

Abstract
Adenosine receptor agonists have cardioprotective, cerebroprotective, and antiinflammatory properties. We report that a carbocyclic modification of the ribose moiety incorporating ring constraints is a general approach for the design of A1 and A3 receptor agonists having favorable pharmacodynamic properties. While simple carbocyclic substitution of adenosine agonists greatly diminishes potency, methanocarba-adenosine analogues have now defined the role of sugar puckering in stabilizing the active adenosine receptor-bound conformation and thereby have allowed identification of a favored isomer. In such analogues a fused cyclopropane moiety constrains the pseudosugar ring of the nucleoside to either a Northern (N) or Southern (S) conformation, as defined in the pseudorotational cycle. In binding assays at A1, A2A, and A3 receptors, (N)-methanocarba-adenosine was of higher affinity than the (S)-analogue, particularly at the human A3 receptor (N/S affinity ratio of 150). (N)-Methanocarba analogues of various N6-substituted adenosine derivatives, including cyclopentyl and 3-iodobenzyl, in which the parent compounds are potent agonists at either A1 or A3 receptors, respectively, were synthesized. The N6-cyclopentyl derivatives were A1 receptor-selective and maintained high efficacy at recombinant human but not rat brain A1 receptors, as indicated by stimulation of binding of [35S]GTP-γ-S. The (N)-methanocarba-N6-(3-iodobenzyl)adenosine and its 2-chloro derivative had Ki values of 4.1 and 2.2 nM at A3 receptors, respectively, and were highly selective partial agonists. Partial agonism combined with high functional potency at A3 receptors (EC50 < 1 nM) may produce tissue selectivity. In conclusion, as for P2Y1 receptors, at least three adenosine receptors favor the ribose (N)-conformation.
In work designed to develop potent and selective agents, the structure–activity relationships of adenosine derivatives as ligands (principally agonists) at the four subtypes of adenosine receptors (A1, A2A, A2B, and A3) have been explored extensively. Adenosine receptor agonists1,2 are being studied for their potential use as antiarrhythmic,3 antinociceptive,4 and antilipolytic5,6 agents (A1 subtype); as cerebroprotective7 and cardioprotective8 agents (A1 and A3 subtypes); and as hypotensive9 and antipsychotic10 agents (A2A subtype).
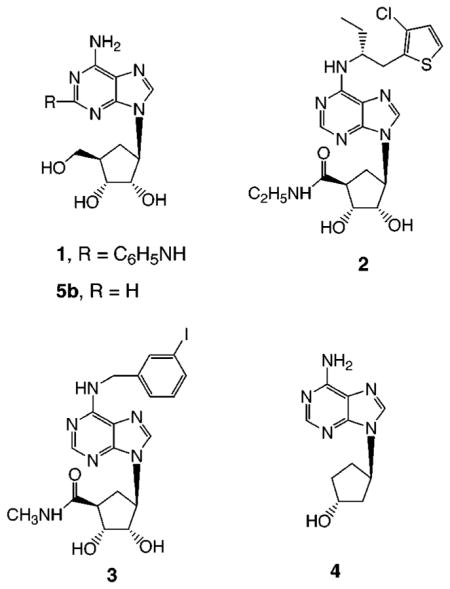
In general, for adenosine agonists, numerous modifications of the N6-position with cycloalkyl and other hydrophobic moieties provide selectivity for A1 receptors, although the affinities of these N6-substituted adenosine derivatives (e.g. N6-cyclopentyl) at A3 receptors are often intermediate between their respective A1 and A2A affinities.1 Structurally, few ribose modifications, other than amide substitution at the 5′-position, are tolerated in adenosine agonists. An intact furanose moiety is present in most of the potent adenosine agonists previously developed. Adenine riboside derivatives are subject to scission of the glycosidic bond and other pathways of metabolic degradation in vivo.5 Alternately, carbocyclic modifications of the ribose moiety have been introduced in order to design nonglycosylic adenosine agonists and thereby potentially increase selectivity and biological stability. In previous studies of adenosine analogues it was found that many adenosine derivatives having carbocyclic modifications of the ribose ring (compounds 1–4, 5b) bind to adenosine receptors, but usually only with reduced affinity.11–16 The simplest of such 9-cyclopentyladenosine derivatives, aristeromycin, 5b, was reported to have hypotensive properties related to A2A receptor activation.11 This led to the design of (1R,2S,3R,5R)-3-[6-amino-2-(phenylamino)-9H-purin-9-yl]-5-(hydroxymethyl)-1,2-cyclopentanediol (CGS 23321), 1, which also relaxed porcine coronary smooth muscle through activation of adenosine A2 receptors.12 [1S-[1a,2b,3b,4a(S*)]]-4-[7-[[2-(3-chloro-2-thienyl)-1-methylpropyl]amino]-3H-imidazo[4,5-b]pyridin-3-yl]cyclopentanecarboxamide (AMP 579), 2, activated A1 and A2 adenosine receptors and, in the heart, induced coronary dilation without causing endocardial steal and decreased postischemic myocardial infarct size.13 (±)-9-[2α,3α-Dihydroxy-4β-(N-methylcarbamoyl)cyclopent-1β-yl)]-N6-(3-iodobenzyl)adenine (MRS 582), 3, is 17 700-fold less potent in binding to rat A3 adenosine receptors than the corresponding 4′-oxygen analogue, N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine (IB-MECA).14 The carbocyclic derivative 9-[(1R,3R)-trans-cyclopentan-3-ol]adenine hydrochloride (MDL 201,449), 4, is a weak adenosine agonist that inhibits synthesis of TNFα (tumor necrosis factor) in murine bone-marrow-derived macrophages and has therapeutic potential for treatment of inflammatory diseases.15,16
In the present study we have explored the effects at adenosine receptors of the “methanocarba” nucleoside modification introduced by Marquez and co-workers17–24 as a complex carbocyclic modification which maintains a fixed conformation. In methanocarba analogues a fused cyclopropane ring constrains the accompanying cyclopentane moiety to mimic the conformation of a rigid furanose ring held in either a Northern (N) or Southern (S) conformation. These analogues adopt envelope, as opposed to twist, conformations. This has allowed us to focus on the impact of pseudorotation of the ribose moiety on both receptor affinity and efficacy of adenosine agonists, since often only one isomeric form of methanocarba adenosine retains high affinity and selectivity at a given binding site.
Results
Chemical Synthesis
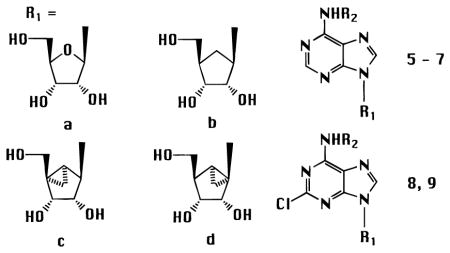
The optically active methanocarbocyclic adenosine analogues (Table 1), in which a fused cyclopropane ring constrains the cyclopentane ring into a rigid (N)-envelope conformation, were synthesized by the general approach (Scheme 1) of Marquez and coworkers.18 The (N)-methanocarba analogues of various N6-substituted adenosine derivatives, including cyclopentyl and 3-iodobenzyl, were prepared. The parent adenosine analogues are potent agonists at either A1 (cyclopentyl) or A3 (3-iodobenzyl) receptors.1 The N6-(3-iodobenzyl) substitution is present in the selective A3 agonist IB-MECA and also promotes A3 receptor affinity in the 5′-hydroxy series.25 2,6-Dichloropurine, 10, was condensed with the cyclopentyl derivative, 11,18 using the Mitsunobu reaction, followed by substitution at the 6-position and deprotection to give 8c or 9c. These intermediates allowed the incorporation, in the adenosine series of the (N)-configuration, of the 2-chloro substitution, of interest for its structure-dependent affinity-enhancing effects at both A1 and A3 receptors.1,26 The 2-chloro substitution of compound 8c could also be removed by catalytic reduction to give 6c. An N6-(3-iodobenzyl) group was introduced in either aristeromycin, 5b, or (N)-methanocarba-adenosine, 5c, by the Dimroth rearrangement,25 to give 7b (Scheme 2) and 7c (Scheme 1). A sample of the antipodal (S)-methanocarba-adenosine, 5d, was synthesized in racemic form by Marquez and co-workers by an intramolecular carbene addition reaction.23
Table 1.
Affinities of Adenosine (a), Simple Carbocyclic (b), and Methanocarba-adenosine ((N)-conformation, c, and (S)-conformation, d) Derivatives in Radioligand Binding Assays at Rat A1, Rat A2A, Human A2B, and Human A3 Receptors, Unless Notedg
| ||||||
|---|---|---|---|---|---|---|
| compound | R2 | rA1a | rA2Ab |
Ki (nM) or % displacement
|
A1/A3 | |
| hA2Bb | hA3c | |||||
| 5a | H | estd 10d | estd 30d | <10% at 100 μM | estd 1000 (r)d,e | 100 |
| 5b | H | 6260 ± 730 | 2150 ± 950 | 47300 ± 10600 | 20000 ± 7900 (r)e | 0.31 |
| 5c | H | 1680 ± 80 | 22500 ± 100 (h)e,f | 35 ± 2% at 50 μMf | 404 ± 70f | 4.2 |
| 5d (racemic) | H | 15% at 100 μM | >100000 (h)e,f | 20 ± 4% at 50 μMf | 62500 ± 2900f | >1 |
| 6a | CP | 1.50 ± 0 51 | 857 ± 163 | 21200 ± 4300 | 274 ± 20240 (r)e | 0.0055 |
| 6c | CP | 5.06 ± 0.51 | 6800 ± 1800 | 139000 ± 19000 | 170 ± 51 | 0.030 |
| 7a | IB | 20.0 ± 8.5 | 17.5 ± 0.5 | 3570 ± 100 | 9.5 ± 1.4 (r)e | 2.1 |
| 7b | IB | 25900 ± 1600 | <10% at 100 μM | nd | 1960 ± 370 | 13 |
| 7c | IB | 69.2 ± 9.8 | 601 ± 236 | 12100 ± 1300 | 4.13 ± 1.76 | 17 |
| 8a | CP | 1.33 ± 0.19 | 605 ± 154 | 20400 ± 1200 | 237 (r)e | 0.0056 |
| 8c | CP | 8.76 ± 0.81 | 3390 ± 520 | 27 ± 7% at 100 μM | 466 ± 58 | 0.019 |
| 9a | IB | 18.5 ± 4.7 | 38.5 ± 2.0 | 5010 ± 1400 | 1.41 ± 0.17 (r)e | 13 |
| 9c | IB | 141 ± 22 | 732 ± 207 | 41000 ± 7000 | 2.24 ± 1.45 | 63 |
Displacement of specific [3H]R-PIA binding to A1 receptors in rat brain membranes, expressed as Ki ± SEM (n = 3–5), unless noted.
Displacement of specific [3H]CGS 21680 binding to A2A receptors in rat striatal membranes, expressed as Ki ± SEM (n = 3–6), and at A2B receptors expressed in HEK-293 cells vs [3H]ZM241,385, unless noted.
Displacement of specific [125I]AB-MECA binding at human A3 receptors expressed in HEK cells, in membranes, expressed as Ki ± SEM (n = 3–4), unless noted.
Reference 41.
Ki values were determined in radioligand binding assays at recombinant human A2A receptors expressed in HEK-293 cells vs [3H]ZM241385 or [125I]AB-MECA binding at rat A3 receptors expressed in CHO cells.
Measured in the absence of ADA.
nd, not determined; 6c, MRS 1781; 7c, MRS 1743; 8c, MRS 1761; 9c, MRS 1760. g CP = cyclopentyl; IB = 3-iodobenzyl.
Scheme 1.

Synthesis of N6-Substituted (N)-Methanocarba-adenosine Derivatives Optimized for Interaction with A1 (CP = cyclopentyl) or A3 (IB = 3-iodobenzyl) Receptorsa
a Reagents: (a) DEAD, Ph3P; (b) MeOH, rt; (c) BCl3; (d) H2/Pd; (e) 3-iodobenzyl bromide, 50 °C, DMF, 2 days; (f) NH4OH, MeOH, 80 °C, 3 days.
Scheme 2.

Synthesis of an N6-Substituted Aristeromycin Derivative by the Dimroth Rearrangementa
a Reagents: (a) 3-iodobenzyl bromide, 80 °C, DMF, 3 days; (b) NH4OH, MeOH, 80 °C, 1 h.
Biological Activity
A pair of methanocarba analogues of adenosine, 5c18 and 5d,23 corresponding to (N)-and (S)-conformations of ribose, were tested in binding assays (Table 1) at four subtypes of adenosine receptors.25,27 The more synthetically challenging (S)-isomer (5d) was available only as the racemate and therefore was tested as such.23 At rat A1, rat A2A, and human A3 subtypes, the (N)-analogue proved to be of much higher affinity than the (S)-analogue. At the human A2B receptor, binding was carried out using [3H]ZM 241,385 (4-(2-[7-amino-2-{furyl}{1,2,4}triazolo{2,3-a}{1,3,5}-triazin-5-ylaminoethyl)phenol);27 however, the affinity was too weak to establish selectivity for a specific isomer. Affinity of (N)-methanocarba-adenosine, 5c, vs adenosine, 5a, was particularly enhanced at the A3 receptor subtype (Figure 1), for which the ratio of affinities of (N)- to (S)-analogues was 150-fold. Although a relatively poor substrate for adenosine deaminase (ADA),22 the binding curve for 5c was shifted to the right in the presence of ADA; therefore, Table 1 reports the affinity values for 5c and 5d obtained in the absence of ADA. The (S)-conformer, 5d, as the 2′-deoxy analogue,22 behaved as an even worse substrate of ADA (100-fold less), which would explain why the curves in the presence and absence of ADA for 5d are virtually the same (Figure 1). Aristeromycin, 5b, bound weakly to adenosine receptors, with slight selectivity for the A2A subtype. Compound 5c was more potent than aristeromycin, 5b, in binding to A1 (4-fold) and A3 (4500-fold) adenosine receptors.
Figure 1.

Representative competition curves for inhibition of binding of [125I]AB-MECA (N6-(4-amino-3-iodobenzyl)-adenosine-5′-N-methyluronamide) by (N)-methanocarba-adenosine, 5c (circles), and (S)-methanocarba-adenosine, 5d (racemic) (diamonds), at human A3 receptors expressed in CHO cells at 25 °C, in the presence (solid symbols) or absence (open symbols) of 2 IU/mL ADA. Structures show ring pucker envelope conformations.
Compounds 6c and 8c are patterned after A1 receptor-selective agonists, while compounds 7c and 9c are patterned after A3 receptor-selective agonists. Compounds 6 and 7 are unsubstituted at the 2-position, while compounds 8 and 9 contain the potency-enhancing 2-chloro substituent.26 The N6-cyclopentyl (N)-methanocarba derivative, 6c, based on CPA (N6-cyclopentyl-adenosine), 6a, maintained high selectivity for A1 receptors, although the affinity of 6c at rat A1 receptors was 3-fold less than for 6a.
In one series it was possible to compare ribose, cyclopentyl, and (N)-methanocarba derivatives having the same N6-substitution. The N6-(3-iodobenzyl) (N)-methanocarba derivative, 7c, with a Ki value of 4.1 nM, was 2.3-fold more potent at A3 receptors than the ribose-containing parent, 7a. Thus, the selectivity of 7c for human A3 versus rat A1 receptors was 17-fold. The aristeromycin analogue, 7b, was relatively weak in binding to adenosine receptors.
Among 2-chloro-substituted derivatives, the (N)-methanocarba analogue, 8c, was less potent at A1 and A2A receptors than its parent 2-chloro-N6-cyclopentyl-adenosine, 8a, and roughly equipotent at A3 receptors. Thus, 8c was 53-fold selective in binding to rat A1 vs human A3 receptors. The (N)-methanocarba analogue, 9c, of 2-chloro-N6-(3-iodobenzyl)adenosine, 9a, had Ki values of 141, 732, and 2.2 nM at A1, A2A, and A3 receptors, respectively. Thus, the 2-chloro group slightly enhanced affinity at A3 receptors, while reducing affinity at A1 receptors.
Replacement of ribose with the (N)-methanocarba moiety best preserved receptor binding affinity at the A3 subtype, at which differences were small. At A1 receptors, the loss of affinity upon substitution of ribose with an (N)-methanocarba ring for structures 6–9 was between 3- and 8-fold. At A2A receptors the loss of affinity was between 6- and 34-fold.
The agonist-induced stimulation of binding of guanine nucleotides to activated G-proteins has been used as a functional assay for a variety of receptors, including adenosine receptors.28,29 Binding of [35S]GTP-γ-S was studied in membranes prepared from CHO (Chinese hamster ovary) cells stably expressing human A1 or A3 receptors (Table 2). The nonselective adenosine agonist NECA (5′-N-ethyluronamidoadenosine) caused a concentration-dependent increase in the level of the guanine nucleotide bound (Figure 2A). Compound 6c was highly selective and a full agonist at human A1 but not rat A1 receptors. Both 7c and 9c stimulated the binding of [35S]GTP-γ-S (Figure 2B); however, the maximal stimulation was significantly less than that produced by either NECA or N6-(3-iodobenzyl)adenosine (Figure 2), 7a, both being full A3 agonists. Compounds 7c and 9c resulted in relative stimulation of [35S]GTP-γ-S binding of only 45% and 22%, respectively, indicating that the efficacy of the (N)-methanocarba analogue at A3 receptors was further reduced upon 2-chloro modification. The potency of compounds 7c and 9c, indicated by the EC50 values in this functional assay, was greater than the potencies of either NECA or compound 7a (Table 2). Thus, the (N)-methanocarba N6-(3-iodobenzyl) analogues appear to be highly potent and selective partial agonists at human A3 receptors.
Table 2.
Effect of Ligands To Stimulate [35S]GTP-γ-S Binding to Membranes of Cells Expressing the Cloned hA1AR or hA3AR or in Rat Cerebral Cortical Membranes Containing the A1AR
| ligand | cloned hA1AR EC50 (nM)a | % maximal stimulationc | rA1AR EC50 (nM)a | % maximal stimulationc | cloned hA1AR EC50 (nM)a | % maximal stimulationc |
|---|---|---|---|---|---|---|
| NECA | nd | nd | 155 ± 15 | 100 | ||
| 6a | 4.15 ± 0.90 | 100 | 20.3 ± 13.1 | 100 | 7980 ± 60 | 100 |
| 6c | 21.5 ± 2.3 | 102 ± 1 | 100 ± 17 | 75 ± 6 | >10000 | 14 ± 2% at 10 μM |
| 7a | 43.1 ± 10.4 | 91 ± 1 | 340 ± 98 | 95 ± 4 | 5.16 ± 0.71 | 100 |
| 7b | >10000 | 5 ± 2% at 10 μM | ndb | >10000 | 14 ± 2% at 10 μM | |
| 7c | 218 ± 18 | 86 ± 2 | 940 ± 114 | 55 ± 5 | 0.70 ± 0.16 | 45.3 ± 6.8 |
| 8c | 31.2 ± 3.3 | 97 ± 1 | 145 ± 35 | 96 ± 2 | >10000 | 15 ± 5% at 10 μM |
| 9c | 142 ± 24 | 91 ± 1 | 684 ± 75 | 48 ± 3 | 0.67 ± 0.19 | 22.0 ± 2.8 |
EC50 for stimulation of basal [35S]GTP-γ-S (0.1 nM) binding by agonists in membranes from transfected CHO cells (±SEM), n = 5–10. Basal and 100% maximal levels of binding correspond to (fmol/mg of protein) 490 ± 60 and 1520 ± 80 (hA1), 340 ± 20 and 1010 ± 70 (rA1), and 240 ± 14 and 580 ± 49 (hA3).
nd, not determined.
Figure 2.
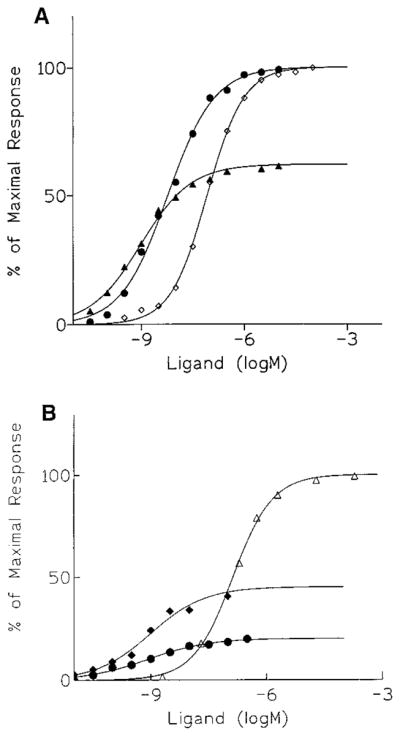
Concentration–response curves for stimulation of binding of [35S]GTP-γ-S by (A) NECA (◇), 7a (●), or 7c (▲); or (B) NECA (△), 7c (◆), or 9c (●), in membranes prepared from CHO cells stably expressing human brain A3 receptors.
Discussion
Nearly all of the thousands of known adenosine agonists are derivatives of adenosine.1,2 Although molecular modeling of adenosine agonists has been carried out,30 there has been little direct evidence for a conformational preference of the ribose ring in the receptor binding site. The furanose ring of nucleosides and nucleotides in solution is known to exist in a rapid, dynamic equilibrium between a range of Northern (2E → 3T2 → 3E) and opposing Southern (2E → 2T3 → 3E) conformations as defined in the pseudorotational cycle.39,40 For methanocarba analogues, the bicyclo-[3.1.0]hexane ring can constrain the cyclopentane ring into an (N)-, 2′-exo (2E) envelope pucker, and an (S)-, 3′ exo (3E) form, as shown in Figure 1. These two extreme forms of ring pucker usually define biologically active conformations.19 The energy difference between (N)- and (S)-conformations, which is roughly 4 kcal/mol, could explain the difference between micromolar and nanomolar binding affinities. We have now applied this approach to nucleosides binding to P1 (adenosine) receptors and found that one of the fixed ring-envelope conformations is highly favored at the receptor binding site.
A pair of methanocarba-adenosine analogues, 5c and 5d, was prepared to explore the role of sugar puckering in ligand recognition. The (S)-methanocarba analogue of adenosine, 5d, was only weakly active in binding to adenosine receptors, presumably because of an unfavorable conformation that decreases receptor binding. In contrast, the methanocarba analogues constrained in the (N)-conformation, e.g. 5c–9c, displayed high receptor affinity, particularly at the A3 receptor. In binding assays at A1, A2A, and A3 receptors, (N)-methanocarba-adenosine, 5c, proved to be of higher affinity than the (S)-analogue, 5d, with an N/S affinity ratio of 150 at the human A3 receptor.
As previously reported14 and confirmed in this study, the SAR of adenosine agonists indicates that the ribose ring oxygen may be substituted with carbon; however, much affinity is lost. As with the N6-(3-iodobenzyl) aristeromycin derivative, 7b, simple carbocyclic substitution of the ribose moiety of otherwise potent N6-substituted adenosine agonists greatly diminishes affinity.
In comparison to the ribose analogues, the (N)-methanocarba N6-substituted adenosine agonists were of comparable affinity at A3 receptors, but less potent at A1, A2A, and A2B receptors. The (N)-methanocarba-N6-cyclopentyl derivatives were A1 receptor-selective and maintained high efficacy at human recombinant but not rat brain A1 receptors, as indicated by stimulation of binding of [35S]GTP-γ-S. This may be related to either species differences or heterogeneity of G proteins, since the degree of agonist efficacy of a given compound may be highly dependent on the receptor-associated G protein.32–34 (N)-Methanocarba-N6-(3-iodobenzyl)adenosine and the 2-chloro derivative had Ki values of 4.1 and 2.2 nM at A3 receptors, respectively, and were selective partial agonists. As for the ribose parents, additional 2-chloro substitution was favorable for receptor selectivity. However, unlike the ribose forms, efficacy was reduced in N6-(3-iodobenzyl) analogues, such that 7c and 9c proved to be partial A3 receptor agonists.
Depending on the desired biological activity, partial receptor agonists may be more desirable than full agonists as therapeutic agents, due to the possibility of reduced side effects in the former. Partial agonists may display in vivo specificity for sites at which spare receptors are present,32 and the drug would therefore behave with apparent “full” efficacy.31,35 Thus, compounds 7c and 9c, which combine partial agonism and high functional potency at A3 receptors (EC50 < 1 nM), should be explored for tissue-selective effects.
At least three of the four adenosine receptors favor the (N)-conformation of methanocarba-adenosine. Thus, the biological potency appears to be correlated to ring puckering, which in turn would influence the orientation of the hydroxyl groups (Figure 1) and the base within the receptor binding site. The 2′- and 3′-hydroxyl groups are important for affinity of adenosine analogues, and the 2′-hydroxyl is particularly important for activation of the receptor.31 From energy considerations, it is expected that the rotation about the hydroxymethyl group is relatively free for both 5c and 5d; however, rotation about the pseudoglycosyl bond is more restricted than for conventional nucleosides.19 The adenine moiety is thought to be present in an anti-conformation in the adenosine receptor binding site.30 The relative contributions of the barrier to pseudoglycosyl bond rotation and ring pucker in the recognition of 5c have not been explored. Franchetti et al.,37 using a different type of ribose-modified nucleoside analogue, also found an indication of higher affinity in binding to A1 receptors for the (N)-conformation. The 2′-C-methyl-was more potent than the 3′-C-methylribosyl analogue of adenosine, suggesting that an anti-conformation having an (N)-puckered furanose ring was preferred at the receptor.
An additional element associated with the presence of the bicyclo[3.1.0]hexane itself could play a role. Indeed, the presence of the fused cyclopropane moiety has the potential for causing repulsive steric interactions by the presence of the extra “CH2” below the plane of the ring. Although this property is common to both rigid pseudorotational antipodes, the distance between the “CH2” groups from the superimposed pseudosugar structures is estimated to be approximately 1.7 Å.19 Therefore, this group appears to be shifted more toward the base in the case of (S)-methanocarba-adenosine, and it too might contribute to its diminished biological activity.
For another member of the GPCR superfamily, the P2Y1 receptor, we recently reported that the ribose (N)-conformation of adenine nucleotides also appears to be preferred at the receptor binding site.36 Therefore, at least some of the P1 and P2 purinoceptors appear to share a preference for the (N)-conformation. This may indicate a common motif38 of binding of nucleoside moieties to a variety of GPCRs. The insights of this conformational preference may be utilized in simulated docking of adenosine agonists in a putative receptor binding site, to design even more potent and selective agents.
At the binding site of ADA, the (N)-isomer is also preferred, although the carbocyclic adenosine analogues are relatively poor substrates.22 N6-Substituted analogues, such as 6c–9c, would not be expected to be substrates for ADA. In other systems effective discrimination between conformational antipodes is evident.19,21 For example, HIV reverse transcriptase has shown almost exclusive preference for the (N)-isomer of AZT, and only the (N)-isomer of thymidine was demonstrated to have effective antiherpes activity.19 In recent experiments kinases in general have shown preference for the opposite conformation (Victor E. Marquez, Staffan Eriksson, Riad Agbaria, and D. G. Johns, unpublished results). This could serve as the basis for distinguishing the activities of adenosine analogues at receptors and in metabolic processes.
Since the methanocarba analogues are lacking a glycosylic bond, it will be important to determine whether the in vivo half-life of adenosine analogues is prolonged by this modification. The half-life of 6a, for example, is 24 min in whole blood and, following infusion in conscious rats, only 8.2 min.5 These data are typical of N6-substituted adenosine agonists, although the reasons for this short half-life were not established. Major likely factors include 5′-phosphorylation and renal excretion of uncharged nucleosides, as well as possible degradation.
In conclusion, we have found that the introduction of a methano carbocyclic modification of the ribose ring of purine agonists represents a general approach for the design of adenosine agonists with favorable pharmacodynamic properties, especially with respect to A1 and A3 receptors. The present study has identified new pharmacological probes of A1 and A3 receptors that are selective and either full or partial agonists and are now being tested in disease models. The effect of the absence of the glycosylic bond on pharmacokinetic properties should now be studied. This approach could be applied to the development of cardioprotective, cerebroprotective, and antiinflammatory agents acting through A1 and A3 receptors.24
Materials and Methods
Chemical Synthesis
Nucleosides and synthetic reagents were purchased from Sigma Chemical Co. (St. Louis, MO) and Aldrich (St. Louis, MO). 2,6-Dichloropurine was obtained from Sigma. m-Iodobenzyl bromide was purchased from Aldrich (St. Louis, MO). Optically active (1′R,2′R,3′S,4′R,5′S)-4-(6-aminopurin-9-yl)-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (5c) was synthesized as described.18 4-(6-Aminopurin-9-yl)-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (11) and racemic rel-(1′R,2′S,3′R,4′R,5′S)-1-(6-aminopurin-9-yl)-4-(hydroxymethyl)-bicyclo[3.1.0]hexane-2,3-diol (5d) were synthesized according to Marquez and colleagues (manuscript in preparation23). Compounds 7a and 9a were synthesized as described.25,26
Proton nuclear magnetic resonance spectroscopy was performed on a Varian GEMINI-300 spectrometer, and all spectra were obtained in CDCl3. Chemical shifts (δ) relative to tetramethylsilane are given. FAB (fast atom bombardment) mass was performed with a JEOL SX102 spectrometer using 6 kV Xe atoms. Elemental analysis was performed by Atlantic Microlab Inc. (Norcross, GA). NMR and mass spectra were consistent with the assigned structures. The determination of purity was performed with a Hewlett-Packard 1090 HPLC system using an SMT OD-5-60 C18 analytical column (250 mm × 4.6 mm, Separation Methods Technologies, Inc., Newark, DE) in two different linear gradient solvent systems. One solvent system (A) was 0.1 M triethylammonium acetate buffer:CH3CN in ratios of 95:5 to 40:60 for 20 min with flow rate 1 mL/min. The other (B) was MeOH:CH3CN, 95:5 to 40: 60, in 20 min with flow rate 1 mL/min. Peaks were detected by UV absorption using a diode array detector.
(1′R,2′R,3′S,4′R,5′S)-4-[6-Cyclopentylamino)purin-9-yl]-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (6c)
A solution of 8c (4 mg, 0.01 mmol) in methanol (0.5 mL) was hydrogenated at atmospheric pressure over 10% Pd/C (1 mg) to furnish the product 6c (83% yield). 1H NMR (CD3OD): δ 0.7–0.8 (m, 1H, 6′CHH), 1.46–1.88 (m, 10H, 6′CHH, 5′CH, 4CH2), 2.01–2.20 (m, 1H, NCH), 3.34 (d, 1H, J = 9.77 Hz, CH2O), 3.88 (d, 1H, J = 6.84 Hz, 3′CH), 4.26 (d, 1H, J = 9.77 Hz, CH2O), 4.66–4.98 (m, 2H, 2′CH, 1′CH), 8.28 (s, 1H, 2CH), 8.5 (s, 1H, 8CH). HRMS (FAB): calcd 346.1879, found 346.1879. HPLC showed 90% purity with retention times (min) of 11.17 (A) and 2.16 (B).
(1′R,2′R,3′R,4′R)-2,3-Dihydroxy-4-(hydroxymethyl)-1-(6-(3-iodobenzylamino)purin-9-yl)cyclopentane (7b)
A mixture of aristeromycin (3.5 mg, 0.013 mmol) and 3-iodobenzyl bromide (12 mg, 0.039 mmol) in anhydrous DMF (1 mL) was heated at 80 °C for 3 days. The solvent was removed under vacuum, and the excess 3-iodobenzyl bromide was removed from the reaction mixture by suspending the residue in ether, followed by decantation of the supernatant ether phase. The residue was dried and dissolved in methanol (1 mL), and ammonium hydroxide (0.5 mL) was added. The mixture was heated at 80 °C in a closed tube for 1 h. Solvent was removed under vacuum, and the residue obtained was purified by flash column chromatography using 7/3 chloroform/methanol to furnish 3.0 mg (47%) of the product, mp. 85 °C. HPLC showed 93% purity with retention times (min) of 17.53 (A) and 2.31 (B). MS (CI): m/z 482 (M+ + 1). HRMS: (FAB): calcd 481.0611, found 481.0610. 1H NMR(CD3OD): δ 1.86–1.96 (m, 1H, 5′CHH), 2.14–2.30 (m, 1H, 5′CHH), 2.38–2.48 (m, 1H, 4′CH), 3.67 (d, 2H, J = 5.77 Hz, CH2O), 3.96–4.06 (m, 1H, 3′CH), 4.43–4.48 (m, 1H, 2′CH), 4.73–4.82 (m, 1H, 1′CH), 5.26 (s, 2H, ArCH2), 7.12 (t, 1H, J = 7.82 Hz, ArH), 7.32 (d, 1H, J = 7.82 Hz, ArH), 7.66 (d, 1H, J = 7.82 Hz, ArH), 7.73 (s, 1H, ArH), 8.06 (s, 1H, 2CH). 8.08 (s, 1H, 8CH).
(1′R,2′R,3′S,4′R,5′S)-1-(Hydroxymethyl)-4-{6-[(3-iodophenylmethyl)amino]purin-9-yl}bicyclo[3.1.0]hexane-2,3-diol (7c)
To a solution of 4-(6-aminopurin-9-yl)-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (5c, 20 mg, 0.0721 mmol) in DMF (0.5 mL) was added m-iodobenzyl bromide (64 mg, 0.216 mmol), and the mixture was stirred at 50 °C for 2 days. DMF was then removed under a stream of N2. Dry ether (1 mL) and 0.5 mL of acetone were added to the resulting syrup and the syrup solidified. The solvents were removed by decantation, and again ether was added and removed. The solid was dried and dissolved in 1 mL of MeOH. NH4OH (1.5 mL) was added, and the mixture was stirred at 80 °C for 3 days. After cooling to room temperature, the solvents were removed under reduced pressure, and the residue was purified by preparative TLC (silica 60; 1000 μm; Analtech, Newark, DE; ethyl acetate–i-PrOH–H2O (8:2:1)) to give 26 mg (73% yield) of the product (7c). 1H NMR (CDCl3): δ 0.82 (t, J = 6.0 Hz, 1 H, 6′CHH), 1.41 (t, J = 4.8 Hz, 1H, 6′CHH), 1.72 (dd, J = 8.5, 6.0 Hz, 1H, 5′CH), 3.36 (d, J = 10.8 Hz, 1H, CH2O), 4.05 (d, J = 6.9 Hz, 1H, CH2O), 4.33 (m, 1H, 3′CH), 4.80–4.88 (m, 3 H, CH2N, 1′CH), 5.21 (d, J = 6.9 Hz, 1H, 2′CH), 6.25 (m, br, 1H, NH), 7.07 (t, J = 7.8 Hz, 1H, Ar), 7.35 (d, J = 7.8 Hz, 1H, Ar), 7.61 (d, J = 7.8 Hz, 1H, Ar), 7.74 (s, 1H), 7.93 (s, 1H, 2CH), 8.33 (s, 1H, 8CH). MS (FAB): m/z 494 (M+ + 1). Anal CHN.
(1′R,2′R,3′S,4′R,5′S)-4-[2-Chloro-6-(cyclopentylamino)-purin-9-yl]-1-(hydroxymethyl)bicyclo[3.1.0]hexane-2,3-diol (8c)
To a solution of 15 (36 mg, 0.076 mmol) in anhydrous dichloromethane was added BCl3 (1 M solution in dichloromethane, 0.23 mL, 0.23 mmol) at 0 °C. The reaction mixture was warmed to room temperature and stirred for 10 min. To this mixture was added methanol (1 mL) followed by ammonium hydroxide (0.5 mL). The mixture was concentrated under vacuum, and the residue obtained was purified by flash column chromatography using 9/1 chloroform/methanol as eluent to furnish 14 mg of the product 8c (48% yield) as a solid, mp. 130 °C. HPLC showed 99% purity with retention times (min) of 14.56 (A) and 2.19 (B). 1H NMR (CDCl3): δ 0.65–0.9 (m, 1H, 6′CHH), 1.1–1.4 (m, 2H, 6′CHH, 5′CH), 1.4–1.9 (m, 8H, 4CH2), 2.0–2.2 (m, 1H, N6CH), 3.34 (d, 1H, J = 7.2 Hz, CH2O), 3.97 (d, 1H, J = 4.6 Hz, 3′CH), 4.25 (d, 1H, J = 7.2 Hz, CH2O), 4.687 (s, 1H, 1′CH), 5.11 (d, 1H, J = 4.6, 2′CH), 7.85 (s, 1H, 8CH). HRMS (FAB): calcd 380.1489, found 380.1498.
(1′R,2′R,3′S,4′R,5′S)-4-{2-Chloro-6-[(3-iodophenylmethyl)amino]purin-9-yl}-1-(hydroxymethyl)bicyclo[3.1.0]-hexane-2,3-diol (9c) was synthesized by the same method as 8c in 53% yield, mp 121 °C. HPLC showed 98% purity with retention times (min) of 17.59 (A) and 2.17 (B). 1H NMR (CD3-OD): δ 0.70–0.78 (m, 1H, 6′CHH), 1.50–1.63 (m, 2H, 6′CHH, 5′CH), 3.33 (d, 1H, J = 11.72 Hz, CH2O), 3.88 (d, 1H, J = 6.84 Hz, 3′CH), 4.26 (d, 1H, J = 11.72 Hz, CH2O), 4.71–4.83 (m, 2H, 1′CH, 2′CH), 7.1 (t, 1H, J = 7.82 Hz, ArH), 7.40 (d, 1H, J = 7.82 Hz, ArH), 7.61 (d, 1H, 7.82 Hz, ArH), 7.78 (s, 1H, ArH), 8.54 (s, 1H, 8CH). HRMS (FAB): calcd 528.0299, found 528.0295.
(1′R,2′R,3′S,4′R,5′S)-4-(2,6-Dichloropurin-9-yl)-1-[(phenylmethoxy)methyl]bicyclo[3.1.0]hexane-2,3-(O-isopropylidene) (12)
To a solution of triphenylphosphine (260 mg, 1 mmol) in anhydrous THF (2 mL) was added DEAD (diethyl azadicarboxylate, 0.16 mL, 1 mmol) dropwise at 0 °C, and stirring was continued for 20 min. To this solution was added a solution of 2,6-dichloropurine in THF (4 mL), followed by the addition of 11 (145 mg, 0.5 mmol) in THF (4 mL). The reaction mixture was warmed to room temperature, and stirring was continued for 6 h. The solvent was evaporated under vacuum, and the residue obtained was purified by flash chromatography using 7/3 petroleoum ether/ethyl acetate as eluent to furnish 141 mg of the product (12) (70% yield) as a gum. 1H NMR (CDCl3): δ 1.0 (m, 1H, 6′CHH), 1.24 (s, 3H, CH3), 1.27–1.38 (m, 1H, 6′CHH), 1.55 (s, 3H, CH3), 1.62 (dd, 1H, J = 4.88, 9.77 Hz, 5′CH), 3.34 (d, 1H, J = 9.77 Hz, CH2O), 3.97 (d, 1H, J = 9.77 Hz, CH2O), 4.50 (d, 1H, J = 6.84 Hz, 3′CH), 4.57–4.68 (qAB, 2H, J = 12.7 Hz, ArCH2), 5.17 (s, 1H, 1′CH), 5.32 (d, 1H, J = 6.84 Hz, 2′H), 7.2–7.4 (m, 5H, Ar), 8.63 (s, 1H, 8CH).
(1′R,2′R,3′S,4′R,5′S)-4-[2-Chloro-6-(cyclopentylamino)purin-9-yl]-1-[(phenylmethoxy)methyl]bicyclo[3.1.0]hexane-2,3-(O-isopropylidene) (15)
To a solution of 12 (42 mg, 0.105 mmol) in methanol (2 mL) was added cyclopentylamine (52 μL, 0.53 mmol) at room temperature, and stirring was continued for 6 h for complete reaction. Solvent was removed under vacuum, and the residue obtained was purified by flash column chromatography using 7/3 petroleum ether/ethyl acetate as eluent to furnish 45 mg of the product 15 (90% yield) as a gum. 1H NMR (CDCl3): δ 0.92–0.96 (m, 1H, 6′CHH), 1.14–1.01 (m, 1H, 6′CHH), 1.23 (s, 3H, CH3), 1.42–1.81 (m, 9H, 5′CH, 4CH2), 1.54 (s, 3H, CH3), 2.08–2.21 (m, 1H, N6CH), 3.44 (d, 1H, J = 9.76 Hz, CH2O), 3.90 (d, 1H, J = 9.76 Hz, CH2O), 4.51 (d, 1H, J = 6.84 Hz, 3′CH), 4.57–4.67 (qAB, 2H, J = 12.7 Hz, ArCH2), 5.04 (s, 1H, 1′CH), 5.32 (d, 1H, J = 6.84 Hz, 2′CH), 7.2–7.4 (m, 5H, Ar), 8.18 (s, 1H, 8CH).
(1′R,2′R,3′S,4′R,5′S)-4-{2-Chloro-6-[(3-iodophenylmethyl)amino]purin-9-yl}-1-[(phenylmethoxy)methyl]bicyclo-[3.1.0]hexane-2,3-(O-isopropylidene) (16) was synthesized in 70% yield by the same method as for 15, except using 3-iodobenzylamine hydrochloride and 2 equiv of triethylamine. 1H NMR (CDCl3): δ 0.87–0.91 (m, 1H, 6′CHH), 1.10–1.29 (m, 1H, 6′CHH), 1.17 (s, 3H, CH3), 1.42–1.56 (m, 1H, 5′CH), 1.47 (s, 3H, CH3), 3.37 (d, 1H, J = 9.77 Hz, CH2O), 3.84 (d, 1H, J = 9.77 Hz, CH2O), 4.44 (d, 1H, J = 6.84 Hz, 3′CH), 4.50–4.60 (qAB, 2H, J = 11.72 Hz, ArCH2), 4.70 (bs, 1H, NH), 4.98 (s, 1H, 1′CH), 5.24 (d, 1H, J = 6.84 Hz, 2′CH), 7.0 (t, 1H, J = 7.82 Hz, ArH), 7.2–7.34 (m, 6H, ArH), 7.55 (d, 1H, J = 7.82, ArH), 7.65 (s, 1H, ArH), 8.08 (s, 1H, 8CH).
Pharmacological Analyses
Materials
F-12 (Ham’s) medium, fetal bovine serum (FBS), and penicillin/streptomycin were from Gibco BRL (Gaithersburg, MD). [125I]AB-MECA (2000 Ci/mmol) and [35S]guanosine 5′-(γ-thio)triphosphate (1000–1500 Ci/mmol) were from DuPont NEN (Boston, MA). Adenosine deaminase (ADA) was from Boehringer Mannheim (Indianapolis, IN). All other materials were from standard local sources and of the highest grade commercially available.
Cell Culture and Membrane Preparation
CHO cells stably transfected with either human A1 or A3 receptors (gift of Dr. Gary Stiles and Dr. Mark Olah, Duke University Medical Center) were cultured as monolayers in medium supplemented with 10% fetal bovine serum. Cells were washed twice with 10 mL of ice-cold phosphate buffered saline, lysed in lysis buffer (10 mM Tris-HCl buffer, pH 7.4, containing 2 mM MgCl2 and 5 mM EDTA), and homogenized in a Polytron homogenizer in the presence of 0.2 IU/mL ADA, centrifuged at 3000g for 10 min, followed by centrifugation of the supernatant at 40 000g for 15 min. The pellet was washed once with the lysis buffer and recentrifuged at 40 000g for 15 min. The final pellets were resuspended in 50 mM Tris-HCl buffer, pH 7.4, containing 10 mM MgCl2 and 1 mM EDTA and stored at –70 °C.
Radioreceptor Binding
Determination of binding to adenosine A1, A2A, and A2B receptors was carried out as reported.26,27 Determination of A3 adenosine receptor binding was carried out using [125I]AB-MECA.29 Briefly, aliquots of crude transfected CHO cell membranes (approximately 10 μg protein/tube) were incubated with 0.5 nM [125I]AB-MECA, 10 mM MgCl2, 2 units/mL adenosine deaminase, and 50 mM Tris-HCl (pH 7.4) at 25 °C for 60 min. The total volume of the reaction mixture was 125 μL. Bound and free ligands were separated by rapid filtration of the reaction mixture through Whatman GF/B glass filters. The filters were immediately washed with two 5-mL portions of ice-cold 50 mM Tris-HCl buffer (pH 7.4). The radioactivity bound to the filters was determined in a Beckman γ-counter. Specific binding was defined as the amount of the radioligand bound in the absence of competing ligand minus the amount of that bound in the presence of 100 μM NECA. Ki values were calculated using the Kd for [125I]AB-MECA binding of 0.56 nM.
Determination of [35S]GTP-γ-S Binding
[35S]GTP-γ-S binding was determined by the method of Lorenzen et al.28 The incubation mixture contained in a total volume of 125 μL, 50 mM Tris-HCl (pH 7.4), 1 mM EDTA, 10 mM MgCl2, 10 μM guanosine 5′-diphosphate, 1 mM dithiothreitol, 100 mM NaCl, 0.2 units/mL adenosine deaminase, 0.16 nM [35S]GTP-γ-S, and 0.5% bovine serum albumin. The CHO cell membranes expressing A128 or A329 receptors were preincubated with the above-mentioned assay mixture at 30 °C for 1 h and further incubated for 1 h after the addition of [35S]GTP-γ-S. Incubations were terminated by rapid filtration of the samples through glass fiber filters (Whatman GF/B), followed by two 5-mL washes of the same buffer. After transferring the filters into a vial containing 3 mL of scintillation cocktail, the radioactivity was determined in a scintillation counter.
Data Analysis
Analyses of saturation binding assays and concentration–response curves were carried out using Graph-Pad Prism (GraphPad Software Inc., San Diego, CA). Comparisons between groups were carried out using the unpaired Student’s t test.
Supplementary Material
Acknowledgments
We thank Gilead Sciences (Foster City, CA) for financial support to R.G.R.
Footnotes
Supporting Information Available: Analysis for 6c, 7b,c, 8c, and 9c. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Jacobson KA, Knutsen LJS. P1 and P2 purine and pyrimidine receptors. In: Abbracchio MP, Williams M, editors. Handbook of Experimental Pharmacology. in press. [Google Scholar]
- 2.Baraldi PG, Cacciari B, Romagnoli R, Spalluto G. A1 and A3 adenosine receptor agonists: An overview. Exp Opin Therap Patents. 1999;9:515–527. [Google Scholar]
- 3.Belardinelli L, Lu J, Dennis D, Martens J, Shryock JC. The cardiac effects of a novel A1-adenosine receptor agonist in guinea pig isolated heart. J Pharmacol Exp Ther. 1994;271:1371–1382. [PubMed] [Google Scholar]
- 4.Sawynok J. Adenosine receptor activation and nociception. Eur J Pharmacol. 1998;347:1–11. doi: 10.1016/s0014-2999(97)01605-1. [DOI] [PubMed] [Google Scholar]
- 5.van Schaick EA, Kulkarni C, von Frijtag Drabbe Künzel JK, Mathôt RAA, Cristalli G, IJzerman AP, Danhof M. Time course of action of three adenosine A1 receptor agonists with differing lipophilicity in rats: Comparison of pharmacokinetic, hemodynamic and EEG effects. Naunyn-Schmiedeberg’s Arch Pharmacol. 1997;356:827–837. doi: 10.1007/pl00005124. [DOI] [PubMed] [Google Scholar]
- 6.Wagner H, Milavec-Krizman M, Gadient F, Menninger K, Schoeffter P, Tapparelli C, Pfannkuche HJ, Fozard JR. General pharmacology of SDZ WAG 994, a potent selective and orally active adenosine A1 receptor agonist. Drug Dev Res. 1995;34:276–288. [Google Scholar]
- 7.von Lubitz DKJE, Lin RCS, Popik P, Carter MF, Jacobson KA. Adenosine A3 receptor stimulation and cerebral ischemia. Eur J Pharmacol. 1994;263:59–67. doi: 10.1016/0014-2999(94)90523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stambaugh K, Jacobson KA, Jiang JL, Liang BT. 1997 A novel cardioprotective function of adenosine A1 and A3 receptors during prolonged stimulated ischemia. Am J Physiol. 1997;273:H501–H505. doi: 10.1152/ajpheart.1997.273.1.H501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Casati C, Monopoli A, Dionisotti S, Zocchi C, Bonizzoni E, Ongini E. Repeated administration of selective adenosine A1 and A2 receptor agonists in the spontaneously hypertensive rat: tolerance develops to A1-mediated hemodynamic effects. J Pharmacol Exp Ther. 1994;268:1506–1511. [PubMed] [Google Scholar]
- 10.Dixon DA, Fenix LA, Kim DM, Raffa RB. Indirect modulation of dopamine D2 receptors as potential pharmacotherapy for schizophrenia: I. Adenosine agonists. Ann Pharmacother. 1999;33:480–488. doi: 10.1345/aph.18215. [DOI] [PubMed] [Google Scholar]
- 11.Dunham EW, Vince R. Hypotensive and renal vasodilator effects of carbocyclic adenosine (aristeromycin) in anesthetized spontaneously hypertensive rats. J Pharmacol Exp Ther. 1986;238:954–959. [PubMed] [Google Scholar]
- 12.Balwierczak JL, Krulan CM, Wang ZC, Chen J, Jeng AY. Effects of adenosine A2 receptor agonists on nucleoside transport. J Pharmacol Exp Ther. 1989;251:279–287. [PubMed] [Google Scholar]
- 13.McVey MJ, Smits GJ, Cox BF, Kitzen JM, Clark KL, Perrone MH. Cardiovascular pharmacology of the adenosine A1/A2-receptor agonist AMP 579: Coronary hemodynamic and cardioprotective effects in the canine myocardium. J Cardiovasc Pharmacol. 1999;33:703–710. doi: 10.1097/00005344-199905000-00005. [DOI] [PubMed] [Google Scholar]
- 14.Siddiqi SM, Jacobson KA, Esker JL, Melman N, Tiwari KN, Secrist JA, Schneller SW, Cristalli G, Johnson CA, IJzerman AP. Search for new purine- and ribose-modified adenosine analogues as selective agonists and antagonists at adenosine receptors. J Med Chem. 1995;38:1174–1188. doi: 10.1021/jm00007a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bradshaw M, Rutherford MS, Hoeper BJ, McWhinney CD, Borcherding DR, Schook LB, Edwards CK., III Specific transcriptional inhibition of bone marrow-derived macrophage tumor necrosis factor-alpha gene expression protein production using novel enantiomeric carbocyclic nucleoside analogues. J Pharmacol Exp Ther. 1995;273:1506–1518. [PubMed] [Google Scholar]
- 16.McWhinney CD, Dudley MW, Bowlin TL, Peet NP, Schook L, Bradshaw M, De M, Borcherding DR, Edwards CK., III Activation of adenosine A3 receptors on macrophages inhibits tumor necrosis factor-alpha. Eur J Pharmacol. 1996;310:209–216. doi: 10.1016/0014-2999(96)00272-5. [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez JB, Marquez VE, Nicklaus MC, Mitsuya H, Barchi JJ., Jr Conformationally locked nucleoside analogues. Synthesis of dideoxycarbocyclic nucleoside analogues structurally related to neplanocin C. J Med Chem. 1994;37:3389–3399. doi: 10.1021/jm00046a024. [DOI] [PubMed] [Google Scholar]
- 18.Jeong LS, Marquez VE, Yuan CS, Borchardt RT. 4′,1′a-Methanocarbocyclic adenosine analogues as potential inhibitors of S-adenosylhomocysteine hydrolase. Heterocycles. 1995;41:2651–2656. [Google Scholar]
- 19.Marquez VE, Siddiqui MA, Ezzitouni A, Russ P, Wang J, Wagner RW, Matteucci MD. Nucleosides with a twist. Can fixed forms of sugar ring pucker influence biological activity in nucleosides and oligonucleotides? J Med Chem. 1996;39:3739–3747. doi: 10.1021/jm960306+. [DOI] [PubMed] [Google Scholar]
- 20.Ezzitouni A, Russ P, Marquez VE. (1S, 2R)-[(Benzyloxy)-methyl]cyclopent-3-enol. A versatile synthon for the preparation of 4′, 1′a-methano, 1′, 1′a-methano carbocyclic nucleosides. J Org Chem. 1997;62:4870–4873. [Google Scholar]
- 21.Marquez VE, Ezzitouni A, Russ P, Siddiqui MA, Ford H, Jr, Feldman RJ, Mitsuya H, George C, Barchi JJ., Jr HIV-1 Reverse Transcriptase Can Discriminate Between Two Conformationally Locked Carbocyclic AZT triphosphate Analogues. J Am Chem Soc. 1998;120:2780–2789. [Google Scholar]
- 22.Marquez VE, Russ P, Alonso R, Siddiqui MA, Shin KJ, George C, Nicklaus M, Dai F, Ford H., Jr Conformationally restricted nucleosides. The reaction of adenosine deaminase with substrates built on a bicyclo[3.1.0]hexane template. Nucleosides Nucleotides. 1999;18:521–530. doi: 10.1080/15257779908041487. [DOI] [PubMed] [Google Scholar]
- 23.Shin KJ, Moon HR, George C, Marquez VE. Construction of the bicyclo[3.1.0]hexane template of a conformationally locked carbocyclic adenosine via an olefin ketocarbene cycloaddition. J Org Chem. 2000;65:2172–2178. doi: 10.1021/jo9917691. [DOI] [PubMed] [Google Scholar]
- 24.Jacobson KA. A3 adenosine receptors: Novel ligands and paradoxical effects. Trends Pharmacol Sci. 1998;19:184–191. doi: 10.1016/s0165-6147(98)01203-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gallo-Rodriguez C, Ji XD, Melman N, Siegman BD, Sanders LH, Orlina J, Fischer B, Pu Q, Olah ME, van Galen PJM, Stiles GL, Jacobson KA. Structure–activity relationships of N6-benzyladenosine-5′-uronamides as A3-selective adenosine agonists. J Med Chem. 1994;37:636–646. doi: 10.1021/jm00031a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim HO, Ji X, Siddiqi SM, Olah ME, Stiles GL, Jacobson KA. 2-Substitution of N6-benzyladenosine-5′-uronamides enhances selectivity for A3-adenosine receptors. J Med Chem. 1994;37:3614–3621. doi: 10.1021/jm00047a018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ji XD, Jacobson KA. Use of the triazolotriazine [3H]-ZM241385 as a radioligand at recombinant human A2B adenosine receptors. Drug Design Discov. 1999;16:217–226. [PMC free article] [PubMed] [Google Scholar]
- 28.Lorenzen A, Guerra L, Vogt H, Schwabe U. Interaction of full and partial agonists of the A1 adenosine receptor with receptor/G protein complexes in rat-brain membranes. Mol Pharmacol. 1996;49:915–926. [PubMed] [Google Scholar]
- 29.Jacobson KA, Park KS, Jiang JL, Kim YC, Olah ME, Stiles GL, Ji XD. Pharmacological characterization of novel A3 adenosine receptor-selective antagonists. Neuropharmacology. 1997;9:1157–1165. doi: 10.1016/s0028-3908(97)00104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siddiqi SM, Pearlstein RA, Sanders LH, Jacobson KA. Comparative molecular field analysis of selective A3 adenosine agonists. Bioorg Med Chem. 1995;3:1331–1343. doi: 10.1016/0968-0896(95)00116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van der Wenden EM, Carnielli M, Roelen HCPF, Lorenzen A, von Frijtag Drabbe Künzel JK, IJzerman AP. 5′-Substituted adenosine analogues as new high-affinity partial agonists for the adenosine A1 receptor. J Med Chem. 1998;41:102–108. doi: 10.1021/jm970508l. [DOI] [PubMed] [Google Scholar]
- 32.Kenakin T. Differences between natural and recombinant G protein-coupled receptor systems with varying receptor/G protein stoichiometry. Trends Pharmacol Sci. 1997;18:456–464. doi: 10.1016/s0165-6147(97)01136-x. [DOI] [PubMed] [Google Scholar]
- 33.Yang Q, Lanier SM. Influence of G protein type on agonist efficacy. Mol Pharmacol. 1999;56:651–656. doi: 10.1124/mol.56.3.651. [DOI] [PubMed] [Google Scholar]
- 34.Clark RB, Knoll BJ, Barber R. Partial agonists and G protein-coupled receptor desensitization. Trends Pharmacol Sci. 1999;20:279–286. doi: 10.1016/s0165-6147(99)01351-6. [DOI] [PubMed] [Google Scholar]
- 35.Pilla M, Perachon S, Sautel F, Garrido F, Mann A, Wermuth CG, Schwartz JC, Everitt BJ, Sokoloff P. Selective inhibition of cocaine-seeking behaviour by a partial dopamine D3 receptor agonist. Nature. 1999;400:371–375. doi: 10.1038/22560. [DOI] [PubMed] [Google Scholar]
- 36.Nandanan E, Jang SY, Moro S, Kim H, Siddiqi MA, Russ P, Marquez V, Busson R, Herdewijn P, Harden TK, Boyer JL, Jacobson KA. Synthesis, biological activity, and molecular modeling of ribose-modified adenosine bisphosphate analogues as P2Y1 receptor ligands. J Med Chem. 2000;43:829–842. doi: 10.1021/jm990249v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Franchetti P, Cappellacci L, Marchetti S, Trincavelli L, Martini C, Mazzoni MR, Lucacchini A, Grifantini M. 2′-C-Methyl analogues of selective adenosine receptor agonists: Synthesis binding studies. J Med Chem. 1998;41:1708–1715. doi: 10.1021/jm9707737. [DOI] [PubMed] [Google Scholar]
- 38.Moro S, Guo D, Camaioni E, Boyer JL, Harden KT, Jacobson KA. Human P2Y1 receptor: Molecular modeling site-directed mutagenesis as tools to identify agonist antagonist recognition sites. J Med Chem. 1998;41:1456–1466. doi: 10.1021/jm970684u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Altona C, Sundaranlingam M. Conformational Analysis of the Sugar Ring in Nucleosides and Nucleotides. A New Description Using the Concept of Pseudorotation. J Am Chem Soc. 1972;94:8205–8212. doi: 10.1021/ja00778a043. [DOI] [PubMed] [Google Scholar]
- 40.Saenger W. Principles of Nucleic Acid Structure. Springer-Verlag; New York: 1984. [Google Scholar]
- 41.Jacobson KA, Kim HO, Siddiqi SM, Olah ME, Stiles GL, von Lubitz DKJE. A3 adenosine receptors: design of selective ligands and therapeutic prospects. Drugs Future. 1995;20:689–699. doi: 10.1358/dof.1995.020.07.531583. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.