-α level compared to the LPS control group (& p<0.05). In contrast, there was no significant difference of IL-10 between IKKβ-wt + LPS and the LPS group (Table 1). These results demonstrated that AAV-IKKβ-dn blocks the NF-κB signaling pathway in mouse macrophages, resulting in decreased levels of pro-inflammatory cytokines such as TNF-α and IL-10. We observed that AAV-IKKβ-dn decreased TNF-α by a 2-to 3 -fold margin, while IL-10 was decreased as much as 10-to 15 -fold.
Table 1.
The levels of TNF-α and IL-10 in medium 24 hours after LPS stimulation
| TNF-α, ng/ml | IL-10, ng/ml | |
|---|---|---|
| LPS | 14046 ± 2000 (n=2) | 1681 ± 276 (n=2) |
| IKKwt + LPS | 17565 ± 175 (n=2) | 979 ± 276 (n=2) |
| IKKdn + LPS | 5041 ± 262 (n=4) * # | 97 ± 38 (n=4) * # |
Notice:
p<0.05, IKKdn + LPS vs IKKwt + LPS
p<0.05, IKKdn + LPS vs LPS
Efficiency of gene transfer in vitro
Before the in vivo gene transfer experiment, we observed successful AAV-mediated gene transfer with dominant-negative forms of IKKs (IKKα-dn and IKKβ-dn) and IKK wild-type controls (IKKα and β) into 293 cells, a human embryonic kidney cell line. Since the given mutations in dominant-negative forms of IKKα-dn and IKKβ-dn were closed to N-terminus, the antibody against C-terminus of IKKα or IKKβ was used to test gene expression of IKKα (dn-and wt) or IKKβ (dn- and wt). As depicted in Figure 1A, in initial study, Western blot analysis showed highly efficient transductions were found in infected cells 48 hours post-infection respectively by AAV2-IKKα-dn, AAV2-IKKα-wt, AAV2-IKKβ-dn, and AAV2-IKKβ-wt, no gene expression was in PBS control group. No significant difference was found between IKKβ-dn and IKKβ-wt in infected cells; however, the level of IKKα-wt was higher than IKKα-dn in infected cells due to the antibody against C-terminus of wild-type IKKα.
Figure 1. IKKs gene transfer efficiency and decreased nuclear NF-κB in treated muscle.

(A) In vitro gene transfer of IKKα-(dn and wt) and IKKβ-(dn and wt) in AAV constructs. Studies were performed to confirm gene transfer efficiencies of AAV2-IKKα-dn, AAV2-IKKα-wt, AAV2-IKKβ-dn, and AAV2-IKKβ-wt in infected-293 cells 48 hours post-treatment. Western blot analysis by the use of anti-C-terminuses of IKKα and anti-antibodies shows high levels of IKKα-(dn and wt) as well as IKKβ-(dn and wt) expressions (MWs of 85 kD and 87 kD). (B) One month after intramuscular gene transfer with four different AAV2 vectors, the cryosections of GAS muscles treated at 2 months of age and TA muscles treated at 11 months of age were determined the efficiencies of gene transfer by Western blot analysis. The increased levels of IKKα-(dn and wt) and IKKβ-(dn and wt) were found in AAV2-IKKα-dn, AAV2-IKKα-wt, AAV2-IKKβ-dn, and AAV2-IKKβ-wt treated young and old mdxmuscles compared to PBS treated mdx muscle at same age. (C) The electrophoretic mobility shift assay (EMSA) demonstrates the level of nuclear NF-κB in the gastrocnemius (GAS) muscle of both 1 and 2 month-old young mdx mice and the tibialis anterior (TA) muscle of 11 month-old mdx mice treated by AAV2-IKKα-dn, AAV2-IKKα-wt, AAV2-IKKβ-dn, AAV2-IKKβ-wt, and PBS. A total of 45 μg of nuclear extract from each mouse was analyzed at 1 month post -treatment.
AAV gene transfer of IKKα-dn and IKKβ-dn reduces nuclear NF-κB in mdx muscle
To evaluate the AAV-mediated effects of both IKKα-dn and IKKβ-dn on the treatment of dystrophic muscle wasting, we locally treated skeletal muscle of both young and old mdx mice, and analyzed the levels of nuclear NF-κB at 1 month after a single intramuscular AAV injection. The efficiencies of AAV-mediated gene transfer in muscles were determined by a Western blot analysis1 month post-treatment. As shown in Figure 1B, there were marked increases in IKKα-(dn and wt) and IKKβ-(dn and wt) activities in gastrocnemius (GAS) treated at 2 months of age and tibialis anterior (TA) muscles treated at 11 months of age when compared to PBS treated controls. To determine whether the IKKα-dn and IKKβ-dn could inhibit IKK/NF-κB pathway by decreasing the level of nuclear NF-κB, we also tested the level of nuclear NF-κB 1 month after treatment. As expected, the levels of nuclear NF-κB in gastrocnemius (GAS) and tibialis anterior (TA) muscles treated by AAV-IKKα-dn and IKKβ-dn at different age groups (1, 2 and 11 months) were much lower compared to AAV-IKKα-wt, AAV-IKKβ-wt and PBS controls in age matched mdx mice (Figure 1C). This indicates that not only IKKβ-dn, but also IKKα-dn decreased the levels of nuclear NF-κB in locally treated dystrophic muscle. In contrast, the levels of nuclear NF-κB in young and old mdx mice treated with a vector carrying either wild-type IKKα or wild-type IKKβ were similar to the PBS control group.
No effect on muscle regeneration of AAV-IKKα-dn and AAV-IKKβ-dn treated young mdx muscle
We investigated the therapeutic effects of the AAV-mediated gene transfer of IKKα-dn and IKKβ-dn on muscle regeneration in 1 and 2 month-old mdx mice (young mice). At 1 month post -treatment, the dystrophic muscle regeneration, as measured by the number of embryonic myosin heavy chain (eMyHC) positive myofibers, was greater in mdx mice treated at 1 month of age compared with mdx mice treated at 2 months of age; however, there was no difference observed between treated and untreated mdx at the same age (Figure 2A and Table 2). These results suggest that abnormal muscle regeneration is higher in dystrophic mice at 2 months of age compared to 3 months of age, likely by the induction of satellite cells into myofibers, which is consistent with previous studies.26
Figure 2. Myogenic regeneration in young and old mdx mice.

Tissue sections were immunostained for embryonic myosin heavy chain (eMyHC) expression (red), to assay levels of muscle regeneration. The first panel from the left represents tissues from negative control mdx mice treated with PBS. The next two panels represent the tissues treated by AAV2-IKKα-dn and AAV2-IKKα-wt. Finally, the last two panels represent the tissues treated by AAV2-IKKβ-dn and AAV2-IKKβ-wt. (A) At 1 month post -treatment, gastrocnemius (GAS) muscles were isolated from AAV vector-treated young mdx mice at 1 and 2 months of age. (B) GAS and tibialis anterior (TA) muscle tissues of old mdx mice, treated at 11 months of age, were collected at 1 month post-treatment. All panels are shown at 100x magnification.
Table 2.
Comparisons of regeneration and necrosis in young and old mdx mice
| Vectors | n | Age at vector injection | Months post injection | % of regenerative fibers * | % of necro-fibers* |
|---|---|---|---|---|---|
| PBS | 4 | 1 month | 1 month | 30.8 (460/1495) | 25.6 (349/1363) |
| IKKα-dn | 4 | 1 | 1 | 29.9 (556/1859) | 19.7 (265/1343) |
| IKKα-wt | 4 | 1 | 1 | 28.4 (450/1584) | 25.2 (371/1471) |
| IKKβ-dn | 4 | 1 | 1 | 29.3 (319/1087) | 17.0 (184/1081) |
| IKKβ-wt | 4 | 1 | 1 | 27.1 (431/1593) | 22.5 (318/1412) |
|
| |||||
| PBS | 4 | 2 | 1 | 19.6 (300/1532) | 24.8 (280/1129) |
| IKKα-dn | 4 | 2 | 1 | 20.5 (421/2058) | 14.8 (120/810) |
| IKKα-wt | 4 | 2 | 1 | 18.6 (362/1948) | 26.7 (379/1420) |
| IKKβ-dn | 4 | 2 | 1 | 20.2 (310/1534) | 13.1 (146/1116) |
| IKKβ-wt | 4 | 2 | 1 | 22.5 (229/1017) | 23.2 (254/1094) |
|
| |||||
| PBS | 4 | 11 | 1 | 1.4 (20/1401) | 34.0 (437/1284) |
| IKKα-dn | 4 | 11 | 1 | 4.3 (87/2026) | 15.5 (339/2186) |
| IKKα-wt | 4 | 11 | 1 | 0.8 (9/1095) | 35.4 (394/1113) |
| IKKβ-dn | 4 | 11 | 1 | 9.8 (118/1203) | 15.3 (312/2033) |
| IKKβ-wt | 4 | 11 | 1 | 0.9 (12/1399) | 34.4 (521/1514) |
Percentage: positive IF staining vs all numbers in visible area including both negative and positive myofibers in gastrocnemius (GAS) and tibialis anterior (TA) muscles.
Improved muscle regeneration in AAV-IKKα-dn and AAV-IKKβ-dn treated old mdx muscle
To investigate whether muscle regeneration could be improved in old mdx mice even when the regenerative capacity of progenitor or myogenic cells becomes exhausted,27 we examined the therapeutic effects of AAV-IKKα-dn or AAV-IKKβ-dn gene transfer in 11 month old mdx mice. Interestingly, there was increased muscle regeneration in both GAS and TA muscles of older mdx mice treated by IKKα-dn and IKKβ-dn, in contrast to AAV-IKKα-wt, AAV-IKKβ-wt and PBS-treated controls (Figure 2B and Table 2). These results demonstrate a therapeutic effect of NF-κB inhibition evidenced by enhances regeneration of dystrophic skeletal muscle tissue in old mdx mice.
Decreased muscle fiber necrosis in IKKα-dn and IKKβ-dn treated young and old mdx muscle
Chronic inflammation is a feature of the dystrophic muscle pathology in DMD and likely exacerbates the dystrophic muscle wasting process. To determine whether AAV-IKKα-dn or AAV-IKKβ-dn treated muscle demonstrates a decrease in the level of muscle fiber necrosis, sections were incubated with fluorescently-labeled mouse IgG. At one month post treatment, a significant decrease in myofiber necrosis was found in muscles treated with either AAV-IKKα-dn or AAV-IKKβ-dn at the age of 1, 2 and 11 months. In contrast, muscle treated with AAV-IKKα-wt, AAV-IKKβ-wt or PBS showed extensive myofiber necrosis (Figure 3 and Table 2).
Figure 3. AAV-IKKs-dn inhibits muscle necrosis.

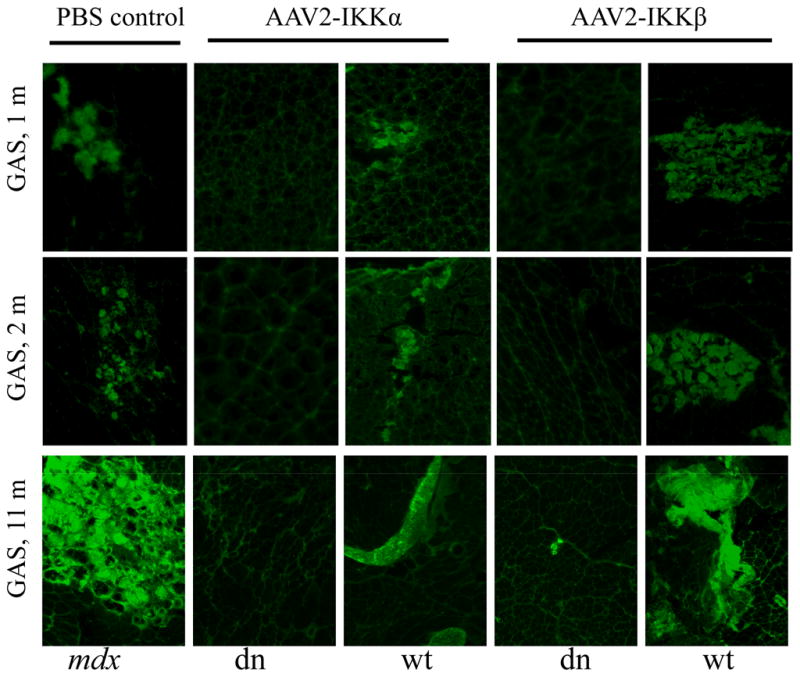
Cryosections of muscle, from mice treated at 1, 2 and 11 months of age and sacrificed 1 month after gene transfer, were incubated with fluorescently-labeled mouse IgG. From left to right, many IgG positive necrotic (green) were found in GAS muscles treated by PBS (first panel), AAV2-IKKα-wt (third panel) and AAV2-IKKβ-wt (fifth panel) in both 1, 2 and 11 month-old groups. In contrast, the GAS muscle treated by AAV2-IKKα-dn (second panel) and AAV2-IKKβ-dn (fourth panel) showed only few necrotic myofibers. All panels are shown at 100x magnification.
Discussion
Progressive muscle degeneration and wasting leads to pre-mature death in DMD patients.1,2 The accumulation of fibrotic and necrotic tissue in dystrophic muscle exacerbates the wasting process. Fibrosis is the final result of chronic inflammation mediated, in part, by the pathological activation of NF-κB, in both muscle-infiltrating immune cells and damaged skeletal muscle fibers.19 In our previous studies, AAV-mediated delivery of a mini-dystrophin gene resulted in efficient and functional recovery of dystrophic skeletal muscle in mdx mice; however, this approach was not sufficient to reverse the muscle wasting process or restore a normal life-span in a severe murine model, the dKO mouse.9–11 Despite noted successes in pre-clinical mouse studies, AAV mini-dystrophin gene transfer has limitations of partial transduction of muscle fibers and incomplete reversal of the dystrophic phenotype in muscle tissue. Thus, the development of methods to improve regeneration and block chronic inflammation in dystrophic muscle would not only offer direct therapeutic potential for DMD, but could also enhance the efficacy of gene therapy approaches.
In prior studies, blockade of the NF-κB signaling pathway with pharmacological agents has demonstrated success in delaying progression of the pathological features of dystrophy in mdx mice. 17,28 Small molecules, including the use of an IKK inhibitory peptide, used to block pathological activation of NF-κB have improved muscle regeneration. However, the use of small molecules requires chronic, systemic treatment and raises concerns for potential systemic side effects. In contrast, a local gene transfer approach with gene transduction and transgene expression limited to muscle could impart a greater therapeutic effect with minimal adverse side effects. Therefore, inhibition of IKK/NF-κB signaling by a gene transfer-mediated approach could result in reducing progressive muscle loss and weakness. Tas, S et al. used AAV to successfully deliver IKKβ-dn (K > M) to the synovium, resulting in reduced severity of inflammation in adjuvant-induced arthritis (AA) in vivo and proinflammatory cytokine production in human rheumatoid arthritis (RA) synovial tissues ex vivo.292929 Thus, in this study, we tested AAV-mediated gene transfer of IKKβ-dn on muscle regeneration and necrosis in both young and old mdx mice. Moreover, since the role of IKKα in regulating muscle degeneration and regeneration as well as inflammation has not been characterized, we also examined the effect of IKKα-dn on dystrophic pathology.
The mdx mouse has a nonsense mutation in exon 23 of the dystrophin gene, resulting in premature chain termination of the dystrophin protein.30 Progressive pathological muscle necrosis and regeneration,31,32 are most marked between 1 and 2 months of age. 33 The satellite cell pool is diminished with advancing age of mdx mice due to the ongoing cycles of muscle degeneration and regeneration. 26
Hence, the AAV mediated IKKα-dn and IKKβ-dn treatment was first performed in young mdx mice. As summarized in Table 2, we demonstrated that the level of myofiber necrosis (mice treated beginning at 1 month and 2 months of age) was decreased in the AAV-IKKα-dn (19.7% and 15.0%) and AAV-IKKβ-dn (17.8% and 13.1%) treated muscle of young mdx mice as compared to age-matched control groups treated with PBS (26.9%and 24.5%), AAV-IKKα-wt (25.3% and 26.3%) and AAV-IKKβ-wt (23.9% and 23.3%). This result suggests that the local treatment of the AAV-IKKα-dn and AAV-IKKβ-dn can reduce the course of muscle necrosis.
In contrast to the effect on muscle fiber necrosis, there was no significant increase in muscle regeneration in mdx mice (treated at 1 and 2 months of age) with AAV-IKKα-dn (29.8% and 20.4%) or AAV-IKKβ-dn (29.5% and 20.2%) when compared to controls treated with PBS (30.8% and 19.5%), AAV-IKKα-wt (29.0% and 18.7%) and AAV-IKKβ-wt (27.3% and 22.5%). The absence of an effect on regeneration in mdx mice treated at a young age was surprising because of prior studies showing that 3 times weekly treatment with a peptide inhibitor of IKK, resulted in a significant enhancement of muscle regeneration. 22 One possible explanation for the differing results may lie in the difference between a peptide-based therapeutic that would be expected to act immediately and an AAV vector-based therapeutic that requires time, up to two weeks, for transgene expression.
In contrast to the results in young mdx mice, we observed improved skeletal muscle regeneration in mdx mice treated at 11 months of age. As described in Table 2, there were significant increases in muscle regeneration in mdx mice (treated at 11 months of age) with AAV-IKKα-dn (4.3%) or AAV-IKKβ-dn (9.9%) when compared to controls treated with PBS (1.4%), AAV-IKKα-wt (0.9%), and AAV-IKKβ-wt (0.8%). This result suggests that inhibition of pathological activation of NF-κB is effective at a time when the regenerative capacity of dystrophic muscle is significantly reduced. This result is of potential significant importance for its ultimate utility in the clinical treatment of dystrophic muscle since the dystrophic process in human DMD muscle is already well advanced even early in life.
The ability of dominant negative IKKα and IKK β to reduce necrosis in old mdx muscle was also examined. Reduced levels of necrosis were observed in older mdx mice treated with AAV-IKKα-dn (15.4%) and AAV-IKKβ-dn (15.1%) compared to age-matched control groups treated with PBS (34.0%), AAV-IKKα-wt (35.6%) and AAV-IKKβ-wt (34.9%).
By using the ubiquitous CMV promoter for expression of the therapeutic constructs, the potential for transduction and expression in inflammatory cells, which originate from hematopoietic stem cells, recruited to dystrophic muscle tissue was preserved. Because the inhibition of NF-κB activity could benefit dystrophic muscle through effects on inflammatory cells as well as muscle fibers, the observed therapeutic effects may reflect a combination of both mechanisms.
Although IKKβ participates the “classical” activation of NF-κB, IKKα is a component of “alternative” activation of NF-κB.34 Interestingly, our results suggest that targeting IKKα also inhibits the level of nuclear NF-κB in dystrophic muscle and enhances muscle regeneration. This result is in contrast to a previous study 35 showing that transfection of the IKKα-dn plasmid into the C2C12 cell line had no significant effect on pro-inflammatory gene expression following LPS-stimulation of myotubes. However, our results demonstrate that following AAV-mediated gene transfer to muscle in vivo, IKKα-dn is as effective in improving pathology as IKKβ-dn.
In summary, AAV-mediated gene transfer of dominant negative IKK, either IKKβ or IKKα, to block pathological NF-κB signaling is a promising strategy for the treatment of DMD. The current studies suggest effectiveness as an independent therapeutic, especially when the dystrophic process is more advanced. Furthermore, combination therapy for DMD with mini-dystrophin gene transferor stem cell transplantation and inhibition of NF -κB activation may be synergistic and will be explored in future studies.
Materials and methods
Construction of IKKs and rAAV vector production
The pEF6Bplasmids containing cDNAs of dominant negative IKKα (IKKα-dn), wild-type IKKα (IKKα-wt), dominant negative IKKβ (IKKβ-dn), and wild-type IKKβ (IKKβ-wt) served as templates for PCR to obtain different gene cloning sequences. The cDNA encoding for IKKα-dn had mutations at positions 130 (A → G) and 131 (A → C) which resulted in a point mutation (K > A) compared to IKKα-wt (GenBank, NM_001278.3), another cDNA encoding for IKKβ-dn had a point mutations at positions 529 – 531 (A → G, G → A, and T → A) and 541–542 (T → G and C → A), resulting in the changes of amino acids at both positions (S > E) compared to IKKβ-wt (GenBank, NM_001556.1). The N-terminal primers utilized for amplification of IKKα (dn and wt) and IKKβ (dn and wt) were 5′-GGCCACCATGGAGCGGCCCCCGGGGCand 5′-GGCCACCATGAGCTGGTCACCTTCCC, respectively; which include a kozak box (GCCACC) immediately in 5′ terminus of the start cod on to increase the level of transcription at the N-terminus. The C-terminal primers for IKKα and IKKβ were 5′-GCTCATCATTCTGTTAACCAACTCCAATCand 5′-GCTCATCATGAGGCCTGCTC CAGGC, respectively; both include a stop codon. The purified PCR products were electrophoresed as 2.2 kb bands, purified and treated with PNK enzyme (NEB, MA, USA) to add a phosphate to the N-terminus. The fragments were inserted separately into the XhoI site of the single-stranded AAV vectors shuttle plasmids under the control of the CMV promoter.23 The resulting clones were sequenced and purified twice by CsCl gradient to generate the serotype 2 of rAAV vector using a previously published protocol. 36 The vector titers were approximately 3–5 × 1013 viral genome (v.g.) particles per ml as determined by dot blot assay.
In vitro effects of IKKβ-dn on mouse macrophages
To investigate the effect of IKKβ-dn on cultured non-muscle cells, mouse macrophage RAW cells from American Type Culture Collection (ATCC, VA, USA) were seeded into 6 well-plates and cultured to 80%confluence. Transfections with rAAV plasmid DNAs were performed with a lipofectamine kit (Invitrogen, CA, USA) according to a standard protocol. The medium was changed twice with DMEM containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (1% P/S v/v) at 6 and 24 hours after transfection. Stimulation with lipopolysaccharide (LPS) (Sigma, MO, USA) was achieved by adding 100 ng/ml LPS to the medium for 24 hours. An aliquot of 200 μl of culture medium was removed and the levels of TNF-α and IL-10 were determined using an ELISA kit (R & D systems, MN, USA) according to standard protocol provided by the manufacturer.
Mdx mice and rAAV vector administration
Mdx (C57BL/10ScSn-mdx/J) mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). All animal studies were approved by the University of Pittsburgh Animal Care and Use Committee. Thirty male mdx mice were divided into 1, 2 and 11 month-old age groups (10 mice per age group). In each age group, 10 mice were further distributed into 5 groups to receive a single intramuscular injection of one of four different AAV2 vectors (AAV2-IKKα-dn, AAV2-IKKα-wt, AAV2-IKKβ-dn, and AAV2-IKKβ-wt) at 1 ~ 2 × 10 11 v.g. particles of AAV and phosphate buffered saline (PBS), as a control, at the volume of 30 ~ 50 μl bilaterally in tibialis anterior (TA) and gastrocnemius (GAS) muscles. Therefore, the number of mouse muscles for each group was 4. All mice were sacrificed at one month post-treatment and the muscles were collected for quantitative analysis of NF-κB and muscle histology.
Efficiency of the gene transfer in vitro and in vivo
Before the in vivo study, we tested the gene transfer efficiencies of AAV-vectors in 293 cells. The four groups of cells were transduced with AAV2-IKKα-dn, AAV2-IKKα-wt, AAV2-IKKβ-dn, and AAV2-IKKβ-wt at an m.o.i of 2.5×104 in 6 well -plate, and then were harvested for western blot analysis 48 hours post-transduction according to previously published methods.24,37 Briefly, cells were lysed in 100 μl of radioimmunoprecipitation assay (RIPA) buffer, and 10 μl of samples was performed for Western blot analysis using the standard protocol.37 Western blot was also carried out to determine the gene transfer in vivo, 25–30 cryosections (10 μm) of GAS muscles treated at 2 months of age and TA muscles treated at 11 months of age were lysed in 20 μl of radioimmunoprecipitation assay (RIPA) buffer, and 15 μl of samples was performed for Western blot analysis. The rabbit polyclonal antibody against C-terminus of IKKα and IKKβ (Abcam, Inc., Cambridge, MA, USA) were used because of this antibodies correspond respectively to the highly conserved C-terminus regions of IKKα and IKKβ, both dominant negative and wild-type forms. All in vitro transfection experiments in 293 cells were performed in triplicates. Beta -actin (43 kD) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH, 37 kD) were used as markers to evaluate the amount of protein in each group used for Western blot in vivo and in vivo, respectively.
NF-κB electrophoretic mobility shift assay (EMSA)
For NF-κB electrophoretic mobility shift assays to determine the level of nuclear NF-κB, a double-stranded DNA probe containing the NF-κB binding sequence was radiolabeled with 32P as previously described.38 Briefly, 45 μg of nuclear extract from the indicated hindlimb muscle was incubated in 5x gel shift assay buffer (Promega, WI, USA), followed by the addition of a 32P radiolabeled NF-κB DNA probe. Reaction samples were electrophoresed on a 6% non-denaturing gel at 30 mA, which were then dried and imaged with autoradiographic film.
Analysis of muscle histology
Immunohistochemical analysis for eMyHC was used to evaluate myogenic regeneration. The M.O.M. immunodetection kit (Vector Laboratories, Burlingame, CA, USA) and a mouse monoclonal antibody against eMyHC (F1.652 from the Department of Biological Sciences, University of Iowa, Iowa, USA) were used. Fluorescently-labeled secondary antibody was Cy™ 3 conjugated AffiniPure rabbit anti-mouse IgG (1:500, West Grove, PA, USA). To determine muscle fiber necrosis, muscle sections were incubated with FITC-conjugated horse anti-mouse IgG at 1:100 dilution (Vector Laboratories, Burlingame, CA, USA), according to a published protocol.39
Statistical analysis
The significance of differences between groups of experiments was evaluated by analysis of variance or a two-tailed Student’s t-test where appropriate. All results were expressed as the mean ± SEM. A p value < 0.05 was considered significant.
Acknowledgments
This work was funded by the Department of Defense (W81XWH-06-1-0406 subcontract to BW and W81XWH-06-1-0406 to P.R.C.), the Pittsburgh Foundation, and internal funds from the Department of Orthopaedic Surgery, University of Pittsburgh. The authors take full responsibility for the contents of this paper, which do not represent the views of the Department of Veterans Affairs or the United States Government. The authors wish to thank Jonathan Proto for critical reading of the manuscript.
Footnotes
Conflict of interest statement
The authors declare no conflicts of interest and have received no payment for the preparation of this manuscript.
References
- 1.Hoffman EP, Monaco AP, Feener CC, Kunkel LM. Conservation of the Duchenne muscular dystrophy gene in mice and humans. Science. 1987;238:347–350. doi: 10.1126/science.3659917. [DOI] [PubMed] [Google Scholar]
- 2.Lapidos KA, Kakkar R, McNally EM. The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ Res. 2004;94:1023–1031. doi: 10.1161/01.RES.0000126574.61061.25. [DOI] [PubMed] [Google Scholar]
- 3.Bonilla E, Samitt CE, Miranda AF, Hays AP, Salviati G, DiMauro S, et al. Duchenne muscular dystrophy: deficiency of dystrophin at the muscle cell surface. Cell. 1988;54:447–452. doi: 10.1016/0092-8674(88)90065-7. [DOI] [PubMed] [Google Scholar]
- 4.Koenig M, Kunkel LM. Detailed analysis of the repeat domain of dystrophin reveals four potential hinge segments that may confer flexibility. J Biol Chem. 1990;265:4560–4566. [PubMed] [Google Scholar]
- 5.Koenig M, Monaco AP, Kunkel LM. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. 1988;53:219–228. doi: 10.1016/0092-8674(88)90383-2. [DOI] [PubMed] [Google Scholar]
- 6.Miranda AF, Francke U, Bonilla E, Martucci G, Schmidt B, Salviati G, et al. Dystrophin immunocytochemistry in muscle culture: detection of a carrier of Duchenne muscular dystrophy. Am J Med Genet. 1989;32:268–273. doi: 10.1002/ajmg.1320320231. [DOI] [PubMed] [Google Scholar]
- 7.Senter L, Luise M, Presotto C, Betto R, Teresi A, Ceoldo S, et al. Interaction of dystrophin with cytoskeletal proteins: binding to talin and actin. Biochem Biophys Res Commun. 1993;192:899–904. doi: 10.1006/bbrc.1993.1500. [DOI] [PubMed] [Google Scholar]
- 8.McLoon LK. Focusing on fibrosis: halofuginone-induced functional improvement in the mdx mouse model of Duchenne muscular dystrophy. Am J Physiol Heart Circ Physiol. 2008;294:H1505–1507. doi: 10.1152/ajpheart.00176.2008. [DOI] [PubMed] [Google Scholar]
- 9.Gregorevic P, Allen JM, Minami E, Blankinship MJ, Haraguchi M, Meuse L, et al. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat Med. 2006;12:787–789. doi: 10.1038/nm1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yue Y, Liu M, Duan D. C-terminal-truncated microdystrophin recruits dystrobrevin and syntrophin to the dystrophin-associated glycoprotein complex and reduces muscular dystrophy in symptomatic utrophin/dystrophin double-knockout mice. Mol Ther. 2006;14:79–87. doi: 10.1016/j.ymthe.2006.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang B, Li J, Fu FH, Xiao X. Systemic human minidystrophin gene transfer improves functions and life span of dystrophin and dystrophin/utrophin-deficient mice. J Orthop Res. 2009;27:421–426. doi: 10.1002/jor.20781. [DOI] [PubMed] [Google Scholar]
- 12.Roman-Blas JA, Jimenez SA. NF-kappaB as a potential therapeutic target in osteoarthritis and rheumatoid arthritis. Osteoarthritis Cartilage. 2006;14:839–848. doi: 10.1016/j.joca.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 13.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 14.Peters RT, Maniatis T. A new family of IKK-related kinases may function as I kappa B kinase kinases. Biochim Biophys Acta. 2001;1471:M57–62. doi: 10.1016/s0304-419x(00)00024-x. [DOI] [PubMed] [Google Scholar]
- 15.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 16.Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 17.Messina S, Bitto A, Aguennouz M, Minutoli L, Monici MC, Altavilla D, et al. Nuclear factor kappa-B blockade reduces skeletal muscle degeneration and enhances muscle function in Mdx mice. Exp Neurol. 2006;198:234–241. doi: 10.1016/j.expneurol.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 18.Ladner KJ, Caligiuri MA, Guttridge DC. Tumor necrosis factor-regulated biphasic activation of NF-kappa B is required for cytokine-induced loss of skeletal muscle gene products. J Biol Chem. 2003;278:2294–2303. doi: 10.1074/jbc.M207129200. [DOI] [PubMed] [Google Scholar]
- 19.Kumar A, Boriek AM. Mechanical stress activates the nuclear factor-kappaB pathway in skeletal muscle fibers: a possible role in Duchenne muscular dystrophy. Faseb J. 2003;17:386–396. doi: 10.1096/fj.02-0542com. [DOI] [PubMed] [Google Scholar]
- 20.Baghdiguian S, Richard I, Martin M, Coopman P, Beckmann JS, Mangeat P, et al. Pathophysiology of limb girdle muscular dystrophy type 2A: hypothesis and new insights into the IkappaBalpha/NF-kappaB survival pathway in skeletal muscle. J Mol Med. 2001;79:254–261. doi: 10.1007/s001090100225. [DOI] [PubMed] [Google Scholar]
- 21.Bakkar N, Wang J, Ladner KJ, Wang H, Dahlman JM, Carathers M, et al. IKK/NF-kappaB regulates skeletal myogenesis via a signaling switch to inhibit differentiation and promote mitochondrial biogenesis. J Cell Biol. 2008;180:787–802. doi: 10.1083/jcb.200707179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Acharyya S, Villalta SA, Bakkar N, Bupha-Intr T, Janssen PM, Carathers M, et al. Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J Clin Invest. 2007;117:889–901. doi: 10.1172/JCI30556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang B, Li J, Xiao X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc Natl Acad Sci U S A. 2000;97:13714–13719. doi: 10.1073/pnas.240335297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watchko J, O’Day T, Wang B, Zhou L, Tang Y, Li J, et al. Adeno-associated virus vector-mediated minidystrophin gene therapy improves dystrophic muscle contractile function in mdx mice. Hum Gene Ther. 2002;13:1451–1460. doi: 10.1089/10430340260185085. [DOI] [PubMed] [Google Scholar]
- 25.Wang Z, Kuhr CS, Allen JM, Blankinship M, Gregorevic P, Chamberlain JS, et al. Sustained AAV-mediated Dystrophin Expression in a Canine Model of Duchenne Muscular Dystrophy with a Brief Course of Immunosuppression. Mol Ther. 2007;15:1160–1166. doi: 10.1038/sj.mt.6300161. [DOI] [PubMed] [Google Scholar]
- 26.Heslop L, Morgan JE, Partridge TA. Evidence for a myogenic stem cell that is exhausted in dystrophic muscle. J Cell Sci. 2000;113(Pt 12):2299–2308. doi: 10.1242/jcs.113.12.2299. [DOI] [PubMed] [Google Scholar]
- 27.Anderson JE. The satellite cell as a companion in skeletal muscle plasticity: currency, conveyance, clue, connector and colander. J Exp Biol. 2006;209:2276–2292. doi: 10.1242/jeb.02088. [DOI] [PubMed] [Google Scholar]
- 28.Carlson CG, Samadi A, Siegel A. Chronic treatment with agents that stabilize cytosolic IkappaB-alpha enhances survival and improves resting membrane potential in MDX muscle fibers subjected to chronic passive stretch. Neurobiol Dis. 2005;20:719–730. doi: 10.1016/j.nbd.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 29.Tas SW, Adriaansen J, Hajji N, Bakker AC, Firestein GS, Vervoordeldonk MJ, et al. Amelioration of arthritis by intraarticular dominant negative Ikk beta gene therapy using adeno-associated virus type 5. Hum Gene Ther. 2006;17:821–832. doi: 10.1089/hum.2006.17.821. [DOI] [PubMed] [Google Scholar]
- 30.Sicinski P, Geng Y, Ryder-Cook AS, Barnard EA, Darlison MG, Barnard PJ. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244:1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 31.Bulfield G, Siller WG, Wight PA, Moore KJ. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci USA. 1984;81:1189–1192. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sicinski P, Geng Y, Ryder-Cook AS, Barnard EA, Darlison MG, Barnard PJ. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244:1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 33.Torres LF, Duchen LW. The mutant mdx: inherited myopathy in the mouse. Morphological studies of nerves, muscles and end-plates. Brain. 1987;110(Pt 2):269–299. doi: 10.1093/brain/110.2.269. [DOI] [PubMed] [Google Scholar]
- 34.Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 35.Demoule A, Divangahi M, Yahiaoui L, Danialou G, Gvozdic D, Labbe K, et al. Endotoxin triggers nuclear factor-kappaB-dependent up-regulation of multiple proinflammatory genes in the diaphragm. Am J Respir Crit Care Med. 2006;174:646–653. doi: 10.1164/rccm.200509-1511OC. [DOI] [PubMed] [Google Scholar]
- 36.Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang B, Li J, Qiao C, Chen C, Hu P, Zhu X, et al. A canine minidystrophin is functional and therapeutic in mdx mice. Gene Ther. 2008;15:1099–1106. doi: 10.1038/gt.2008.70. [DOI] [PubMed] [Google Scholar]
- 38.Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS., Jr NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol. 1999;19:5785–5799. doi: 10.1128/mcb.19.8.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Straub V, Rafael JA, Chamberlain JS, Campbell KP. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J Cell Biol. 1997;139:375–385. doi: 10.1083/jcb.139.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]