

Abstract
Protective humoral immune responses result from immunoglobulin (Ig) diversification reactions that proceed through programmed DNA double-strand breaks and mutations in developing or mature B cells. While primary Ig diversity is dependent on V(D)J recombination and the RAG proteins, secondary diversification is achieved through class switch recombination (CSR) and somatic hypermutation (SHM), which require AID (activation induced deaminase). Because aberrant AID activity can result in mutations in non-Ig loci and DNA translocations between the Ig locus and non-Ig genes, the activity of AID must be stringently regulated. AID mRNA expression is regulated transcriptionally by cytokine stimulation and post-transcriptionally by miRNAs. AID activity is regulated by post-translational modifications, subcellular localization, and interaction with other proteins. All of these molecular mechanisms have evolved to specifically induce AID-dependent mutations and DNA double-strand breaks at the Ig loci to promote maximal Ig gene diversification while limiting the access of this mutator to non-Ig regions.
1. INTRODUCTION
1.1. Overview
Mounting an effective humoral immune response requires the production of an almost infinite repertoire of immunoglobulins (Igs) that are capable of recognizing a diverse range of pathogens. In developing B cells, initial variable region diversity is generated through V(D)J recombination, whereby genes that encode the V, D, and J segments are shuffled into proximity by the RAG proteins (RAG1/RAG2) [1, 2]. Although V(D)J recombination generates a vast repertoire of Igs, this genomic reorganization fails to produce Igs with high affinity (Ka 109 M-1) toward their target antigens [3]. In addition, Igs produced solely through V(D)J recombination contain only a Cμ constant coding region (i.e. IgM isotype) and therefore have limited effector functions (e.g. formation of pentameric polymers). In order to generate high affinity Igs with diverse effector functions, mature but naïve IgM-expressing B cells upon encountering antigens in secondary lymphoid organs undergo two additional programmed DNA alteration reactions – class switch recombination (CSR) and somatic hypermutation (SHM) [4-9].
Occurring within the IgH locus, CSR is a DNA deletional-recombination reaction whereby the exons encoding the default Cμ constant coding region are excised to juxtapose a different constant region gene (e.g. Cγ, Cε, or Cα) downstream of the rearranged variable region segment to produce IgG, IgE, or IgA [10]. SHM generates random mutations, and occasionally insertions or deletions, in the variable coding region of the recombined Ig heavy or light chain locus to alter the affinity of the immunoglobulin to its target antigen without affecting its effector function. Another related immunoglobulin diversification reaction, gene conversion (GC), which occurs in chickens, rabbits and sheep, generates templated alterations within the variable regions (reviewed elsewhere [11]). CSR, SHM, and GC are all dependent on AID (activation induced deaminase), a single-strand DNA cytidine deaminase, expressed primarily in activated B cells [12-15]. Cytidine deamination by AID generates G:U mismatches in DNA to initiate a series of reactions that ultimately lead to mutations in the variable region genes during SHM or breaks in the phosphodiester backbone of the DNA during CSR and GC [16-21].
1.2. Molecular Mechanism of CSR and SHM
CSR alters the isotype of the expressed IgH through a programmed DNA double-strand break (DSB) and end-joining reaction between two recombining switch (S) regions. S regions are 1-12 kb G:C rich, repetitive DNA sequences that precede the constant coding sequences (CH) for each Ig isotype [8-10]. The CH genes are organized as cytokine-inducible germline transcriptional units comprising of promoters, IH exons, SH regions, and CH exons [22, 23]. B cells activated for CSR induce germline transcription through Sμ and a downstream switch region, such as Sε, that is poised to undergo recombination. Transcription through these S regions generates ssDNA in R-loop structures [24-27], which are targets of AID-mediated DNA deamination [17]. The deaminated cytosines are removed by UNG (uracil DNA glycosylase), a component of the base-excision repair (BER) machinery, to create abasic sites and subsequently APE-1 (apurinic/apyrimidinic endonuclease) nicks the DNA [16, 28-30]. In an alternative pathway, AID-induced G:U mismatches can be processed by the mismatch repair (MMR) machinery to generate nicks and gaps in the DNA [6, 16, 23] (see also Saribasak and Gearhart, and Chahwan et al. this issue). While the ssDNA of the non-template strand within R-loops is an optimal target of AID activity [17, 20], template strand deamination has been shown to be dependent in part upon the activity of the RNA exosome complex [31]. Overall, AID-mediated DNA deamination at both DNA strands and processing of the deaminated residues lead to the generation of staggered DSBs. Both the non-homologous end-joining (NHEJ) and a poorly characterized alternative-NHEJ pathways ligate DSBs between two distinct S regions to complete CSR [32-37].
Mechanistically, CSR and SHM are linked through the requirement of both transcription and AID [13, 14, 38-48]. However, V regions, unlike S regions, do not contain the G:C-rich sequences that stabilize ssDNA in the form of R-loop structures. In this context, AID interacts with the ssDNA binding protein RPA (Replication Protein A) and the AID•RPA complex, in vitro, binds to and deaminates transcribed DNA, such as V genes, that normally would not form stable R-loop structures [49]. These in vitro studies led to the proposal that during SHM of variable region genes, RPA binds to and stabilizes ssDNA within transcription bubbles, allowing AID-mediated deamination [49]. Consistent with this notion, mutations in AID that abrogate interaction with RPA (see section below on AID phosphorylation) significantly impair SHM without altering AID deaminase activity [50, 51].
The G:U mismatch created following AID deamination of V gene sequences can be processed in several ways to generate the observed SHM pattern [16]. Replication across the G:U mismatch generates transition mutations at G:C base pairs, while replication across a UNG-created abasic site by error-prone DNA polymerases can lead to both transitions and transversions [16]. The G:U mismatch can also be processed by the MMR pathway, which with its associated error-prone DNA polymerases can induce mutations at both G:C and A:T base pairs [7]. Thus, upon inactivation of UNG, transversion mutations at G:C bases are dramatically reduced, while mutations in MMR genes (e.g., Msh2) lead to a significant reduction in A:T mutations [28, 29, 52]. In cells lacking both UNG and Msh2 activities, almost all mutations are transitions at G:C bases occurring via replication across the G:U mismatch [53]. The altered spectrum of SHM and severe defect in CSR observed in UNG and MMR mutant B cells [28, 29, 52-58] provide compelling evidence that AID-generated uridines in DNA serve as obligate intermediates of CSR and SHM.
1.3. Functions of AID outside of immunoglobulin diversification
Although much of the published work on AID has examined its role in Ig diversification, several recent reports suggest that the deaminase activity of AID, in concert with the BER pathway can mediate DNA demethylation during epigenetic reprogramming [59-61]. First, AID and the related DNA deaminase APOBEC1 were shown to deaminate cytosine and 5-methylcytosine (5mC) in vitro [62]. Second, in zebrafish, AID was shown to be in the same genetic pathway with the BER protein MBD4 and a scaffold protein GADD45a to promote DNA demethylation [63]. Third, in primordial germ cells, AID deficiency resulted in a significantly altered methylation profile [61]. Finally, using siRNAs in heterokaryons, AID was shown to be involved in promoter demethylation and induction of pluripotency associated genes Oct4 and Nanog [59]. Taken together, these results provide compelling evidence that AID participates in active DNA demethylation and can partake in nuclear reprogramming towards pluripotency of somatic cells through deamination of 5mC, whereby the AID-induced T:G mismatch can be resolved by DNA glycosylases such as TDG (thymidine DNA glycosylase).
More recently it was shown that over-expression of AID can enhance demethylation of 5-hydroxymethylcytosine (5hmC), which is generated through oxidation of 5mC by the TET proteins [64], to initiate a process of BER that indirectly results in the demethylation of 5mC [65]. Although these remarkable studies have described a role of AID in active or passive demethylation, the physiological significance of AID-induced DNA demethylation of 5mC or 5hmC remains to be determined, as AID-deficient mice develop without any gross abnormalities of major organs [14]. Interestingly, AID-deficient female mice crossed to AID-deficient males produce smaller litters as compared to their wild-type counterparts; however, whether heritable epimutations are responsible for this phenotype remains to be determined [61]. In addition, the molecular mechanism by which AID accesses hypermethylated DNA to deaminate 5mC or 5hmC within the context of ssDNA remains unclear. Nevertheless, these and other reports have sparked extensive scientific inquiry into the function of AID beyond immunoglobulin gene diversification.
2. REGULATION OF AID EXPRESSION AND LOCALIZATION
While Ig loci are the primary targets of AID, several other genes are also mutated by AID [66-75]. These non-Ig targets of AID are of particular relevance to B cell lymphomagenesis, as a number of mature B cell lymphomas harbor mutations and translocations of oncogenes, such as c-Myc and Bcl6, that have been directly linked to AID activity [67, 69-73]. Thus, multiple mechanisms exist to restrict the expression and deamination activity of AID in B cells undergoing CSR or SHM.
2.1. Transcriptional regulation of AID
The discovery of AID through subtractive hybridization as a molecule upregulated in cells undergoing CSR suggested that its expression is modulated [12]. Encoded from the Aicda locus, AID is expressed primarily in activated germinal center B cells [12, 76], even though low levels of AID have been detected in early developing B cells in the bone marrow [77], germ cells, embryonic stem cells, and breast and ovarian tissue [59, 62, 78]. Regulation of Aicda expression has most extensively been studied in mature B cells. Binding sites for multiple transcription factors were identified both at the promoter and at regions upstream and downstream of the transcription start site, suggesting that a balance of activators and repressors regulate Aicda transcription in B cells [78-82] (see Orthwein and Di Noia this issue). The Aicda promoter just upstream of the transcription start site has binding sites for generally expressed activators, such as Sp1, NFκB and HoxC4, and putative binding sites for the estrogen and progesterone receptors [78-86]. In naïve, unactivated B cells, repressor proteins E2f and c-Myb bind to a “silencer” region located within intron 1 of the Aicda locus. E2A proteins and Pax-5 can bind to this intronic sequence to partially alleviate the repression but cannot induce Aicda expression to the extent observed in activated B cells upon cytokine stimulation [82]. The cytokine-induced enhancer upstream of the transcriptional start site is bound by NF-kB, SMAD 3/4, STAT6 and C/EBP to counter the repressive activity of intron 1 to amplify Aicda transcription. Thus, the ubiquitous expression of the repressor proteins and B-cell specific expression of the activators ensures that Aicda expression is induced only in cytokine stimulated B cells (reviewed in [87]).
Estrogen and progesterone have been shown to have opposing regulatory effects on Aicda transcription. B cells stimulated with estrogen upregulate Aicda transcription, which enhances CSR and SHM ex vivo and in vivo, whereas progesterone treatment represses Aicda transcription, CSR, and SHM [78, 79, 81]. A progesterone binding element (PRE) was characterized within the Aicda promoter that functions to repress TNFα-dependent Aicda transcriptional activation without displacing NFκB activation complexes, which are assembled distal to the PRE [81]. Initial analysis of a 1.5kb promoter upstream of the Aicda transcription start site suggested that a putative estrogen response element (ERE) functioned independently and synergistically with the 5’ proximal NFκB sites to regulate Aicda transcription [78]. However, subsequent studies using mice deficient for Hoxc4 demonstrated that estrogen-mediated transcriptional activation of Aicda in B cells requires HoxC4 binding to a conserved HoxC4/Oct-binding site within the 1.5kb promoter [79]. These data suggest that hormonal regulation of AID expression through estrogen and progesterone may function to improve humoral immune responses in females, which may help to protect them and their fetuses (or newborns) during their reproductive cycles, but also implicates dysregulated AID expression in the generation of autoantibodies, autoimmune disorders, or neoplastic transformation of non-B cells that are disproportionately found in females [88, 89].
2.2. Regulation of AID expression by miRNA
The 3’- untranslated region (3’UTR) of AID mRNA binds two miRNAs, miR-155 and miR-181b, and both these miRNAs have been shown to regulate the mRNA and protein expression of AID [90-92]. miR-155 and miR-181b were identified as miRNAs upregulated during CSR. Mutation of the miR-155 binding site in the 3’-UTR of AID mRNA, either in the context of a bacterial artificial chromosome (BAC) transgene or in the endogenous Aicda locus led to enhanced steady-state levels of AID, which increased CSR and IgH-cMyc translocations. Strikingly, there was no increase in SHM at the V region genes even though mutations at Bcl-6 were enhanced in the mutant B cells [90, 91]. Overexpression of miR-181b in primary B cells caused a 50% reduction in CSR to IgG1 with a corresponding decrease in AID mRNA and protein [92]. B cells from miR-155 deficient mice have a 50% reduction in CSR despite normal levels of AID expression, suggesting that miR-155 regulates the expression of additional proteins required for CSR [91, 93]. As miR-181b levels decrease while miR-155 levels increase upon stimulation for CSR, the concerted actions of both miRNAs likely have nonoverlapping roles in regulating AID expression and consequently AID activity at non-Ig sequences, such as Bcl6 and Myc.
2.3. Subcellular localization of AID
The majority of AID is localized to the cytoplasm due to Crm1/Exportin1-dependent active nuclear export of AID through the C-terminal nuclear export sequence (NES) [94-98] (also see Orthwein and Di Noia and Häsler, Rada, and Neuberger this issue). Remarkably, mutations that impair nuclear export also impair CSR without affecting SHM [96, 99, 100] and reduce AID protein stability, which may be a result of increased ubiquitylation of AID in the nucleus (see section on AID ubiquitylation) [97, 98, 100]. Although these data suggest that association of AID with Crm1/exportin1 and the nuclear export machinery is critical for the activation of CSR, AID binding to exportin1 is not a determinant for CSR, as AID chimeric proteins in which the NES of AID is replaced with an NES from another protein cannot reconstitute CSR in AID-deficient B cells [100, 101]. Hence, the relationship between the nuclear export of AID and efficient CSR remains an enigma.
2.4. AID Ubiquitylation and protein degradation
Ubiquitylated forms of AID are detected primarily in the nucleus of cells treated with the proteosome inhibitor MG132 [97]. Surprisingly, no specific lysine residue within the AID polypeptide sequence could be identified as the site of polyubiquitylation, as a mutant form of AID in which all eight lysines were mutated to arginine still had detectable ubiquitylation in the presence of MG132. AID was also found to be ubiquitinylated in the cytoplasm upon pharmacological inhibition of its interaction with Hsp90 using geldanamycin [102]. More recently, REG-γ, a protein that regulates the levels of p21Cip1 independent of ubiquitin [103], was found to interact with nuclear localized AID to promote its degradation [104] (Figure 1). As compared to wild-type cells, REG-γ-deficient B cells displayed an increase in CSR, which is concomitant with an increase in AID protein levels [104]. Regardless of whether AID is degraded in a ubiquitin-dependent or -independent pathway, these reports clearly underscore the importance of regulating the levels of nuclear AID in order to maximize deamination, mutations, and DSBs at Ig loci while minimizing these events at non-Ig loci.
Figure 1.

Mechanisms regulating AID protein levels and subcellular localization. Cytoplasmic AID is stabilized by Hsp90, whereas nuclear AID is degraded by ubiquitylation or interaction with REG-γ. AID enters the nucleus passively through nuclear pores or actively through its putative N-terminal NLS (nuclear localization signal). AID is actively exported from the nucleus through its C-terminal NES sequence, which binds to Exportin1.
3. ASSOCIATION OF AID WITH DNA
Since the discovery of AID and its characterization as a general mutator, significant effort has been focused on elucidating the mechanism that specifically promotes the binding of AID to the Ig locus during CSR and SHM (Figure 2). In B cells, AID exists in a high-molecular weight complex [17] and several interactors have been identified. The ssDNA binding protein, RPA (Replication Protein A), was shown to interact with AID and promote binding and deamination of AID to transcribed variable region genes in vitro [49]. As RPA can bind to single-stranded DNA in transcription bubbles[105], it was proposed that the AID•RPA complex binds to and stabilizes ssDNA of transcribed variable region genes, allowing AID-mediated deamination during SHM [49]. While binding of RPA to variable region genes in germinal center cells undergoing SHM has not been directly demonstrated, a point mutation in AID that abrogates interaction with RPA (see below) significantly impairs SHM [50, 106], supporting the notion that the AID•RPA interaction is critical for SHM. In addition to RPA, other proteins have been shown to regulate AID deamination of V genes during SHM. The germinal center-associated nuclear protein (GANP) is induced in germinal center B cells during an immune response and has been reported to recruit AID to V genes. GANP-deficient B cells undergo normal CSR but are significantly impaired for SHM [107]. GANP binds V region transcripts, and this purportedly enhances AID binding to V region DNA [108].
Figure 2.
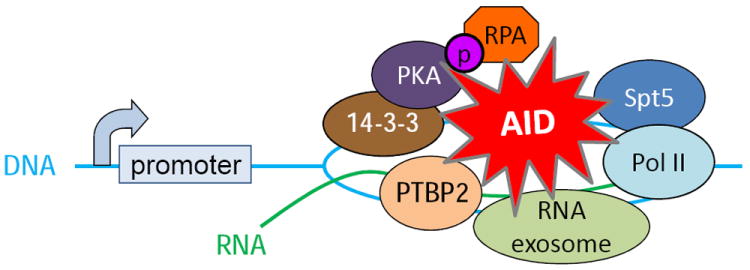
Proteins interacting with AID at S regions during CSR
A screen for proteins that specifically bind to 5′-AGCT-3′ repeat sequences, which are prevalent in S regions, identified 14-3-3 proteins as factors that could recruit AID to S region DNA [109]. Several isoforms of 14-3-3 are upregulated in B cells undergoing CSR. B cells deficient in 14-3-3γ or expressing a dominant negative form of 14-3-3σ are impaired for CSR with an associated reduction in the amount of AID and PKA (protein kinase A), which phosphorylates AID at serine38 (see section 4), bound to activated S regions [109]. These data support a model whereby 14-3-3 proteins function to nucleate AID and its associated proteins at DNA sequences that are targeted for recombination.
The observations that CSR, SHM and mutations of non-Ig genes are tightly linked to transcription of the recombined or mutated DNA sequence [38, 43, 47, 110-113] and that AID interacts with RNA polymerase II (Pol II) [114] suggested that Pol II might facilitate the binding of AID to target DNA sequences. Consistent with this notion, an shRNA screen for effectors of CSR revealed that the Pol II-associated factor Spt5, which binds to stalled Pol II, is required for CSR [115]. Spt5 interacts with AID and depletion of Spt5 markedly reduces recruitment of AID to Ig and non-Ig sequences. ChIP-sequencing analyses showed that Spt5 co-localizes with stalled Pol II and AID and remarkably, occupancy of Spt5 on stalled Pol II sites is predictive of AID-dependent mutations at the corresponding DNA sequence [115]. Spt5 and Pol II are also found throughout the Cμ coding region [115], suggesting that additional mechanisms exist either to dissociate AID from the Spt5-Pol II complex or to limit the ability of AID to deaminate the DNA at Cμ (e.g. lack of R-loops). Interestingly, in S. cerevisiae Spt5 coimunoprecipitates with proteins of the histone chaperone complex FACT [116], which was shown to regulate histone3 lysine4 trimethylation (H3K4me3) and DSB formation at S regions during CSR [117]. These observations demonstrate a critical role for Spt5 and its associated proteins (Pol II, FACT) in regulating AID targeting to specific DNA sequences, and the subsequent generation of DSBs or mutations at these sites, during CSR or SHM [115].
PTBP2 is an AID interacting protein that plays a pivotal role in CSR [118]. PTBP2, and its closely related family member PTBP1, are RNA-binding proteins that participate in alternative pre-mRNA splicing [119]. Expression of PTBP2 was initially thought to be restricted to the brain and is therefore also known as brPTB (brain PTB) and nPTB (neuronal PTB). Subsequently, the protein was detected in testis, liver, heart, lung, skeletal muscle, thymus [120], and recently in B cells [118]. Knock-down of PTBP2 in B cells significantly impairs CSR [118]. The defect in CSR is associated with a reduction in the amount of AID bound to S regions, thereby identifying PTBP2 as a novel AID interactor that influences CSR by recruiting AID to DNA [118].
The mechanism by which PTBP2 targets AID to the Ig locus during CSR is still unknown. Although CSR has been closely linked to both germline transcription and splicing of germline transcripts [121], it is unclear whether components of the splicing machinery or the spliced transcripts themselves (or both) are required for CSR. Current models of CSR posit that transcription generates ssDNA substrates for AID in the context of R-loops [6]. This R-loop dependent model for CSR requires that the primary, unprocessed germline transcript stably hybridizes to the template strand. If splicing occurs co-transcriptionally and efficiently, R-loop formation would be transient. PTBP2 could be recruited to S region DNA, maybe through interactions with other chromatin-associated factors to inhibit splicing, thus enhancing the stability of R-loops and facilitating CSR. While unaltered steady-state levels of spliced germline transcripts in PTBP2 knock-down cells [118] argue against such a role of PTBP2, transient alterations in the half-life of R-loop DNA may not be detectable. Alternatively, the role of PTBP2 in CSR could be independent of its role in splicing. In vitro studies show that PTBP2 binds to sense and antisense S region RNA suggesting that PTBP2 may recruit AID to S regions through its interaction with the S region transcripts [118]. Interestingly, AID has also been shown to interact with CTNNBL1, a poorly characterized protein that has been purified in 14S splicing complexes [122]. Mutants of AID that cannot bind to CTNNBL1 show a significant reduction in CSR and SHM; however, DT40 and CH12F3 cells carrying gene-targeted deletions of CTNNBL1 display low-levels of SHM and normal CSR, respectively [122, 123]. Additional work is required to elucidate the precise function of PTBP2, CTNNBL1, and the splicing of germline transcripts in CSR and SHM.
4. PHOSPHORYLATION OF AID
AID can be phosphorylated at multiple residues—serine 3 (S3), threonine 27 (T27), S38, T140 and tyrosine-184 (Y184) [49, 51, 106, 124, 125] (Figure 3). The S38, T140 and Y184 sites were identified by mass spectrometric analyses of AID purified from activated splenic B cells [51, 124]. Below we summarize the requirement of phosphorylation at each of these residues and describe in depth the possible function of AID phosphorylation at S38.
Figure 3.
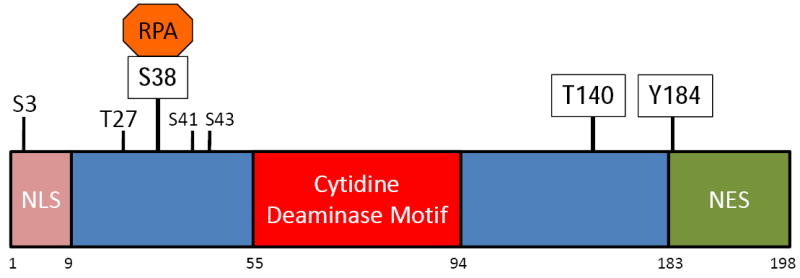
AID phosphorylation sites. The primary structure of AID is shown with the numbers below indicating the amino acid residues that demarcate the putative NLS, the cytidine deaminase catalytic domain, and the NES (nuclear export sequence). Sites of AID phosphorylation are indicated with vertical lines above the primary structure, where boxed amino acids indicate AID phosphorylation sites that were identified in B cells undergoing CSR.
4.1. Phosphorylation at S3
The S3 residue was identified as a site on recombinant AID that could be phosphorylated in vitro by protein kinase C (PKC) [125]. Mutation of S3 to alanine (AIDS3A) does not affect the DNA deaminase activity of AID [125]. Strikingly, expression of a non-phosphorylatable mutant (S3A or S3D) in AID-deficient B cells and fibroblasts increases CSR and SHM, respectively, suggesting that phosphorylation of S3 is an inactivating event [125]. Consistent with the enhanced activity of AIDS3A in CSR, B cells expressing the mutant protein displayed a significant increase in cMyc/Igh translocation frequency relative to B cells expressing wild-type AID protein [125]. Blocking the activity of the serine/threonine phosphatase PP2A with pharmacological inhibitors increases AID phosphorylation at S3 with a concomitant reduction in CSR [125]. Thus, phosphorylation of AID at S3 suppresses AID activity; however, the mechanism through which this phosphorylation regulates AID function has not yet been determined.
4.2. Phosphorylation at T140
Mutation of T140 to alanine in AID does not affect DNA deaminase activity [106]. B cells from mice expressing AIDT140A displayed a mild defect in CSR [106]. Interestingly, AIDT140A mice exhibited a more profound defect in SHM as the mutant B cells displayed a mutation frequency of approximately 40% of wild-type B cells [106]. The mechanism by which T140 phosphorylation modulates AID activity during SHM is not known, but the differential activities of AIDT140A in CSR and SHM is reminiscent of mutations in AID that affect SHM but not CSR (and vice versa) [99, 126]. Thus, phosphorylation of AID at T140 may modulate the ability of AID to interact with an unidentified factor(s) that is required for SHM but not CSR.
4.3. AID phosphorylation sites of unknown function
Whereas mutation of Y184 to alanine does not affect the ability of AID to mediate CSR, mutation of T27 impairs CSR [124, 127]. However, AIDT27A is significantly weaker as a ssDNA deaminase, suggesting that the catalytic site of AID is affected by the mutation [124]. Although the T27 residue can be phosphorylated in vitro with recombinant protein kinase A (PKA) [124, 127], there is no evidence that it is phosphorylated in activated B cells. Likewise, additional phosphorylation sites at S41 and S43 were detected in Sf9 cells expressing AID but whether these residues are functionally relevant during CSR or SHM is not known [128].
4.4. Phosphorylation at S38
Phosphorylation of AID at S38 (AID-pS38) has been studied most extensively. Lying within a PKA consensus phosphorylation site, S38 can be phosphorylated in vitro and in vivo by PKA and in vitro by multiple isoforms of PKC (protein kinase C) [51, 106, 124, 127, 129]. AID-pS38 is indistinguishable from wild-type AID in its ability to deaminate ssDNA [124]. Remarkably, deamination of transcribed V genes in vitro is strongly dependent on phosphorylation at S38 [49, 124, 130]. Biochemical experiments demonstrated that AID-pS38 interacts with RPA [49, 124, 130]. The AID-pS38•RPA complex binds and deaminates V genes [49] and Xenopus laevis S regions [131], which are rich in RGYW motifs but do not form R-loop structures when they are transcribed in vitro by T7 RNA polymerase. These observations led to the suggestion that the AID-pS38•RPA complex, by virtue of the ssDNA binding ability of RPA, can stabilize ssDNA within transcription bubbles at V genes, thereby generating AID substrates during SHM [49]. In addition, because RGYW motifs, and in particular the AGCT sequence, are highly enriched in mammalian S regions, the RPA-dependent mechanism of generating AID substrates was suggested to be important for CSR [49].
Consistent with predictions derived from in vitro activities of pS38-AID, an S38A mutant of AID (AIDS38A) fails to interact with RPA and is severely compromised in its ability to reconstitute CSR when expressed via retroviral transduction into AID-deficient B cells [50, 51, 124, 127]. Unequivocal demonstration that the S38 phosphorylation site is critical for AID function came from gene targeting studies. B cells from mice with an S38A knock-in mutation (AicdaS38A) are substantially impaired in mediating both CSR and SHM [50, 106]. The defect is particularly pronounced in mice haploinsufficient for AicdaS38A where the frequency of CSR and rate of SHM are reduced to near background levels [50, 106].
Several observations suggest that it is the failure to phosphorylate AID at S38, rather than a requirement of the serine residue per se that leads to the CSR and SHM defect in AicdaS38A mice. First, AIDS38A is indistinguishable from wild-type AID in its ability to deaminate ssDNA, indicating that the mutation does not impair the core catalytic activity of the protein [124, 129]. Second, unphosphorylated wild-type AID, or AIDS38A which cannot be phosphorylated by PKA in vitro, does not interact with RPA and cannot mediate deamination of transcribed, double-stranded V region DNA despite retaining full ssDNA deaminase activity [124]. Third, genetic manipulation of PKA subunits that lead to a reduction of PKA activity in B cells decreases levels of AID phosphorylated at S38 and impairs CSR [129]. Finally, second site mutations in AIDS38A that restore RPA binding in the absence of AID phosphorylation significantly rescue the CSR defect of AIDS38A protein [130]. With regards to this latter point, zebra fish AID (zAID), which can partially restore CSR activity in AID-deficient cells, lacks an amino acid equivalent to mammalian S38 [132, 133]; however, zAID binds RPA constitutively, due to an aspartatic acid residue (D44) that mimics the phsophorylated S38 [130]. Remarkably, a D44A mutant of zAID cannot bind RPA nor restore CSR in AID-deficient B cells; however, a double mutant of zAID (G42S, D44A), which has lost the D44-dependent RPA binding but has gained a serine within the PKA consensus sequence that is required for RPA binding to mouse AID, restores RPA binding to zAID only in the presence of PKA and reconstitutes CSR comparably to wild-type zAID [130]. These results strongly suggest that the major, if not exclusive, activity of AID-pS38 is to interact with RPA.
Variable region genes, unlike S regions, are not G:C rich and thus do not form R-loop structures upon transcription. As discussed earlier, the AID-pS38•RPA interaction was revealed in a biochemical screen for factors that would allow AID access to ssDNA in transcribed variable region genes. Since S regions are also rich in AGCT sequences, which serve as SHM hot-spots, it was proposed that the AID-pS38•RPA complex could participate in the binding of AID to S region DNA during CSR [49]. Contrary to this prediction however, AIDS38A, which does not interact with RPA [50, 124, 129], was as competent as wild-type AID in binding S region DNA, suggesting that AID phosphorylation and RPA binding to AID is not required for the association of AID with S region DNA during CSR [129]. However, as predicted from in vitro studies [49], association of RPA with activated S regions is strictly dependent on AID-pS38, as B cells expressing AIDS38A fail to recruit RPA to recombining S regions [129, 134]. Taken together, these observations suggest that RPA participates in CSR but not at the level of AID recruitment to S region DNA.
Given the current data, one can only speculate on the possible roles of RPA in CSR. Bound to S regions, RPA may function downstream of deamination to recruit UNG or MMR proteins, which participate in converting deaminated cytidines to DSBs. RPA may also recruit proteins such as 53BP1 and H2AX to DSBs to promote synapsis between distal broken S regions prior to their ligation during CSR. The known requirements of UNG, mismatch repair proteins, 53BP1 and H2AX in CSR and the reported interactions of these proteins with RPA support a role for RPA downstream of DNA deamination [8]. The failure to recruit RPA to S regions could thus impair conversion of the deaminated residues into DSBs or the interaction between distal DSBs. Either or both defects could be manifested as the marked reduction in CSR observed in AicdaS38A B cells [50, 106]. Ongoing CSR in AicdaS38A mutants probably reflects the ability of unphosphorylated AID to bind and deaminate S regions, with a few of the deaminated residues being converted into nicks and DSBs to effect a productive recombination reaction with a downstream S region in an RPA-independent mechanism. Consistent with this notion, artificially generating DSBs in S regions allows low levels of CSR in the absence of AID [135].
4.5. Protein Kinase A (PKA)
As mentioned earlier, the S38 residue of AID constitutes a consensus PKA phosphorylation site and PKA can phosphorylate AID at this residue [124]. The PKA holoenzyme comprises of two catalytic and two inhibitory, regulatory subunits. Binding of cyclic AMP to the regulatory subunits releases and activates the kinase activity of the catalytic subunits. Chromatin immunoprecipitation (ChIP) experiments in CSR-stimulated B cells demonstrated that the PKA catalytic subunit (PKA-Cα) and regulatory subunit (PKA-RIα) are specifically recruited to S regions [129]. The recruitment of PKA-Cα and PKA-RIα to S regions is independent of AID, as the association is detectable in AID-deficient B cells. Additional ChIP experiments in B cells derived from mice genetically engineered to express a hypomorphic mutant of PKA-RIα, which reduces PKA activity, showed that efficient phosphorylation of AID at S regions is required for wild-type levels of RPA recruitment and CSR [129].
The mechanism by which the PKA subunits are ferried and tethered to the S regions is unclear. PKA may associate with an unknown protein (factor X, Figure 4) which could be a specific transcription factor, an adaptor protein such as 14-3-3 [109] or a completely uncharacterized molecule. Binding sites for the cAMP-response element binding protein (CREB), a well-characterized PKA substrate, have been found in the germline promoters of Sμ and Sγ1 and CREB has been shown to regulate germline transcription downstream of transforming growth factor-β stimulation [136]. The recruitment of PKA to S regions may be mediated by AKAPs (A Kinase Anchoring Proteins) that could coordinate the enzymes and substrates required for CSR to permit efficient activation of AID by juxtaposing AID to its substrate (DNA), its activator (PKA), and its co-factor (RPA). Irrespective of the mechanism of PKA recruitment to S regions, the association of PKA with S regions is consistent with the hypothesis that localized activation of PKA through cAMP microdomains direct PKA activity towards specific substrates. AID phosphorylation and activation by PKA directly at recombining S regions thus establishes a paradigm to direct the specificity of a generally expressed kinase for its substrate through the concerted recruitment of both to a specific subcellular compartment. Accumulating high concentrations of inactive AID on S regions prior to “synchronously” activating the protein by phosphorylation could thus provide the mechanism by which a high density of DSBs are created in a particular S region during CSR [129].
Figure 4.

Model of AID activation and RPA recruitment during class switch recombination. Activation of CSR induces the recruitment of PKA and AID either independently or as a complex through a putative, unidentified targeting factor X. The PKA regulatory subunits (R) inhibit AID phosphorylation and keep AID inactive for CSR until a burst of cAMP induces the release of the catalytic subunits (C), which phosphorylate AID. Phosphorylated AID recruits RPA to S regions to activate the CSR cascade.
5. CONCLUDING REMARKS
Generating mutations across two different chromosomes during SHM and inducing DSBs across several kilobases of sequence on one chromosome during CSR, AID has the unique ability to promote DNA diversification unlike any other characterized protein involved in Ig diversification (e.g. RAG, TdT). Because AID has the power to radically alter the organization of DNA as well as the coding sequences of DNA, mechanisms must exist to limit its activity specifically at the Ig loci. Significant progress has been made in identifying transcriptional, post-transcriptional, and post-translational regulatory mechanisms. In addition, several factors that influence AID binding to DNA have been identified. However, a specificity-factor, if it exists, that dictates how AID is able to precisely induce very high levels of mutations and DSBs at Ig loci remains elusive. Identifying how AID gains access to non-Ig genes, the mechanisms that promote (or restrict) this access and the process through which normal error-free repair by the general DNA repair pathways are subverted to induce mutations and DSBs in the genome during CSR and SHM will constitute the next frontier of investigation on the biology of AID.
HIGHLIGHTS.
AID is a ssDNA deaminase
AID expression is regulated transcriptionally and post-transcriptionally
Subcellular localization and post-translational modifications limit AID activity
AID interacting proteins restrict AID activity primarily to Ig loci
Acknowledgments
This work was supported by grants to J.C. from the US National Institutes of Health (1RO1AI072194-01A2) and the Starr Cancer Research Consortium.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jung D, Alt FW. Unraveling V(D)J recombination: Insights into gene regulation. Cell. 2004;116:299–311. doi: 10.1016/s0092-8674(04)00039-x. [DOI] [PubMed] [Google Scholar]
- 2.Schatz DG, Ji Y. Recombination centres and the orchestration of V(D)J recombination. Nat Rev Immunol. 2011;11:251–263. doi: 10.1038/nri2941. [DOI] [PubMed] [Google Scholar]
- 3.Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem. 2007;76:1–22. doi: 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- 4.Papavasiliou FN, Schatz DG. Somatic hypermutation of immunoglobulin genes: merging mechanisms for genetic diversity. Cell. 2002;109:S35–44. doi: 10.1016/s0092-8674(02)00706-7. [DOI] [PubMed] [Google Scholar]
- 5.Honjo T, Kinoshita K, Muramatsu M. Molecular Mechanism of Class Switch Recombination: Linkage with Somatic Hypermutation. Annu Rev Immunol. 2002;20:165–196. doi: 10.1146/annurev.immunol.20.090501.112049. [DOI] [PubMed] [Google Scholar]
- 6.Chaudhuri J, Alt FW. Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nat Rev Immunol. 2004;4:541–552. doi: 10.1038/nri1395. [DOI] [PubMed] [Google Scholar]
- 7.Li Z, Woo CJ, Iglesias-Ussel MD, Ronai D, Scharff MD. The generation of antibody diversity through somatic hypermutation and class switch recombination. Genes Dev. 2004;18:1–11. doi: 10.1101/gad.1161904. [DOI] [PubMed] [Google Scholar]
- 8.Chaudhuri J, Basu U, Zarrin A, Yan C, Franco S, Perlot T, et al. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv Immunol. 2007;94:157–214. doi: 10.1016/S0065-2776(06)94006-1. [DOI] [PubMed] [Google Scholar]
- 9.Stavnezer J. Complex regulation and function of activation-induced cytidine deaminase. Trends Immunol. 2011;32:194–201. doi: 10.1016/j.it.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Honjo T, Kataoka t. Organization of immunoglobulin heavy chain genes and allelic deletion model. Proc Natl Acad Sci U S A. 1978;75:2140–2144. doi: 10.1073/pnas.75.5.2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arakawa H, Buerstedde JM. Immunoglobulin gene conversion: insights from bursal B cells and the DT40 cell line. Dev Dyn. 2004;229:458–464. doi: 10.1002/dvdy.10495. [DOI] [PubMed] [Google Scholar]
- 12.Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, et al. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem. 1999;274:18470–18476. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- 13.Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2) Cell. 2000;102:565–575. doi: 10.1016/s0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- 14.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 15.Arakawa H, Hauschild J, Buerstedde JM. Requirement of the activation-induced deaminase (AID) gene for immunoglobulin gene conversion. Science. 2002;295:1301–1306. doi: 10.1126/science.1067308. [DOI] [PubMed] [Google Scholar]
- 16.Petersen-Mahrt SK, Harris RS, Neuberger MS. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 2002;418:99–103. doi: 10.1038/nature00862. [DOI] [PubMed] [Google Scholar]
- 17.Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, Alt FW. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 2003;422:726–730. doi: 10.1038/nature01574. [DOI] [PubMed] [Google Scholar]
- 18.Bransteitter R, Pham P, Scharff MD, Goodman MF. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc Natl Acad Sci U S A. 2003;100:4102–4107. doi: 10.1073/pnas.0730835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dickerson SK, Market E, Besmer E, Papavasiliou FN. AID Mediates Hypermutation by Deaminating Single Stranded DNA. J Exp Med. 2003;197:1291–1296. doi: 10.1084/jem.20030481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramiro AR, Stavropoulos P, Jankovic M, Nussenzweig MC. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat Immunol. 2003;4:452–456. doi: 10.1038/ni920. [DOI] [PubMed] [Google Scholar]
- 21.Maul RW, Saribasak H, Martomo SA, McClure RL, Yang W, Vaisman A, et al. Uracil residues dependent on the deaminase AID in immunoglobulin gene variable and switch regions. Nat Immunol. 2011;12:70–76. doi: 10.1038/ni.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manis JP, Tian M, Alt FW. Mechanism and control of class-switch recombination. Trends Immunol. 2002;23:31–39. doi: 10.1016/s1471-4906(01)02111-1. [DOI] [PubMed] [Google Scholar]
- 23.Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. 2008;26:261–292. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tian M, Alt FW. Transcription-induced cleavage of immunoglobulin switch regions by nucleotide excision repair nucleases in vitro. J Biol Chem. 2000;275:24163–24172. doi: 10.1074/jbc.M003343200. [DOI] [PubMed] [Google Scholar]
- 25.Shinkura R, Tian M, Smith M, Chua K, Fujiwara Y, Alt FW. The influence of transcriptional orientation on endogenous switch region function. Nat Immunol. 2003;4:435–441. doi: 10.1038/ni918. [DOI] [PubMed] [Google Scholar]
- 26.Yu K, Lieber MR. Nucleic acid structures and enzymes in the immunoglobulin class switch recombination mechanism. DNA Repair (Amst) 2003;2:1163–1174. doi: 10.1016/j.dnarep.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 27.Yu K, Roy D, Bayramyan M, Haworth IS, Lieber MR. Fine-structure analysis of activation-induced deaminase accessibility to class switch region R-loops. Mol Cell Biol. 2005;25:1730–1736. doi: 10.1128/MCB.25.5.1730-1736.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Imai K, Slupphaug G, Lee WI, Revy P, Nonoyama S, Catalan N, et al. Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat Immunol. 2003;4:1023–1028. doi: 10.1038/ni974. [DOI] [PubMed] [Google Scholar]
- 29.Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, Neuberger MS. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr Biol. 2002;12:1748–1755. doi: 10.1016/s0960-9822(02)01215-0. [DOI] [PubMed] [Google Scholar]
- 30.Guikema JE, Linehan EK, Tsuchimoto D, Nakabeppu Y, Strauss PR, Stavnezer J, et al. APE1- and APE2-dependent DNA breaks in immunoglobulin class switch recombination. J Exp Med. 2007;204:3017–3026. doi: 10.1084/jem.20071289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Basu U, Meng FL, Keim C, Grinstein V, Pefanis E, Eccleston J, et al. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell. 2011;144:353–363. doi: 10.1016/j.cell.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Casellas R, Nussenzweig A, Wuerffel R, Pelanda R, Reichlin A, Suh H, et al. Ku80 is required for immunoglobulin isotype switching. Embo J. 1998;17:2404–2411. doi: 10.1093/emboj/17.8.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manis JP, Dudley D, Kaylor L, Alt FW. IgH class switch recombination to IgG1 in DNA-PKcs-deficient B cells. Immunity. 2002;16:607–617. doi: 10.1016/s1074-7613(02)00306-0. [DOI] [PubMed] [Google Scholar]
- 34.Manis JP, Gu Y, Lansford R, Sonoda E, Ferrini R, Davidson L, et al. Ku70 is required for late B cell development and immunoglobulin heavy chain class switching. J Exp Med. 1998;187:2081–2089. doi: 10.1084/jem.187.12.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449:478–482. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- 36.Boboila C, Yan C, Wesemann DR, Jankovic M, Wang JH, Manis J, et al. Alternative end-joining catalyzes class switch recombination in the absence of both Ku70 and DNA ligase 4. J Exp Med. 2010;207:417–427. doi: 10.1084/jem.20092449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee-Theilen M, Matthews AJ, Kelly D, Zheng S, Chaudhuri J. CtIP promotes microhomology-mediated alternative end joining during class-switch recombination. Nat Struct Mol Biol. 2011;18:75–79. doi: 10.1038/nsmb.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Betz AG, Milstein C, Gonzalez-Fernandez A, Pannell R, Larson T, Neuberger MS. Elements regulating somatic hypermutation of an immunoglobulin kappa gene: critical role for the intron enhancer/matrix attachment region. Cell. 1994;77:239–248. doi: 10.1016/0092-8674(94)90316-6. [DOI] [PubMed] [Google Scholar]
- 39.Gearhart PJ, Bogenhagen DF. Clusters of point mutations are found exclusively around rearranged antibody variable genes. Proc Natl Acad Sci U S A. 1983;80:3439–3443. doi: 10.1073/pnas.80.11.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lebecque SG, Gearhart PJ. Boundaries of somatic mutation in rearranged immunoglobulin genes: 5’ boundary is near the promoter, and 3’ boundary is approximately 1 kb from V(D)J gene. J Exp Med. 1990;172:1717–1727. doi: 10.1084/jem.172.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Michael N, Martin TE, Nicolae D, Kim N, Padjen K, Zhan P, et al. Effects of sequence and structure on the hypermutability of immunoglobulin genes. Immunity. 2002;16:123–134. doi: 10.1016/s1074-7613(02)00261-3. [DOI] [PubMed] [Google Scholar]
- 42.Pech M, Hochtl J, Schnell H, Zachau HG. Differences between germ-line and rearranged immunoglobulin V kappa coding sequences suggest a localized mutation mechanism. Nature. 1981;291:668–670. doi: 10.1038/291668a0. [DOI] [PubMed] [Google Scholar]
- 43.Peters A, Storb U. Somatic hypermutation of immunoglobulin genes is linked to transcription initiation. Immunity. 1996;4:57–65. doi: 10.1016/s1074-7613(00)80298-8. [DOI] [PubMed] [Google Scholar]
- 44.Rada C, Gonzalez-Fernandez A, Jarvis JM, Milstein C. The 5’ boundary of somatic hypermutation in a V kappa gene is in the leader intron. Eur J Immunol. 1994;24:1453–1457. doi: 10.1002/eji.1830240632. [DOI] [PubMed] [Google Scholar]
- 45.Rothenfluh HS, Taylor L, Bothwell AL, Both GW, Steele EJ. Somatic hypermutation in 5’ flanking regions of heavy chain antibody variable regions. Eur J Immunol. 1993;23:2152–2159. doi: 10.1002/eji.1830230916. [DOI] [PubMed] [Google Scholar]
- 46.Tumas-Brundage K, Manser T. The transcriptional promoter regulates hypermutation of the antibody heavy chain locus. J Exp Med. 1997;185:239–250. doi: 10.1084/jem.185.2.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Winter DB, Sattar N, Mai JJ, Gearhart PJ. Insertion of 2 kb of bacteriophage DNA between an immunoglobulin promoter and leader exon stops somatic hypermutation in a kappa transgene. Mol Immunol. 1997;34:359–366. doi: 10.1016/s0161-5890(97)00073-4. [DOI] [PubMed] [Google Scholar]
- 48.Yelamos J, Klix N, Goyenechea B, Lozano F, Chui YL, Gonzalez Fernandez A, et al. Targeting of non-Ig sequences in place of the V segment by somatic hypermutation. Nature. 1995;376:225–229. doi: 10.1038/376225a0. [DOI] [PubMed] [Google Scholar]
- 49.Chaudhuri J, Khuong C, Alt FW. Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature. 2004;430:992–998. doi: 10.1038/nature02821. [DOI] [PubMed] [Google Scholar]
- 50.Cheng HL, Vuong BQ, Basu U, Franklin A, Schwer B, Astarita J, et al. Integrity of the AID serine-38 phosphorylation site is critical for class switch recombination and somatic hypermutation in mice. Proc Natl Acad Sci U S A. 2009;106:2717–2722. doi: 10.1073/pnas.0812304106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McBride KM, Gazumyan A, Woo EM, Barreto VM, Robbiani DF, Chait BT, et al. Regulation of hypermutation by activation-induced cytidine deaminase phosphorylation. Proc Natl Acad Sci U S A. 2006;103:8798–8803. doi: 10.1073/pnas.0603272103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Di Noia J, Neuberger MS. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature. 2002;419:43–48. doi: 10.1038/nature00981. [DOI] [PubMed] [Google Scholar]
- 53.Rada C, Di Noia JM, Neuberger MS. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol Cell. 2004;16:163–171. doi: 10.1016/j.molcel.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 54.Rada C, Ehrenstein MR, Neuberger MS, Milstein C. Hot spot focusing of somatic hypermutation in MSH2-deficient mice suggests two stages of mutational targeting. Immunity. 1998;9:135–141. doi: 10.1016/s1074-7613(00)80595-6. [DOI] [PubMed] [Google Scholar]
- 55.Ehrenstein MR, Neuberger MS. Deficiency in Msh2 affects the efficiency and local sequence specificity of immunoglobulin class-switch recombination: parallels with somatic hypermutation. Embo J. 1999;18:3484–3490. doi: 10.1093/emboj/18.12.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schrader CE, Edelmann W, Kucherlapati R, Stavnezer J. Reduced isotype switching in splenic B cells from mice deficient in mismatch repair enzymes. J Exp Med. 1999;190:323–330. doi: 10.1084/jem.190.3.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schrader CE, Vardo J, Stavnezer J. Role for mismatch repair proteins Msh2, Mlh1, and Pms2 in immunoglobulin class switching shown by sequence analysis of recombination junctions. J Exp Med. 2002;195:367–373. doi: 10.1084/jem.20011877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schrader CE, Vardo J, Stavnezer J. Mlh1 can function in antibody class switch recombination independently of Msh2. J Exp Med. 2003;197:1377–1383. doi: 10.1084/jem.20022190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010;463:1042–1047. doi: 10.1038/nature08752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hajkova P, Jeffries SJ, Lee C, Miller N, Jackson SP, Surani MA. Genome-wide reprogramming in the mouse germ line entails the base excision repair pathway. Science. 2010;329:78–82. doi: 10.1126/science.1187945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, et al. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010;463:1101–1105. doi: 10.1038/nature08829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morgan HD, Dean W, Coker HA, Reik W, Petersen-Mahrt SK. Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: implications for epigenetic reprogramming. J Biol Chem. 2004;279:52353–52360. doi: 10.1074/jbc.M407695200. [DOI] [PubMed] [Google Scholar]
- 63.Rai K, Huggins IJ, James SR, Karpf AR, Jones DA, Cairns BR. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell. 2008;135:1201–1212. doi: 10.1016/j.cell.2008.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, et al. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell. 2011;147:95–106. doi: 10.1016/j.cell.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, et al. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451:841–845. doi: 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- 68.Pasqualucci L, Neumeister P, Goossens T, Nanjangud G, Chaganti RS, Kuppers R, et al. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature. 2001;412:341–346. doi: 10.1038/35085588. [DOI] [PubMed] [Google Scholar]
- 69.Ramiro AR, Jankovic M, Callen E, Difilippantonio S, Chen HT, McBride KM, et al. Role of genomic instability and p53 in AID-induced c-myc-Igh translocations. Nature. 2006 doi: 10.1038/nature04495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ramiro AR, Jankovic M, Eisenreich T, Difilippantonio S, Chen-Kiang S, Muramatsu M, et al. AID is required for c-myc/IgH chromosome translocations in vivo. Cell. 2004;118:431–438. doi: 10.1016/j.cell.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 71.Pasqualucci L, Bhagat G, Jankovic M, Compagno M, Smith P, Muramatsu M, et al. AID is required for germinal center-derived lymphomagenesis. Nat Genet. 2008;40:108–112. doi: 10.1038/ng.2007.35. [DOI] [PubMed] [Google Scholar]
- 72.Robbiani DF, Bothmer A, Callen E, Reina-San-Martin B, Dorsett Y, Difilippantonio S, et al. AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell. 2008;135:1028–1038. doi: 10.1016/j.cell.2008.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Robbiani DF, Bunting S, Feldhahn N, Bothmer A, Camps J, Deroubaix S, et al. AID produces DNA double-strand breaks in non-Ig genes and mature B cell lymphomas with reciprocal chromosome translocations. Mol Cell. 2009;36:631–641. doi: 10.1016/j.molcel.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shen HM, Peters A, Baron B, Zhu X, Storb U. Mutation of BCL-6 gene in normal B cells by the process of somatic hypermutation of Ig genes. Science. 1998;280:1750–1752. doi: 10.1126/science.280.5370.1750. [DOI] [PubMed] [Google Scholar]
- 75.Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell. 2011;147:107–119. doi: 10.1016/j.cell.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Crouch EE, Li Z, Takizawa M, Fichtner-Feigl S, Gourzi P, Montano C, et al. Regulation of AID expression in the immune response. J Exp Med. 2007;204:1145–1156. doi: 10.1084/jem.20061952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Han JH, Akira S, Calame K, Beutler B, Selsing E, Imanishi-Kari T. Class switch recombination and somatic hypermutation in early mouse B cells are mediated by B cell and Toll-like receptors. Immunity. 2007;27:64–75. doi: 10.1016/j.immuni.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pauklin S, Sernandez IV, Bachmann G, Ramiro AR, Petersen-Mahrt SK. Estrogen directly activates AID transcription and function. J Exp Med. 2009;206:99–111. doi: 10.1084/jem.20080521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mai T, Zan H, Zhang J, Hawkins JS, Xu Z, Casali P. Estrogen receptors bind to and activate the HOXC4/HoxC4 promoter to potentiate HoxC4-mediated activation-induced cytosine deaminase induction, immunoglobulin class switch DNA recombination, and somatic hypermutation. The Journal of biological chemistry. 2010;285:37797–37810. doi: 10.1074/jbc.M110.169086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Park SR, Zan H, Pal Z, Zhang J, Al-Qahtani A, Pone EJ, et al. HoxC4 binds to the promoter of the cytidine deaminase AID gene to induce AID expression, class-switch DNA recombination and somatic hypermutation. Nature immunology. 2009;10:540–550. doi: 10.1038/ni.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pauklin S, Petersen-Mahrt SK. Progesterone inhibits activation-induced deaminase by binding to the promoter. Journal of immunology. 2009;183:1238–1244. doi: 10.4049/jimmunol.0803915. [DOI] [PubMed] [Google Scholar]
- 82.Tran TH, Nakata M, Suzuki K, Begum NA, Shinkura R, Fagarasan S, et al. B cell-specific and stimulation-responsive enhancers derepress Aicda by overcoming the effects of silencers. Nat Immunol. 2010;11:148–154. doi: 10.1038/ni.1829. [DOI] [PubMed] [Google Scholar]
- 83.Dedeoglu F, Horwitz B, Chaudhuri J, Alt FW, Geha RS. Induction of activation-induced cytidine deaminase gene expression by IL-4 and CD40 ligation is dependent on STAT6 and NFkappaB. Int Immunol. 2004;16:395–404. doi: 10.1093/intimm/dxh042. [DOI] [PubMed] [Google Scholar]
- 84.Gonda H, Sugai M, Nambu Y, Katakai T, Agata Y, Mori KJ, et al. The balance between Pax5 and Id2 activities is the key to AID gene expression. J Exp Med. 2003;198:1427–1437. doi: 10.1084/jem.20030802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sayegh CE, Quong MW, Agata Y, Murre C. E-proteins directly regulate expression of activation-induced deaminase in mature B cells. Nat Immunol. 2003;4:586–593. doi: 10.1038/ni923. [DOI] [PubMed] [Google Scholar]
- 86.Yadav A, Olaru A, Saltis M, Setren A, Cerny J, Livak F. Identification of a ubiquitously active promoter of the murine activation-induced cytidine deaminase (AICDA) gene. Mol Immunol. 2006;43:529–541. doi: 10.1016/j.molimm.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 87.Lee-Theilen M, Chaudhuri J. Walking the AID tightrope. Nat Immunol. 2010;11:107–109. doi: 10.1038/ni0210-107. [DOI] [PubMed] [Google Scholar]
- 88.Maul RW, Gearhart PJ. Women, autoimmunity, and cancer: a dangerous liaison between estrogen and activation-induced deaminase? J Exp Med. 2009;206:11–13. doi: 10.1084/jem.20080086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Petersen-Mahrt SK, Coker HA, Pauklin S. DNA deaminases: AIDing hormones in immunity and cancer. J Mol Med (Berl) 2009;87:893–897. doi: 10.1007/s00109-009-0496-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Teng G, Hakimpour P, Landgraf P, Rice A, Tuschl T, Casellas R, et al. MicroRNA-155 is a negative regulator of activation-induced cytidine deaminase. Immunity. 2008;28:621–629. doi: 10.1016/j.immuni.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dorsett Y, McBride KM, Jankovic M, Gazumyan A, Thai TH, Robbiani DF, et al. MicroRNA-155 suppresses activation-induced cytidine deaminase-mediated Myc-Igh translocation. Immunity. 2008;28:630–638. doi: 10.1016/j.immuni.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.de Yebenes VG, Belver L, Pisano DG, Gonzalez S, Villasante A, Croce C, et al. miR-181b negatively regulates activation-induced cytidine deaminase in B cells. J Exp Med. 2008;205:2199–2206. doi: 10.1084/jem.20080579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vigorito E, Perks KL, Abreu-Goodger C, Bunting S, Xiang Z, Kohlhaas S, et al. microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells. Immunity. 2007;27:847–859. doi: 10.1016/j.immuni.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Brar SS, Watson M, Diaz M. Activation-induced cytosine deaminase (AID) is actively exported out of the nucleus but retained by the induction of DNA breaks. J Biol Chem. 2004;279:26395–26401. doi: 10.1074/jbc.M403503200. [DOI] [PubMed] [Google Scholar]
- 95.McBride KM, Barreto V, Ramiro AR, Stavropoulos P, Nussenzweig MC. Somatic hypermutation is limited by CRM1-dependent nuclear export of activation-induced deaminase. J Exp Med. 2004;199:1235–1244. doi: 10.1084/jem.20040373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ito S, Nagaoka H, Shinkura R, Begum N, Muramatsu M, Nakata M, et al. Activation-induced cytidine deaminase shuttles between nucleus and cytoplasm like apolipoprotein B mRNA editing catalytic polypeptide 1. Proc Natl Acad Sci U S A. 2004;101:1975–1980. doi: 10.1073/pnas.0307335101. Epub 2004 Feb 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Aoufouchi S, Faili A, Zober C, D’Orlando O, Weller S, Weill JC, et al. Proteasomal degradation restricts the nuclear lifespan of AID. J Exp Med. 2008;205:1357–1368. doi: 10.1084/jem.20070950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Patenaude AM, Orthwein A, Hu Y, Campo VA, Kavli B, Buschiazzo A, et al. Active nuclear import and cytoplasmic retention of activation-induced deaminase. Nat Struct Mol Biol. 2009;16:517–527. doi: 10.1038/nsmb.1598. [DOI] [PubMed] [Google Scholar]
- 99.Ta VT, Nagaoka H, Catalan N, Durandy A, Fischer A, Imai K, et al. AID mutant analyses indicate requirement for class-switch-specific cofactors. Nat Immunol. 2003;4:843–848. doi: 10.1038/ni964. [DOI] [PubMed] [Google Scholar]
- 100.Geisberger R, Rada C, Neuberger MS. The stability of AID and its function in class-switching are critically sensitive to the identity of its nuclear-export sequence. Proc Natl Acad Sci U S A. 2009;106:6736–6741. doi: 10.1073/pnas.0810808106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ellyard JI, Benk AS, Taylor B, Rada C, Neuberger MS. The dependence of Ig class-switching on the nuclear export sequence of AID likely reflects interaction with factors additional to Crm1 exportin. Eur J Immunol. 2011;41:485–490. doi: 10.1002/eji.201041011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Orthwein A, Patenaude AM, Affar el B, Lamarre A, Young JC, Di Noia JM. Regulation of activation-induced deaminase stability and antibody gene diversification by Hsp90. J Exp Med. 2010;207:2751–2765. doi: 10.1084/jem.20101321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen X, Barton LF, Chi Y, Clurman BE, Roberts JM. Ubiquitin-independent degradation of cell-cycle inhibitors by the REGgamma proteasome. Mol Cell. 2007;26:843–852. doi: 10.1016/j.molcel.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Uchimura Y, Barton LF, Rada C, Neuberger MS. REG-gamma associates with and modulates the abundance of nuclear activation-induced deaminase. J Exp Med. 2011;208:2385–2391. doi: 10.1084/jem.20110856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wold MS. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- 106.McBride KM, Gazumyan A, Woo EM, Schwickert TA, Chait BT, Nussenzweig MC. Regulation of class switch recombination and somatic mutation by AID phosphorylation. J Exp Med. 2008 doi: 10.1084/jem.20081319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kuwahara K, Fujimura S, Takahashi Y, Nakagata N, Takemori T, Aizawa S, et al. Germinal center-associated nuclear protein contributes to affinity maturation of B cell antigen receptor in T cell-dependent responses. Proc Natl Acad Sci U S A. 2004;101:1010–1015. doi: 10.1073/pnas.0307609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Maeda K, Singh SK, Eda K, Kitabatake M, Pham P, Goodman MF, et al. GANP-mediated recruitment of activation-induced cytidine deaminase to cell nuclei and to immunoglobulin variable region DNA. J Biol Chem. 2010;285:23945–23953. doi: 10.1074/jbc.M110.131441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xu Z, Fulop Z, Wu G, Pone EJ, Zhang J, Mai T, et al. 14-3-3 adaptor proteins recruit AID to 5’-AGCT-3’-rich switch regions for class switch recombination. Nat Struct Mol Biol. 2010;17:1124–1135. doi: 10.1038/nsmb.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yancopoulos GD, DePinho RA, Zimmerman KA, Lutzker SG, Rosenberg N, Alt FW. Secondary genomic rearrangement events in pre-B cells: VHDJH replacement by a LINE-1 sequence and directed class switching. Embo J. 1986;5:3259–3266. doi: 10.1002/j.1460-2075.1986.tb04637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Stavnezer-Nordgren J, Sirlin S. Specificity of immunoglobulin heavy chain switch correlates with activity of germline heavy chain genes prior to switching. Embo J. 1986;5:95–102. doi: 10.1002/j.1460-2075.1986.tb04182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Stavnezer J, Radcliffe G, Lin YC, Nietupski J, Berggren L, Sitia R, et al. Immunoglobulin heavy-chain switching may be directed by prior induction of transcripts from constant-region genes. Proc Natl Acad Sci U S A. 1988;85:7704–7708. doi: 10.1073/pnas.85.20.7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fukita Y, Jacobs H, Rajewsky K. Somatic hypermutation in the heavy chain locus correlates with transcription. Immunity. 1998;9:105–114. doi: 10.1016/s1074-7613(00)80592-0. [DOI] [PubMed] [Google Scholar]
- 114.Nambu Y, Sugai M, Gonda H, Lee CG, Katakai T, Agata Y, et al. Transcription-coupled events associating with immunoglobulin switch region chromatin. Science. 2003;302:2137–2140. doi: 10.1126/science.1092481. [DOI] [PubMed] [Google Scholar]
- 115.Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah-Sobrinho C, et al. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell. 2010;143:122–133. doi: 10.1016/j.cell.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lindstrom DL, Squazzo SL, Muster N, Burckin TA, Wachter KC, Emigh CA, et al. Dual roles for Spt5 in pre-mRNA processing and transcription elongation revealed by identification of Spt5-associated proteins. Mol Cell Biol. 2003;23:1368–1378. doi: 10.1128/MCB.23.4.1368-1378.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Stanlie A, Aida M, Muramatsu M, Honjo T, Begum NA. Histone3 lysine4 trimethylation regulated by the facilitates chromatin transcription complex is critical for DNA cleavage in class switch recombination. Proc Natl Acad Sci U S A. 2010;107:22190–22195. doi: 10.1073/pnas.1016923108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nowak U, Matthews AJ, Zheng S, Chaudhuri J. The splicing regulator PTBP2 interacts with the cytidine deaminase AID and promotes binding of AID to switch-region DNA. Nat Immunol. 2011;12:160–166. doi: 10.1038/ni.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 120.Polydorides AD, Okano HJ, Yang YY, Stefani G, Darnell RB. A brain-enriched polypyrimidine tract-binding protein antagonizes the ability of Nova to regulate neuron-specific alternative splicing. Proc Natl Acad Sci U S A. 2000;97:6350–6355. doi: 10.1073/pnas.110128397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lorenz M, Jung S, Radbruch A. Switch transcripts in immunoglobulin class switching. Science. 1995;267:1825–1828. doi: 10.1126/science.7892607. [DOI] [PubMed] [Google Scholar]
- 122.Conticello SG, Ganesh K, Xue K, Lu M, Rada C, Neuberger MS. Interaction between antibody-diversification enzyme AID and spliceosome-associated factor CTNNBL1. Mol Cell. 2008;31:474–484. doi: 10.1016/j.molcel.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 123.Han L, Masani S, Yu K. Cutting edge: CTNNBL1 is dispensable for Ig class switch recombination. J Immunol. 2010;185:1379–1381. doi: 10.4049/jimmunol.1001643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Basu U, Chaudhuri J, Alpert C, Dutt S, Ranganath S, Li G, et al. The AID antibody diversification enzyme is regulated by protein kinase A phosphorylation. Nature. 2005;438:508–511. doi: 10.1038/nature04255. [DOI] [PubMed] [Google Scholar]
- 125.Gazumyan A, Timachova K, Yuen G, Siden E, Di Virgilio M, Woo EM, et al. Amino-terminal phosphorylation of activation-induced cytidine deaminase suppresses c-myc/IgH translocation. Mol Cell Biol. 2011;31:442–449. doi: 10.1128/MCB.00349-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Barreto V, Reina-San-Martin B, Ramiro AR, McBride KM, Nussenzweig MC. C-terminal deletion of AID uncouples class switch recombination from somatic hypermutation and gene conversion. Mol Cell. 2003;12:501–508. doi: 10.1016/s1097-2765(03)00309-5. [DOI] [PubMed] [Google Scholar]
- 127.Pasqualucci L, Kitaura Y, Gu H, Dalla-Favera R. PKA-mediated phosphorylation regulates the function of activation-induced deaminase (AID) in B cells. Proc Natl Acad Sci U S A. 2006;103:395–400. doi: 10.1073/pnas.0509969103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pham P, Smolka MB, Calabrese P, Landolph A, Zhang K, Zhou H, et al. Impact of phosphorylation and phosphorylation-null mutants on the activity and deamination specificity of activation-induced cytidine deaminase. J Biol Chem. 2008;283:17428–17439. doi: 10.1074/jbc.M802121200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Vuong BQ, Lee M, Kabir S, Irimia C, Macchiarulo S, McKnight GS, et al. Specific recruitment of protein kinase A to the immunoglobulin locus regulates class-switch recombination. Nat Immunol. 2009;10:420–426. doi: 10.1038/ni.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Basu U, Wang Y, Alt FW. Evolution of phosphorylation-dependent regulation of activation-induced cytidine deaminase. Mol Cell. 2008;32:285–291. doi: 10.1016/j.molcel.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zarrin AA, Alt FW, Chaudhuri J, Stokes N, Kaushal D, Du Pasquier L, et al. An evolutionarily conserved target motif for immunoglobulin class-switch recombination. Nat Immunol. 2004;5:1275–1281. doi: 10.1038/ni1137. [DOI] [PubMed] [Google Scholar]
- 132.Barreto VM, Pan-Hammarstrom Q, Zhao Y, Hammarstrom L, Misulovin Z, Nussenzweig MC. AID from bony fish catalyzes class switch recombination. J Exp Med. 2005;202:733–738. doi: 10.1084/jem.20051378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wakae K, Magor BG, Saunders H, Nagaoka H, Kawamura A, Kinoshita K, et al. Evolution of class switch recombination function in fish activation-induced cytidine deaminase, AID. Int Immunol. 2006;18:41–47. doi: 10.1093/intimm/dxh347. [DOI] [PubMed] [Google Scholar]
- 134.Yamane A, Resch W, Kuo N, Kuchen S, Li Z, Sun HW, et al. Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nat Immunol. 2011;12:62–69. doi: 10.1038/ni.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zarrin AA, Del Vecchio C, Tseng E, Gleason M, Zarin P, Tian M, et al. Antibody class switching mediated by yeast endonuclease-generated DNA breaks. Science. 2007;315:377–381. doi: 10.1126/science.1136386. [DOI] [PubMed] [Google Scholar]
- 136.Lin YC, Shockett P, Stavnezer J. Regulation of transcription of the germline immunoglobulin alpha constant region gene. Curr Top Microbiol Immunol. 1992;182:157–165. doi: 10.1007/978-3-642-77633-5_19. [DOI] [PubMed] [Google Scholar]