

Abstract
Immunoglobulin heavy chain (Igh) class switch recombination (CSR) requires targeted introduction of DNA double strand breaks (DSBs) into repetitive “switch” region DNA elements in the Igh locus and subsequent ligation between distal DSBs. Both canonical non-homologous end-joining (C-NHEJ) that seals DNA ends with little or no homology, and a poorly defined alternative-end joining (A-NHEJ) process that requires microhomology ends for ligation have been implicated in CSR. Here we show that the DNA end-processing factor CtIP is required for microhomology-directed A-NHEJ during CSR. Additionally, we demonstrate that microhomology joins that are enriched upon depletion of C-NHEJ component Ku70 require CtIP. Finally, we show that CtIP binds to switch region DNA in an AID-dependent fashion. Our results establish CtIP as a bona fide component of microhomology-dependent A-NHEJ and unmask a hitherto unrecognized physiological role of microhomology-mediated end-joining in a C-NHEJ proficient environment.
During an immune response, mature B-lymphocytes undergo CSR, a deletional-recombination reaction that exchanges the Cµ constant region gene (CH) of the expressed Igh for one of a set of downstream constant CH genes, such as Cγ, Cε or Cα. The B cell thus alters from producing IgM to one expressing a different effector antibody molecule such as IgG, IgE or IgA1. CSR occurs between transcribed, repetitive 1–12 kb long DNA elements termed switch (S) regions that precede each of the CH genes1. Activation-induced cytidine deaminase (AID), an essential enzyme for CSR2,3, deaminates cytidines to uridines within transcribed S regions to initiate a cascade of reactions that generates staggered DSBs4. Synapsis and end-joining of DSBs between two distinct S regions completes CSR.
The end-joining phase of CSR utilizes general DNA repair processes5. C-NHEJ, which seals DNA ends with little (1–3 nucleotides) or no homology, is a major DSB repair pathway in somatic cells6 and was thought to be essential for CSR7. However, recent studies have shown that mutations in several core C-NHEJ components including Ku70, DNA ligase IV and XRCC4 still allowed substantial CSR8–11. The mutant cells though had a striking alteration in the nature of the switch junctions. While the majority of junctions in normal B cells were either blunt or had 1–3 base pairs of microhomology, those in the C-NHEJ mutants displayed a significant trend towards increased microhomology8–11. Thus, CSR proceeds through a robust A-NHEJ pathway that displays a significant bias towards microhomology joins.
In addition to CSR, A-NHEJ has also been observed in a few other instances. First, several reporter substrates that measure joining of microhomologous DNA sequences have revealed the existence of A-NHEJ12–15. Second, while C-NHEJ is essential for end-joining of DSBs generated by RAG proteins during V(D)J recombination, certain RAG mutations unmasked an A-NHEJ reaction that utilized microhomologous sequences for end-joining of reporter recombination substrates16,17. Finally, interchromosomal translocations involving the Igh locus frequently observed in C-NHEJ mutant B cells appear to use the A-NHEJ pathway9,18,19. This process is predicted to involve a DNA end resection step to expose short single-stranded DNA stretches homologous to the other end being joined. Whether all or a subset of microhomology-mediated end joining constitute A-NHEJ is a matter of debate20 but it is clear that A-NHEJ preferably utilizes microhomology sequences. In this study, we have referred to all microhomology-mediated end-joining as A-NHEJ.
The factors required for A-NHEJ have not been elucidated; however, the end-processing proteins Mre11 and CtIP are thought to be involved12–15,21,22. CtIP was originally identified as an interactor of the transcriptional co-repressor molecule CtBP and was thus thought to modulate transcription23. It was subsequently shown to participate in cell cycle checkpoint control23 and DNA repair by homologous recombination (HR) through its ability to bind BRCA1 in a phosphorylation-dependent fashion13,24–26. Additionally, CtIP and its yeast functional homologue Sae227 have been shown to be involved in resection of DSBs during homologous recombination, either acting directly as a nuclease and/or enhancing the nuclease activity of Mre1125,26,28–32. Recently, studies using reporter substrates have demonstrated that CtIP participates in A-NHEJ12,13,15, although the role of CtIP in HR and A-NHEJ are distinct as unlike that for HR, A-NHEJ does not require phosphorylation-dependent interaction with BRCA113.
The overall model that emerged from these studies is that CtIP promotes processing of DSBs to reveal segments of homology that could be utilized for HR-based repair or stretches of microhomology for A-NHEJ. However, the majority of these studies, especially those that examined the role of CtIP in A-NHEJ, relied on the use of artificial substrates, which could potentially have a dominant effect on the nature of the end-joining reaction20. In this study, we have used CSR as a physiological reaction to query the role of CtIP in A-NHEJ and demonstrate that CtIP plays a major role in microhomology mediated end-joining in normal as well as in C-NHEJ deficient cells.
RESULTS
CtIP knock-down impairs CSR
To determine the role of CtIP in CSR, we stably knocked-down CtIP expression in the murine B cell line CH12 using short hairpin RNAs (shRNA). CH12 cells, upon stimulation with a combination of anti-CD40, interleukin-4 (IL-4) and transforming growth factor β (TGF-β) (henceforth referred to as CIT) undergo CSR from IgM to IgA at a high rate2. Two shRNAs (CtIP-1 and CtIP-2) directed against the coding sequence of CtIP mRNA robustly depleted CtIP protein from CH12 cells compared to cells transduced with control “scrambled” shRNA (Fig. 1a). Control and CtIP knock-down cells were stimulated with CIT for 72 hours and CSR to IgA was measured by flow cytometry. Over the course of at least 14 independent experiments (Supplementary Table 1), CtIP knock-down displayed a 40–60% defect in CSR compared to control cells (Fig. 1b and Supplementary Table 1).
Figure 1.
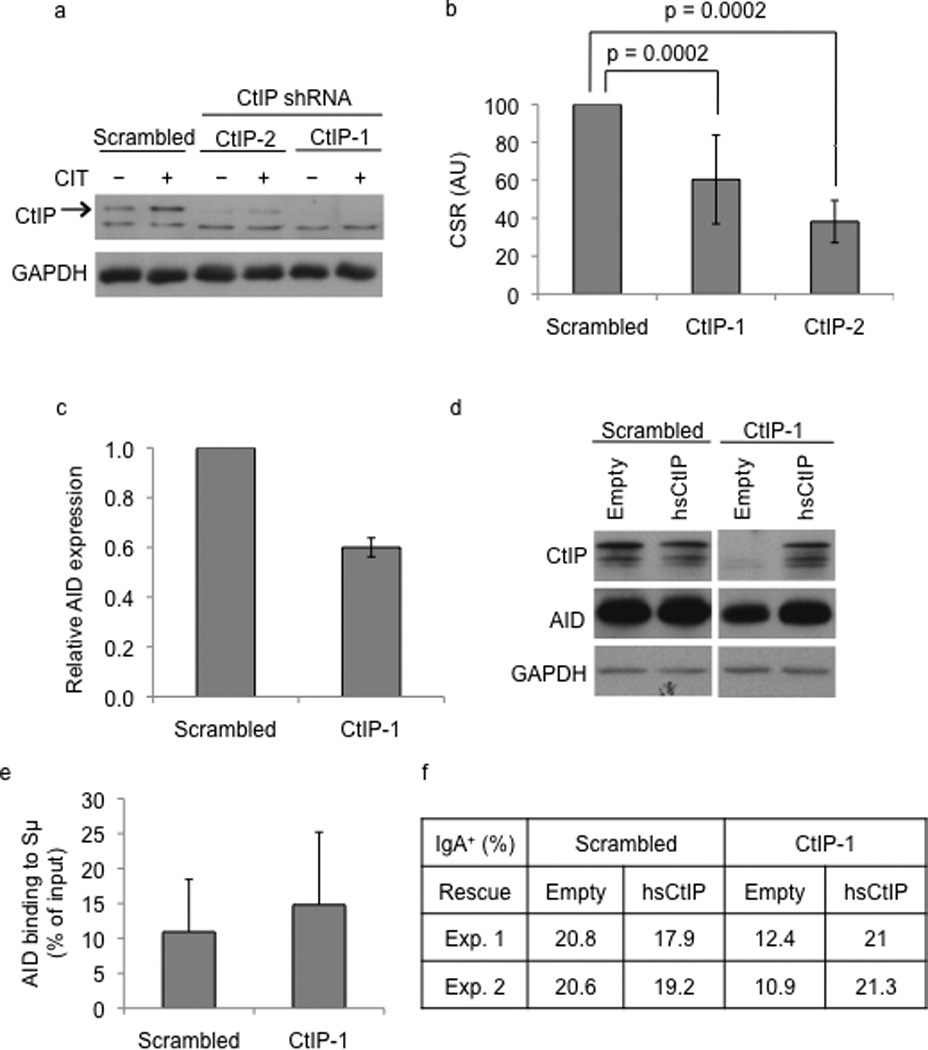
CtIP knock-down alters CSR in CH12 cells. (a) Extracts derived from unstimulated (−) or CIT stimulated (+) CH12 cells expressing two different shRNAs (CtIP-1 or CtIP-2) against CtIP or control “scrambled” shRNA were analyzed by western blotting using antibodies against CtIP or GAPDH (loading control). The arrow indicates the polypeptide corresponding to CtIP. (b) CSR to IgA was measured by flow cytometry. CSR frequency in scrambled shRNA infected cells was assigned an arbitrary unit (AU) of 100. The data is a mean of 14 independent experiments with error bars depicting standard deviation from the mean. (c) Levels of AID transcripts of scrambled or CtIP knock-down cells were measured 48h after CIT stimulation by quantitative real-time PCR. The values are the mean of three independent experiments and the error bars represent standard deviation from the mean. (d) Empty lentiviral vector or one harboring human CtIP (hsCtIP) cDNA was introduced into control or CtIP knock-down cells and western blot was performed to determine expression of CtIP. (e) ChIP was performed from CIT-stimulated CH12 cells with anti-AID and the amount of AID bound to Sµ was determined by real-time quantitative PCR. The values represent the average of 3 independent experiments with error bars representing standard deviation from the mean. (f) CSR was measured by flow cytometry in hsCtIP or empty expression vector transduced CtIP knock-down cells.
The reduction in CSR in CtIP knock-down cells was not accompanied by any significant defect in cell proliferation or marked alterations in the steady-state levels of germline switch transcripts (Supplementary Fig. 1). On the other hand, quantitative real-time PCR (Fig. 1c) and western blot analysis (Fig. 1d, Supplementary Fig. 2) showed that both AID mRNA and protein levels were reduced in CtIP-depleted cells. CtIP is a known transcriptional regulator23 and since AID expression is a function of multiple activators and inhibitors33, it is possible that CtIP regulates AID expression. Thus, reduced AID levels might contribute to the CSR defect observed in CtIP depleted cells. However, chromatin immunoprecipitation (ChIP) experiments showed that the amount of AID associated with Sµ was similar between control and CtIP knock-down cells (Fig. 1e). Significantly, when CtIP expression was restored in CtIP depleted cells through lentiviral transduction of shRNA-resistant human CtIP cDNA (Fig. 1d), both CSR (Fig. 1f) and AID expression (Fig. 1d) were rescued to control levels. The complementation experiment indicated that the CSR defect and reduction of AID expression in CtIP depleted cells were not due to non-specific off-target effects of the shRNAs.
CtIP knock-down alters end-joining during CSR
The nature of the switch junctions following CSR is an ideal readout for the end-joining processes that ligate DSBs. We thus cloned and sequenced Sµ-Sα junctions from control and CtIP-depleted cells stimulated with CIT for 72 hours. Consistent with previous studies1, the majority of Sµ-Sα junctions in control cells had microhomology between 0–3 nucleotides, but a substantial number (approximately 24%) of junctions showed microhomology of 4 nucleotides or more (Fig. 2a–b, Supplementary Fig. 3). The Sµ-Sα junctions in the CtIP-depleted cells were markedly different from the control cells (Fig. 2a). Approximately 11.5% of the junctions in the CtIP knock-down cells displayed microhomology length of 4 nucleotides or more (Fig. 2a–b). Remarkably, there was a significant alteration in frequency of junctions with direct (blunt) joins with no microhomology. The number of junctions with blunt joins increased from around 15% for control cells to approximately 40% for CtIP knock-down cells. Overall, the average microhomology length showed a significant (p<0.04 by the Mann-Whitney test) decrease from 2.4 nucleotides observed for control cells to 1.4 nucleotides for CtIP-depleted cells. Finally, restoring CtIP expression shifted the nature of microhomology-mediated joins to levels approaching that observed for control cells (Fig. 2c). Taken together, these results provide evidence for a CtIP-dependent pathway that utilizes microhomology for the end-joining phase of CSR.
Figure 2.
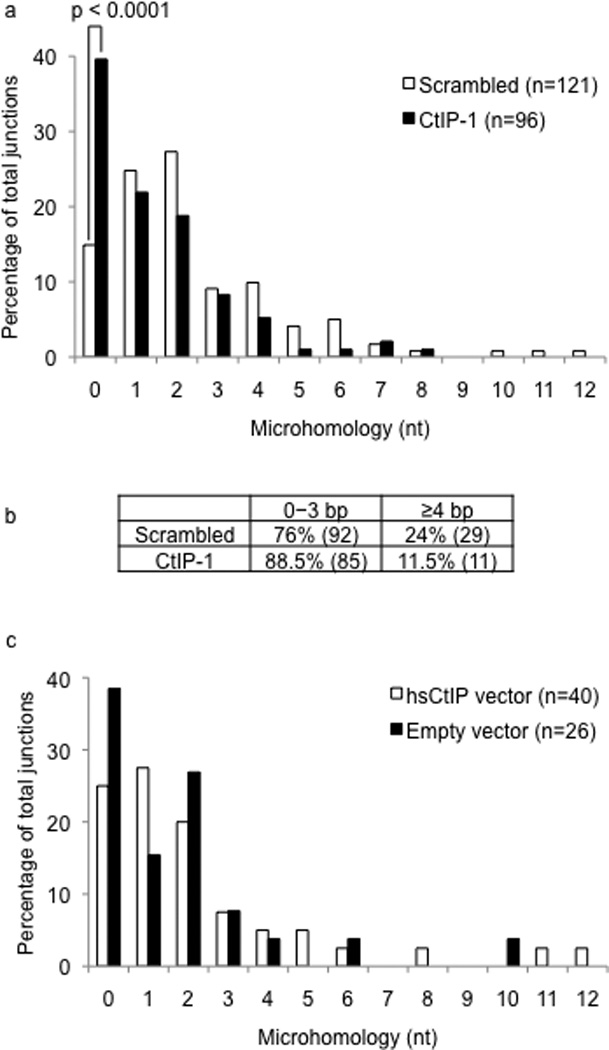
CtIP knock-down alters end-joining during CSR. (a) Sµ-Sα junctions from CIT-stimulated CH12 cells infected with control or CtIP-1 shRNA were analyzed. Sequence data was compiled from six independent experiments. (b) The spectrum of junctions in control versus CtIP knock-down cells was tabulated. The difference in the percentage of junctions with microhomology of 4 nucleotides or more was statistically significant (p=0.02). (c) Sµ-Sα junctions in CtIP knock-down cells transduced with empty vector or vector encoding hsCtIP were analyzed.
C-NHEJ deficiency predisposes CSR to an end-joining pathway that utilizes microhomologous sequences at the ends8–11. To determine if CtIP is responsible for the microhomology dependent joins in cells compromised for C-NHEJ, we generated a stable knock-down of Ku70 in CH12 cells (Fig. 3a). Ku70-depletion led to a marked and significant reduction in CSR (Fig. 3b) although it is not clear if impaired CSR is partially an effect of reduced AID expression in the Ku70-depleted cells (Supplementary Fig. 2). As predicted from previous reports11, Ku70 depletion leads to a marked shift towards switch junctions with microhomology of 4 nucleotides or more (approximately 30%) compared to control cells (approximately 14%, Supplementary Fig. 2). When CtIP was knocked-down in Ku70-depleted cells (Fig. 3c), there was a significant additive effect on CSR compared to cells with depletion of Ku70 alone (Fig. 3d). Strikingly, depletion of CtIP in the Ku70 knock-down cells reduced the number of junctions with extended microhomology and shifted the overall extent of microhomology to that observed for CtIP knock-down cells (Fig. 3e, f). Thus, while the average length of microhomology in Ku70 knock-down cells was 3.3 nucleotides, combined depletion of Ku70 and CtIP reduced the average down to 1.8 nucleotides. These results strongly suggested that CtIP mediates microhomology-mediated end-joining observed in C-NHEJ deficient cells.
Figure 3.
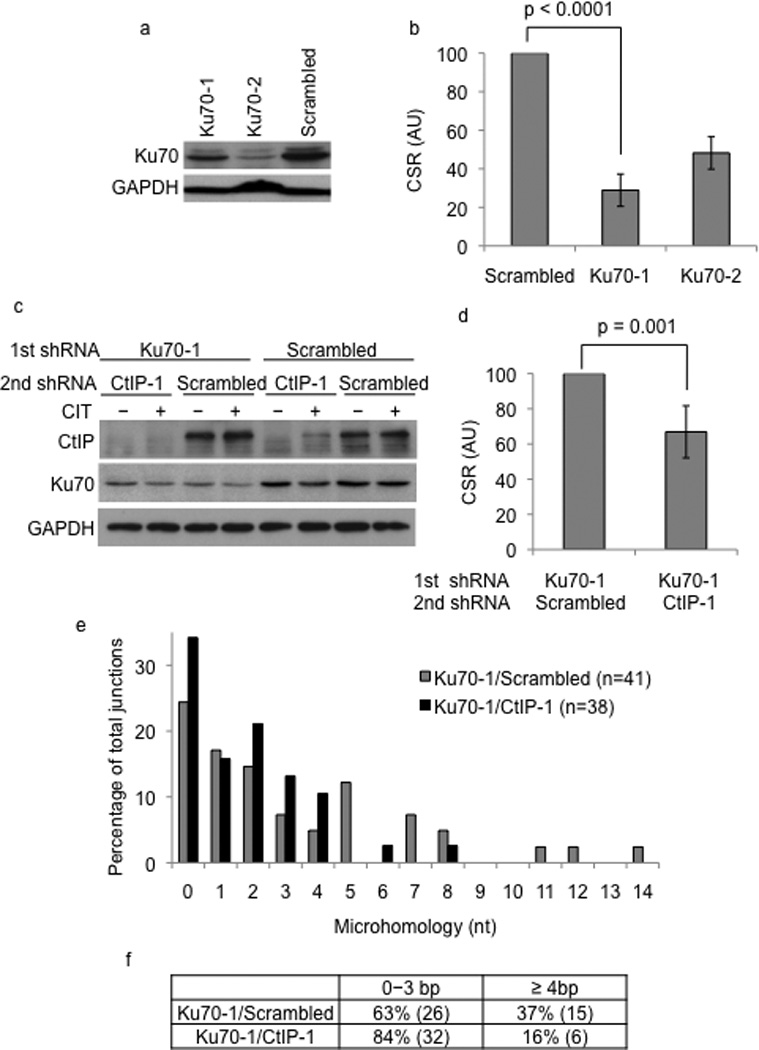
CtIP knock-down alters end-joining in Ku70-deficient cells. (a) CIT-stimulated CH12 cells transduced with scrambled or shRNAs (Ku70-1, Ku70-2) against Ku70 were analyzed by western blotting using Ku70 or GAPDH (loading control) antibodies. (b) CSR to IgA in Ku70-1 or Ku70-2 shRNA transduced cells was measured by flow cytometry. CSR frequency in cells transduced with scrambled shRNA was assigned an arbitrary unit (AU) of 100. The data is a mean of at least three independent experiments with error bars indicating standard deviation from the mean. (c) Ku70 knock-down or control cells were transduced with scrambled or CtIP-1 shRNA and expression of Ku70, CtIP and GAPDH were determined by western blot analysis. (d) CH12 cells with the indicated shRNAs were stimulated with CIT for 72 hours and CSR to IgA was measured by flow cytometry. CSR frequency in cells transduced with scrambled shRNA was assigned an AU of 100. The data is a mean of nine independent experiments with error bars representing standard deviation from the mean. (e) Sµ-Sα junctions from Ku70-1/scrambled and Ku70-1/CtIP-1 cells were cloned, sequenced and the percentage of cells with the indicated lengths of microhomology was plotted. (f) The distribution of microhomology at the Sµ-Sα junctions is tabulated. The difference in the number of junctions with microhomology of 4 nucleotides or more between the Ku70-1/scrambled and Ku70-1/CtIP-1 knock-down cells was statistically significant (p=0.04).
CtIP binds switch region DNA
We carried out ChIP experiments to assess, if in keeping with its ability to participate in the end-joining phase of CSR, CtIP could be detected at S region DNA. CtIP was found to be specifically associated with Sµ in control but not in the CtIP knock-down CH12 cells (Fig. 4a, b). CtIP binding was specific to S region DNA as it was not detected at the Iµ intronic promoter or at p53 genomic locus. Remarkably, there was a 6–8-fold increase in the levels of CtIP bound to Sµ in Ku70 knock-down cells (Fig. 4a, b), suggesting that depletion of Ku70 enhances the ability of CtIP to bind S region DNA. The binding of CtIP to S regions in CH12 cells was dependent on CIT stimulation suggesting that CtIP associates with broken S region DNA (Fig. 4c). This possibility is further supported by the observation that CtIP binding to activated S regions in splenic B cells was dependent on AID. While CtIP could be readily detected at Sµ and Sγ1 in wild type splenic B cells stimulated with anti-CD40 and IL-4, it failed to associate with the S regions in similarly activated AID-deficient splenic B cells (Fig. 4d). Importantly, the observed binding of CtIP to S regions in an AID, and likely DSB-dependent fashion in splenic B cells provides credence to the notion that CtIP participates in CSR in primary B cells.
Figure 4.
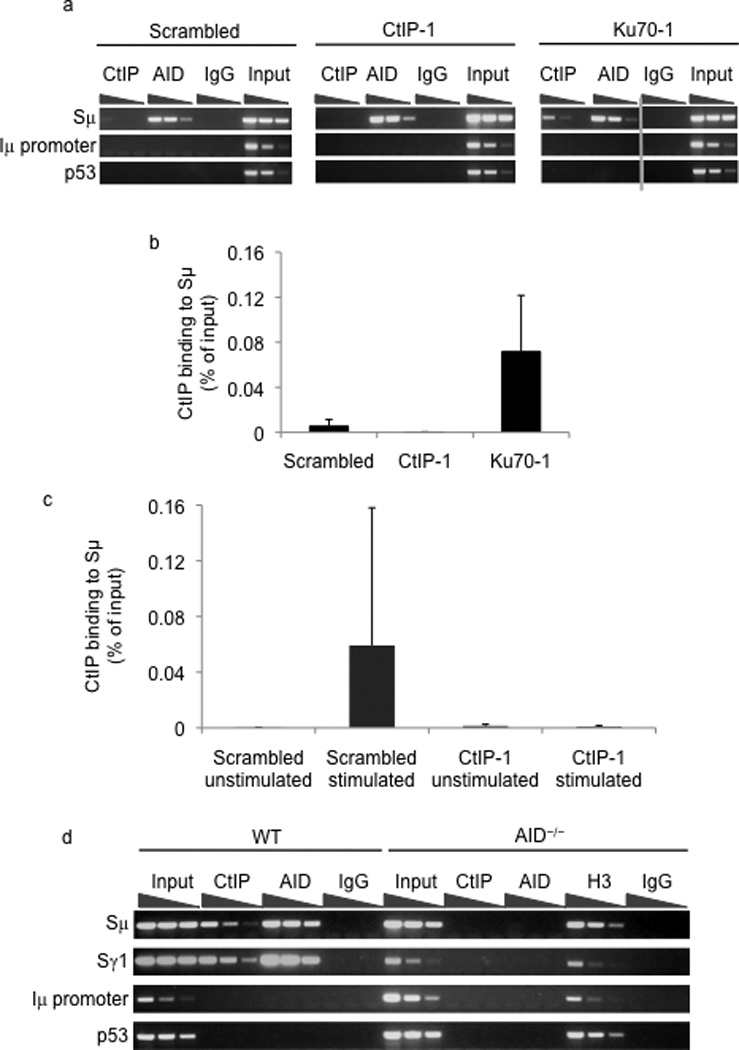
CtIP binds to switch region DNA. (a) ChIP was carried out in CIT-stimulated CH12 cells with AID, CtIP or control non-specific IgG antibodies. DNA from the ChIP samples was diluted 3-fold, amplified by PCR and the presence of Sµ, µpromoter (Iµ promoter) or p53 was determined by analyzing the PCR products on agarose gels. (b) The amount of CtIP bound to Sµ in the indicated cells was measured by real-time quantitative PCR. Data are the mean of three independent experiments with error bars representing standard deviation from the mean. (c) CtIP binding to Sµ in unstimulated or CIT-stimulated CH12 cells was assayed by ChIP and quantified by real-time PCR. The values represent the mean of three independent experiments with error bars representing standard deviation from the mean. (d) Splenic B cells from wild type or AID−/− mice were stimulated with α-CD40 and IL-4 for 48hrs and ChIP was performed with CtIP, AID, H3 and control non-specific IgG antibodies. Immunoprecipitated DNA was diluted 4-fold and amplified by PCR for the presence of Sµ, Sγ1, Iµ promoter or p53.
DISCUSSION
We have used shRNA-mediated knock-down to unequivocally demonstrate that CtIP participates in the end-joining phase of CSR in CH12 cells. While CtIP deficient embryos fail to develop34, CtIP knock-down CH12 cells exhibit no growth defects, possibly due to the residual activity of CtIP in the setting of a knock-down and/or due to a striking induction of the proliferative and anti-apoptotic cytokine IL-6 (Supplementary Fig. 4). Switching in CH12 cells is dependent on AID (Supplementary Fig. 6) and influenced by known factors such as DNA ligase IV10 and there is no evidence that CSR to IgA in CH12 cells is in any way mechanistically different from primary B cells. Supported by the observation that CtIP binds to appropriately activated S regions in primary B cells, it is reasonable to extrapolate our findings in CH12 cells to conclude that CtIP participates in CSR in general.
In the experimental system described here, CtIP levels clearly have an effect on AID expression. This is neither due to a non-specific effect of the shRNAs introduced into the cells, as restoring CtIP expression rescues AID levels, nor an effect on global transcription. Gene expression profiling of CtIP depleted cells by microarray analysis showed that only about 0.25% of the total genes analyzed were altered by a fold change of 2 or more relative to control cells (Supplementary Fig. 4). Whether CtIP regulates AID expression in primary B cells and if this regulation is related to the transcriptional activity of CtIP remains to be determined. Despite repeated attempts, we were unable to restore AID expression to normal levels in CtIP knock-down cells through lentiviral transduction (Supplementary Fig. 5). Thus, we could not formally exclude the possibility that at least a part of the observed defect in CSR upon CtIP depletion is due to reduced AID levels. Still, the decrease in AID expression does not lead to altered levels of AID bound to S region DNA. More importantly, specific knock-down of AID in CH12 cells does not increase the abundance of joins with 0–3 nucleotide microhomology (Supplementary Fig. 6), suggesting that altered junctions in CtIP knock-down cells are not due to levels of AID but rather an effect of the CtIP-dependent end-processing reaction during CSR.
According to the generally accepted model of CSR1, DNA deaminated by AID is processed by the combined activities of base-excision and mismatch repair proteins to generate staggered DNA breaks35. Our results suggest that DSBs thus generated could be bound by Ku and channeled into the C-NHEJ pathway or be processed in a CtIP-dependent microhomology-directed A-NHEJ reaction (Fig. 5). Thus, in Ku70-depleted cells there is increased CtIP binding to S regions (Fig. 4a–b), with an accompanying increase in microhomology joins, while in the absence of CtIP, C-NHEJ is the predominant end-joining pathway operating without the requirement for microhomology at the broken ends.
Figure 5. A model for the role of CtIP in CSR.
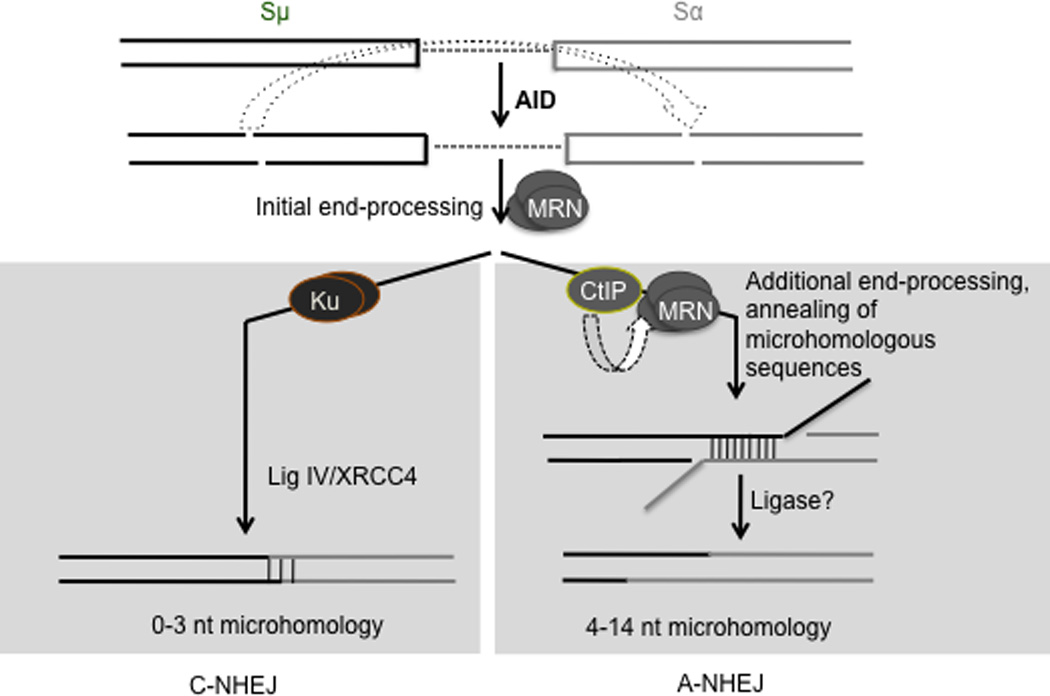
AID activity induces formation of staggered DSBs in two distinct S regions. The DSBs are initially processed by the Mre11/Rad50/Nbs1 (MRN) complex, possibly to generate blunt ends and then channeled into either the C-NHEJ or the A-NHEJ pathway. For C-NHEJ, blunt DSBs are bound by Ku (and other C-NHEJ proteins) and subsequently ligated by DNA ligase IV/XRCC4 that do not require microhomology at the DNA ends. For A-NHEJ, CtIP alone, or in conjunction with MRN, further processes the DNA ends to reveal stretches of microhomology prior to ligation. Since CtIP binding to S regions is enhanced when Ku protein is depleted (fig. 4), Ku could possibly compete with or suppress the A-NHEJ pathway. Additional components of A-NHEJ that participate in CSR, including the ligase that seals the DNA ends are yet to be elucidated.
It has been proposed that CtIP functions in end-processing through its ability to promote the nuclease activity of the Mre11-Rad50-Nbs1(MRN) complex25,26,28,29,31,32. MRN has been implicated in both c-NHEJ and A-NHEJ14,15,21 and mutations in MRN components significantly impair CSR21,36–38. However, in contrast to what we observed for CtIP depletion, Mre11 mutation does not alter the nature of switch junctions21, suggesting that MRN has both CtIP-dependent and CtIP-independent activities during CSR. It is possible that limited resection required for C-NHEJ can be mediated by MRN alone, while extensive processing of ends to expose microhomologous sequences during A-NHEJ relies on CtIP-mediated enhancement of MRN nuclease activity. Alternatively, CtIP might have a role in A-NHEJ that does not require MRN, a notion consistent with the MRN-independent nuclease activity of Sae229 (fig. 5). Further work is clearly required to experimentally examine these possibilities.
C-NHEJ is a distinct pathway with a well-defined set of factors required exclusively for this process. On the other hand, microhomology-mediated A-NHEJ is poorly defined and its function in normal cellular physiology beyond the immune system is yet to be defined. Likewise, the components of this pathway, including the DNA ligases that mediate catalysis of the DNA ends, remain to be identified. Despite these unresolved issues, our studies clearly demonstrate that CtIP is required for the end-joining phase of CSR and thus renders CSR an appropriate model to elucidate the mechanism that promotes end-joining of microhomologous DNA in a physiological setting.
Methods
Cell culture
CH12 cells were grown in RPMI 1640 supplemented with 10% (v/v) fetal bovine serum (FBS, Atlanta biologicals), 100 U ml−1 of penicillin, 0.1 mg ml−1 of streptomycin (Gemini Bio-products), 5% (v/v) NCTC-109 (Gibco), and 10 mM β-mercaptoethanol. To induce CSR, cells were cultured at a density of 0.25 × 106 cells per ml in 1 µg ml−1 anti-CD40 (eBioscience), 12.5 µg ml−1 IL-4 (R&D Systems) and 0.1 ng ml−1 TGF-β (R&D Systems). Mouse splenic B cells were isolated using CD43 Microbeads (Miltenyi Biotec) and cultured at a density of 1 × 106 per ml in RPMI 1640 supplemented with 15% (v/v) FBS, 100 U ml−1 of penicillin, and 0.1 mg ml−1 of streptomycin, 1% (v/v) glutamine and 10 mM β-mercaptoethanol. Stimulation for CSR was achieved with 1 µg ml−1 anti-CD40 and 12.5 µg ml−1 IL-4.
Lentiviral infection of shRNA constructs
The lentiviral MISSION shRNA vectors were obtained from Sigma-Aldrich (Supplementary Table 2). 293T cells were transfected with 12 µg of the shRNA vector, 9 µg of the packaging vector psPAX2 (Addgene) and 3 µg of the envelope vector pMD2.G (Addgene) using Lipofectamine 2000 (Invitrogen). Viral supernatants were harvested 48 hours after transfection. CH12 cells (1 × 106) were transduced with 4 ml of viral supernatant by centrifugation at 2000 rpm for 2 hours at room temperature. Twenty-four hours after transduction, cells were selected with puromycin at a concentration of 3µg ml−1 for 72 hours and then analyzed. For double knock-downs, CH12 cells were transduced with the first shRNA and following 3 days of growth in puromycin, transduced with the second shRNA. Cells were stimulated with CIT 48 hours after second infection.
Expression of proteins in CH12 cells
hsCtIP or mouse AID cDNA was cloned downstream of the EF1α promoter in the lentiviral vector EF.PGK.GFP (Addgene). The vector was transduced into CH12 cells using a protocol similar for transduction of shRNA vectors.
Immunoblotting
Protein extracts for western blotting were prepared by cell lysis in a buffer containing 20mM Tris-HCl (pH 7.5), 5% (v/v) glycerol, 150mM NaCl, 5mM β-mercaptoethanol and 0.5% (v/v) NP-40, followed by high-speed centrifugation. Antibodies against CtIP (T16) and Ku70 (C-19) were purchased from Santa Cruz and AID antibodies were generated as described39. GAPDH (6C5) antibody was purchased from Millipore.
Flow cytometry and SNARF labeling
Cells were stimulated with CIT for 72 hours, stained for surface expression of IgA using FITC-conjugated-anti-IgA antibody (C10-3, BD Pharmingen) or a combination of unconjugated anti-IgA antibody and Alexa Fluor 594 donkey anti-rat antibody (A-21209, Invitrogen). For SNARF labeling, cells were incubated with 9 µM SNARF (carboxylic acid acetate, succinimidyl ester) in 5% (v/v) FBS for 10 min at 37°C. The reaction was quenched with FBS, cells were washed with 2.5% FBS (v/v) and assayed by flow cytometry at indicated time points.
RNA extraction and transcription analysis by standard and real-time PCR
RNA was extracted from cells stimulated with CIT for 48 or 72 hours using Trizol (Invitrogen) according to manufacturer’s protocol. cDNA was generated using the SuperScript III First-Strand Synthesis System (Invitrogen). Amplification was achieved either by standard PCR using a Taq DNA polymerase system (Qiagen) or real-time PCR using iQ SYBR Green supermix (Bio-Rad). Real-time PCR products were analyzed for incorporation of SYBR Green and crossing points were obtained with the CFX96 Real-time system software of Bio-Rad. The comparative Pfaffl method was used to determine fold change in expression between cells transduced with experimental and scrambled control shRNA using β-actin and GAPDH as reference genes. Sequences of all primers are listed in Supplementary Table 2.
Analysis of switch junctions
Genomic DNA was prepared from CH12 cells stimulated with CIT for 72 hours. Sµ-Sα junction DNA was amplified by PCR (38 cycles) using Sµ and Sα primers listed in Supplementary Table 2. The PCR products, which spanned from 1–2 kb, were gel-extracted and cloned into pGEM-Teasy (Promega). DNA from individual clones was sequenced with T7 primer at the MSKCC DNA Sequencing Core Facility.
ChIP assays
ChIP was performed from approximately 1 × 107 cells according to the protocol provided by Upstate Biotech ChIP kit. CtIP and AID antibodies were the same as that used for immunoblotting, the H3 antibody (Ab 1791) was purchased from Abcam and the IgG control (I5006) from Sigma. The immunoprecipitated DNA was amplified by standard or real-time PCR using primer sequences listed in Supplementary Table 2. For quantification of real-time PCR results, the Ct values obtained were plugged into the following equation: Relative quantity=Efficiency (Ct input - Ct ChIP). The resulting value was then divided by 5 (as input was one-fifth used in pulldown) and multiplied by 100 to determine binding as a percentage of input.
Microarray hybridization and analysis
Total RNA was purified from control and CtIP knock-down cells stimulated with CIT for 72 hours. cDNA was generated and hybridized to GeneChIP arrays MOE 430A 2.0 (Affymetrix) at the MSKCC Genomics Core Laboratory. Microarray data was analyzed using the Partek program. Change in expression of candidate genes was confirmed by real-time PCR using 3 independent knock down-cells stimulated for 48 hours. The primer sequences are listed in Supplementary Table 2.
Statistical analysis
A two-tailed paired student’s t-test was employed for analyses of CSR frequency and a two-tailed Fisher’s exact test was applied for analyses of microhomology length at switch junctions.
Supplementary Material
Acknowledgements
We thank Maria Jasin and colleagues for sharing unpublished results and members of the Chaudhuri laboratory for discussion and advice. This work was supported by grants from the Damon Runyon Cancer Research Fund, Alfred Bressler Foundation, Sloan Kettering Institute and National Institutes of Health (1R01AI072194-01A2).
Footnotes
Author Contributions. M. L., A.M., and D.K. performed experiments; A.M., and S.Z. devised protocols for shRNA-mediated knock-down in CH12 cell lines; M.L., and J.C. devised experiments, analyzed data and wrote the manuscript.
References
- 1.Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. 2008;26:261–292. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muramatsu M, et al. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 3.Revy P, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2) Cell. 2000;102:565–575. doi: 10.1016/s0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- 4.Delker RK, Fugmann SD, Papavasiliou FN. A coming-of-age story: activation-induced cytidine deaminase turns 10. Nat Immunol. 2009;10:1147–1153. doi: 10.1038/ni.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zarrin AA, et al. Antibody class switching mediated by yeast endonuclease generated DNA breaks. Science. 2007;315:377–381. doi: 10.1126/science.1136386. [DOI] [PubMed] [Google Scholar]
- 6.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaudhuri J, Alt FW. Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nat Rev Immunol. 2004;4:541–552. doi: 10.1038/nri1395. [DOI] [PubMed] [Google Scholar]
- 8.Soulas-Sprauel P, et al. Role for DNA repair factor XRCC4 in immunoglobulin class switch recombination. J Exp Med. 2007;204:1717–1727. doi: 10.1084/jem.20070255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan CT, et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449:478–482. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- 10.Han L, Yu K. Altered kinetics of nonhomologous end joining and class switch recombination in ligase IV--deficient B cells. J Exp Med. 2008;205:2745–2753. doi: 10.1084/jem.20081623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boboila C, et al. Alternative end-joining catalyzes class switch recombination in the absence of both Ku70 and DNA ligase 4. J Exp Med. 2010;207:417–427. doi: 10.1084/jem.20092449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008;4:e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yun MH, Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature. 2009;459:460–463. doi: 10.1038/nature07955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie A, Kwok A, Scully R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat Struct Mol Biol. 2009;16:814–818. doi: 10.1038/nsmb.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rass E, et al. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat Struct Mol Biol. 2009;16:819–824. doi: 10.1038/nsmb.1641. [DOI] [PubMed] [Google Scholar]
- 16.Lee GS, Neiditch MB, Salus SS, Roth DB. RAG proteins shepherd double-strand breaks to a specific pathway, suppressing error-prone repair, but RAG nicking initiates homologous recombination. Cell. 2004;117:171–184. doi: 10.1016/s0092-8674(04)00301-0. [DOI] [PubMed] [Google Scholar]
- 17.Corneo B, et al. Rag mutations reveal robust alternative end joining. Nature. 2007;449:483–486. doi: 10.1038/nature06168. [DOI] [PubMed] [Google Scholar]
- 18.Weinstock DM, Brunet E, Jasin M. Formation of NHEJ-derived reciprocal chromosomal translocations does not require Ku70. Nat Cell Biol. 2007;9:978–981. doi: 10.1038/ncb1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simsek D, Jasin M. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nat Struct Mol Biol. 2010;17:410–416. doi: 10.1038/nsmb.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zha S, Boboila C, Alt FW. Mre11: roles in DNA repair beyond homologous recombination. Nat Struct Mol Biol. 2009;16:798–800. doi: 10.1038/nsmb0809-798. [DOI] [PubMed] [Google Scholar]
- 21.Dinkelmann M, et al. Multiple functions of MRN in end-joining pathways during isotype class switching. Nat Struct Mol Biol. 2009;16:808–813. doi: 10.1038/nsmb.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deriano L, Stracker TH, Baker A, Petrini JH, Roth DB. Roles for NBS1 in alternative nonhomologous end-joining of V(D)J recombination intermediates. Mol Cell. 2009;34:13–25. doi: 10.1016/j.molcel.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chinnadurai G. CtIP, a candidate tumor susceptibility gene is a team player with luminaries. Biochim Biophys Acta. 2006;1765:67–73. doi: 10.1016/j.bbcan.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 24.Yu X, Wu LC, Bowcock AM, Aronheim A, Baer R. The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J Biol Chem. 1998;273:25388–25392. doi: 10.1074/jbc.273.39.25388. [DOI] [PubMed] [Google Scholar]
- 25.Chen L, Nievera CJ, Lee AY, Wu X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J Biol Chem. 2008;283:7713–7720. doi: 10.1074/jbc.M710245200. [DOI] [PubMed] [Google Scholar]
- 26.Huertas P, Cortes-Ledesma F, Sartori AA, Aguilera A, Jackson SP. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature. 2008;455:689–692. doi: 10.1038/nature07215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clerici M, Mantiero D, Lucchini G, Longhese MP. The Saccharomyces cerevisiae Sae2 protein promotes resection and bridging of double strand break ends. J Biol Chem. 2005;280:38631–38638. doi: 10.1074/jbc.M508339200. [DOI] [PubMed] [Google Scholar]
- 28.Sartori AA, et al. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lengsfeld BM, Rattray AJ, Bhaskara V, Ghirlando R, Paull TT. Sae2 is an endonuclease that processes hairpin DNA cooperatively with the Mre11/Rad50/Xrs2 complex. Mol Cell. 2007;28:638–651. doi: 10.1016/j.molcel.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee K, Lee SE. Saccharomyces cerevisiae Sae2- and Tel1-dependent single-strand DNA formation at DNA break promotes microhomology-mediated end joining. Genetics. 2007;176:2003–2014. doi: 10.1534/genetics.107.076539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–774. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.You Z, et al. CtIP links DNA double-strand break sensing to resection. Mol Cell. 2009;36:954–969. doi: 10.1016/j.molcel.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tran TH, et al. B cell-specific and stimulation-responsive enhancers derepress Aicda by overcoming the effects of silencers. Nat Immunol. 2010;11:148–154. doi: 10.1038/ni.1829. [DOI] [PubMed] [Google Scholar]
- 34.Chen PL, et al. Inactivation of CtIP leads to early embryonic lethality mediated by G1 restraint and to tumorigenesis by haploid insufficiency. Mol Cell Biol. 2005;25:3535–3542. doi: 10.1128/MCB.25.9.3535-3542.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rush JS, Fugmann SD, Schatz DG. Staggered AID-dependent DNA double strand breaks are the predominant DNA lesions targeted to Sm in immunoglobulin class switch recombination. Int. Immunol. 2004;16:549–557. doi: 10.1093/intimm/dxh057. [DOI] [PubMed] [Google Scholar]
- 36.Lahdesmaki A, Taylor AM, Chrzanowska KH, Pan-Hammarstrom Q. Delineation of the role of the Mre11 complex in class switch recombination. J Biol Chem. 2004;279:16479–16487. doi: 10.1074/jbc.M312796200. [DOI] [PubMed] [Google Scholar]
- 37.Kracker S, et al. Nibrin functions in Ig class-switch recombination. Proc Natl Acad Sci U S A. 2005;102:1584–1589. doi: 10.1073/pnas.0409191102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reina-San-Martin B, Nussenzweig MC, Nussenzweig A, Difilippantonio S. Genomic instability, endoreduplication, and diminished Ig class-switch recombination in B cells lacking Nbs1. Proc Natl Acad Sci U S A. 2005;102:1590–1595. doi: 10.1073/pnas.0406289102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chaudhuri J, et al. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 2003;422:726–730. doi: 10.1038/nature01574. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.