

Abstract
Dependence of basal cell carcinomas and medulloblastomas on the Hedgehog pathway provides an opportunity for targeted or “personalized” therapy. The recent effectiveness and FDA approval of the first Smoothened inhibitors validates this class of agents, but has revealed drug-resistant tumor variants that bypass Smoothened inhibition. Here, we summarize the effectiveness of Hedgehog pathway inhibitors and highlight promising areas for the development of next generation drug antagonists for Hedgehog-dependent cancers.
One of the emerging themes in cancer biology is the dependence of cancer subtypes on certain signaling pathways for continued tumor growth. For example, mutations that activate the Hedgehog (Hh) signaling pathway drive growth of a variety of cancers including basal cell carcinomas (BCCs) and medulloblastomas, along with pancreatic, prostate, and small cell lung cancer that account for up to 25% of all human cancer deaths (Epstein, 2008). BCCs are the most prevalent cancer in the world, and nearly half of all US citizens are likely to develop this cancer before retirement (National Cancer Institute, 2010). Twenty years of extensive research identifying Hh pathway components and their functional roles recently culminated in the newly FDA approved Hh pathway antagonist vismodegib (Erivedge; Genentech/Roche) for the treatment of locally advanced or metastatic BCCs. Although vismodegib and other Smo inhibitors appear effective, treatment-driven tumor evolution has resulted in the outgrowth of tumor cell variants resistant to the drug. This rapid tumor evolution during treatment highlights the continued need to understand how tumors circumvent pathway blockade and identify new therapeutic targets for treating Hh-dependent cancers. In this article, we summarize the successful development of Hh pathway inhibitors and highlight promising areas for the development of next generation drug antagonists for Hh-dependent cancers.
A compelling connection to human cancer
Hh signaling is essential for development of all vertebrates and drives proliferation, migration, and differentiation of progenitor cells to pattern organ development (Varjosalo and Taipale, 2008). Despite the critical nature of Hh signaling, how Hh mediates tumor proliferation remains poorly understood. Hh pathway activation begins when the Hh ligand binds to and inhibits the transmembrane receptor Patched1 (Ptch1), allowing the signal transducer Smoothened (Smo) to activate Gli transcription factors and amplify Hh target gene expression. So far, all of the nuclear events ascribed to Hh occur through the Gli transcription factors, with Gli1 acting predominantly as an activator, Gli3 acting predominantly a repressor, and Gli2 possessing both repressive and activator functions.
Although most of the major components of the Hh pathway have been known from three decades of work in Drosophila melanogaster, the role of the primary cilium, a microtubule-based organelle, as a vertebrate-specific regulator of the pathway has been appreciated only recently (Huangfu et al., 2003). Abnormalities within ciliary components lead to a variety of developmental defects and cancer predisposition syndromes called “ciliopathies,” which include Hh-specific phenotypes (Hildebrandt et al., 2011; Hui and Angers, 2011). The primary cilium is necessary to amplify Hh signal in vertebrates, in addition to regulating Gli processing and activity (Hui and Angers, 2011). In the absence of Hh ligand, ciliary Ptch1 suppresses Smo activity and entry into the cilium, allowing the negative regulator Suppressor of Fused (Sufu) to inhibit Gli activity (Fig. 1 A). Ptch1 transiently exits the primary cilium after inactivation by Hh ligand, allowing entry of active Smo into the cilium that binds to and inhibits Sufu, resulting in Gli transcriptional activity (Fig. 1 B). Ciliary components actively transduce Hh signal, and mutations that disrupt intraflagellar trafficking, such as ift25, activate the pathway through inappropriate accumulation of Smo and Ptch1 along the ciliary shaft and loss of Gli at the ciliary tip (Keady et al., 2012). Moreover, primary cilia regulate Gli processing and inhibition through SuFu, which determines the level of pathway activation (Humke et al., 2010; Tukachinsky et al., 2010).
Figure 1.

Hh pathway activation, inhibition, and mechanisms of drug resistance. (A) Diagram of inactive Hh pathway in vertebrates. (B) Hh ligand activates signaling by binding and inhibiting Ptch1, allowing Smo to suppress Sufu and activate Gli transcription factors to turn on Hh target genes. (C) Common pathway-dependent genetic mutations that lead to Hh-dependent tumors include Ptch1 inactivation, Smo activation, or inappropriate SHH expression. (D) Smo antagonists such as vismodegib suppress Hh activation to prevent tumor growth. (E) Genetic escape pathways that evolve during Smo antagonist treatment include Smo point mutations that prevent Smo–drug interaction or (F) Gli target gene amplification of Gli2 or Ccnd1. (G) Compensatory escape pathways that have evolved include inappropriate activation of S6K1 that prevents Sufu inhibition of Gli and (H) PI3K pathway up-regulation leading to inappropriate Gli activity through currently unknown mechanisms.
Overwhelming data exists for the dependence of BCC and medulloblastoma growth on Hh pathway activation. For instance, BCCs, which are invasive epithelial tumors, originate from activating mutations in the Hh pathway in progenitor cells of the interfollicular epidermis and hair follicle. They retain basal keratinocyte histology, and invade as either branching or nest-like nodular structures. Mutations that inappropriately express SHH, inhibit Patched1, or activate Smo (Fig. 1 C) comprise the bulk of BCC cases (Epstein, 2008; Gomez-Ospina et al., 2012). Induction of Hh target genes in human skin grafts also results in BCC-like changes (Callahan et al., 2004), which indicates that pathway activation is sufficient for tumorigenesis and provides compelling therapeutic targets in these tumors.
The first Smo inhibitor: How effective is it?
The first clues that the Hh pathway could be targeted came from the observation that the plant-derived compound cyclopamine potently inhibits Smo (Chen et al., 2002b) and has efficacy against Hh-dependent tumors. Independent in vitro screens by several groups led to the discovery of other Smo antagonists with distinct molecular structure, with one, vismodegib, recently approved for clinical use (Chen et al., 2002a; Weiss and Korn, 2012). Vismodegib was identified from a small molecule screen for Hh pathway antagonists and achieved its final clinical form after chemical optimization for improved solubility, metabolic stability, and biodistribution (Robarge et al., 2009).
Clinical trials using Smo antagonists in patients reveal heterogeneous responses from BCCs in different patient groups. Patients with basal cell nevus syndrome (BCNS) carry a Ptch1 mutation that predisposes them to develop hundreds of BCCs with relatively little sun exposure. Despite the high tumor burden, Smo blockade using vismodegib appears effective with a surprising 100% (38 of 38) response rate in patients (Tang et al., 2012). Although many lesions existed on each patient, no disease progression or acquired resistance developed during the treatment period (mean of 8 mo), revealing a particularly sensitive tumor population with a slow rate of evolution. Vismodegib treatment appeared both tumoricidal and tumoristatic, as many of the tumors regrew with cessation of the drug.
In contrast, treatment of more invasive tumors demonstrates a lower response rate. Phase I trials treating metastatic or locally advanced BCC found that only about half (19 of 33 patients) displayed tumor regression (Von Hoff et al., 2009; LoRusso et al., 2011), despite having a similar BCC histology to the discussed BCNS patients (Fig. 2). Likewise, patients in a phase II clinical trial showed a response rate of 30% (10 of 33) in metastatic and 43% (27 of 63) in locally advanced BCCs (Sekulic et al., 2012). Finally, more invasive solid tumors such as small cell lung or pancreatic cancers demonstrate little or no responsiveness in early phase clinical trials (LoRusso et al., 2011), despite an inhibition of Hh pathway activity in uninvolved skin from the same patient. Although larger Hh-dependent tumor studies need to be performed, early evidence supports the idea that more invasive tumor subtypes exhibit a much greater ability to resist Smo blockade.
Figure 2.
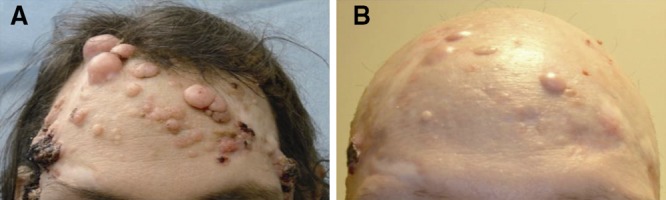
The Smo antagonist vismodegib is an effective BCC therapy. (A) Multiple untreated, but biopsy proven, BCC tumors in an individual with a genetic syndrome leading to Shh overexpression. (B) Vismodegib-treated tumors shrink, but several resistant tumors remain after 1 yr of drug treatment. Image courtesy of A.L.S. Chang.
The rate of acquired resistance represents an alternative measure of tumor evolution. Although one-third of advanced BCC tumors initially respond, persistent vismodegib therapy leads to significant secondary resistance (Chang and Oro, 2012). Of the 28 patients continuously treated with Smo inhibitor, 21% developed at least one tumor regrowth while on the drug, with the mean time to detected regrowth at 56 wk (Fig. 2). These data indicate that more aggressive tumors display higher initial resistance and are more capable of evolving drug-independent growth characteristics. One possibility for the differential response between tumors is that syndromic BCCs are somehow different from sporadic BCCs. Arguing against this idea is the fact that 6 of the 28 patients were BCNS patients that developed locally advanced disease, and 3 of these 6 patients acquired drug resistance during treatment. It is possible that advanced BCCs have higher mutation rates or more tumor cells, leading to a higher chance of resistant mutations that contribute to the ability of the tumor to evolve resistance.
Pathway and compensatory tumor evolution in treated patients
Predicting tumor response to targeted therapies requires an understanding of the how mutations are generated and the evolutionary mechanisms used to bypass treatment. Many recent excellent reviews exist about the molecular basis of the mutator phenotype, where alterations in a mutator gene dramatically increase mutation rates at other loci (Loeb, 2011). This topic will not be discussed further here. However, a key question in drug-induced tumor resistance is: how do tumors bypass the Smo blockade? Advanced tumors can evolve resistance through pathway-dependent genetic mechanisms or through compensatory adaptation. Recent data indicate that pathway and compensatory intratumor heterogeneity exists at the time of diagnosis. Drug-resistant clones, initially present in low numbers, become the dominant clone as they gain growth advantage in the treated tumor (Diaz et al., 2012; Misale et al., 2012). Understanding common escape mechanisms may help prescreen patients to determine which targeted therapy or combination of targeted therapies is most likely to be effective.
Pathway-dependent genetic alterations discovered in resistant tumors from patients and animal models directly affect Hh pathway members. Initial studies in medulloblastoma suggest that vismodegib resistance stems from genetic alterations at the level of, or downstream from, Smo. Resistance can originate from Smo point mutations that ablate Smo–drug interaction while maintaining Hh pathway activation (Yauch et al., 2009). These mutations occur in the ligand-binding pocket of Smo (Fig. 1 E). Other genetic alterations that lead to resistance come from gene duplications of Gli2 or Hh target gene cyclin D1 (Fig. 1 F) that bypass the requirement of Smo to inappropriately maintain or increase Gli target gene induction (Buonamici et al., 2010; Dijkgraaf et al., 2011). These mutations promote high Hh pathway activation in the presence of Smo antagonists and mediate resistant tumor growth.
Compensatory alterations outside of the canonical Hh pathway have also been found that mediate tumor resistance. A compensatory alteration is one where elevated Hh activation occurs in the absence of direct genetic mutation or copy number variation of Hh pathway members, and is epigenetic in nature. One example is the up-regulation of the phosphoinositide 3-kinase (PI3K) pathway (Buonamici et al., 2010). PI3K and its downstream effectors were enriched in resistant versus sensitive medulloblastoma tumors, which suggests that PI3K signaling may in part promote tumor resistance by up-regulating Hh pathway activation (Fig. 1 H). As PI3K signaling can potentiate Gli-dependent transcription induced by low levels of Hh (Riobó et al., 2006), PI3K signaling may work best to promote tumor resistance when Smo antagonists suppress Hh pathway activation. In support of this hypothesis, PI3K or PI3K-mTOR inhibitors in combination with a Smo antagonist can delay tumor resistance (Buonamici et al., 2010).
Similarly, in esophageal cancers resistant to Smo inhibitors, the activity of mTOR pathway component S6 kinase 1 (S6K1) was found to be elevated (Wang et al., 2012). S6K1 phosphorylates Gli1, releasing Gli1 from Sufu inhibition to activate Gli-dependent transcription (Fig. 1 G). S6K1 renders Gli1-expressing tumors partially Smo independent, as inhibition from Sufu is derepressed. Interestingly, S6K1 is inappropriately activated in esophageal adenocarcinoma and some medulloblastomas (Dijkgraaf et al., 2011; Wang et al., 2012), providing a partial explanation for drug resistance.
Racing against tumor evolution
Developing effective targeted therapies to dispatch tumors before they evolve resistance requires knowledge of available escape pathways. This is especially critical given that resistant clones are likely present in small numbers at the time of treatment initiation. Given the pathway and compensatory alterations in Hh-dependent tumors thus far, dual targeting of the most downstream component of the pathway, and the compensatory pathways, will likely generate optimal therapies. Gli transcription factors ultimately transduce the signal from the Hh ligand; moreover, escape pathways that bypass Smo still activate Gli. Targeting Gli directly or the signaling components that activate Gli could prove quite successful as the next level of therapy.
Several Gli inhibitors have been identified that have impressive efficacy. Through a screen of the National Institutes of Health compound library, molecules such as GANT58 and GANT61 were found to block Gli transcriptional activity (Lauth et al., 2007). These drugs inhibit Hh signaling with a similar IC50 to cyclopamine and suppress Gli1-positive human prostate cancer xenografts. Moreover, similar screens using a library of previously FDA-approved drugs identified arsenic trioxide (ATO) as a Gli antagonist (Kim et al., 2010). ATO blocks accumulation of Gli2 to primary cilia, with longer incubation times reducing steady-state Gli2 protein levels, resulting in suppression of medulloblastoma growth in mouse models. As ATO is already in clinical use for acute promyelocytic leukemia, this may be a useful therapy for resistant BCCs in the near future.
Identifying and targeting modulators of Gli activity may also show promise in preventing or treating resistant tumors. New pathways that regulate Gli such as the aforementioned S6K1 may hold the key to delay tumor resistance. Because S6K1 appears to regulate Gli1 activity downstream of mTOR, and Smo inhibitors regulate Gli activity and nuclear localization, combination therapy may prevent tumor cells containing this compensatory alteration from growing. Indeed, in vitro data from esophageal adenocarcinoma xenografts suggest that combination therapy with mTOR inhibitors and vismodegib work synergistically (Wang et al., 2012). These data provide strong preclinical support for the use of combined therapy to delay growth of Smo antagonist–resistant tumors.
The blueprint to develop the first Hh pathway inhibitors came from impressive efforts from the Hh and cancer communities, beginning with identification of Hh pathway components and their roles in cancer and continuing with intense screening and medicinal chemistry that refined drug targets for optimal human use. The challenge for the future is to better understand common pathway-dependent genetic and compensatory escape pathways that evolve from clonal populations within tumors and design combination therapies to block them before further evolution takes place. Through improved genomics, cell biology, and medicinal chemistry, this may be a race medical research can win.
Acknowledgments
This work was funded by National Institutes of Health grants R01ARO46786 and R01ARO52785. A.E. Oro and A.L.S. Chang received funds for clinical trials from Genentech/Roche, Infinity, and Novartis. Illustrations were provided by Neil Smith, www.neilsmithillustration.co.uk.
Footnotes
Abbreviations used in this paper:
- ATO
- arsenic trioxide
- BCC
- basal cell carcinoma
- BCNS
- basal cell nevus syndrome
- Hh
- Hedgehog
- PI3K
- phosphoinositide 3-kinase
References
- Buonamici S., Williams J., Morrissey M., Wang A., Guo R., Vattay A., Hsiao K., Yuan J., Green J., Ospina B., et al. 2010. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med. 2:51ra70 10.1126/scitranslmed.3001599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan C.A., Ofstad T., Horng L., Wang J.K., Zhen H.H., Coulombe P.A., Oro A.E. 2004. MIM/BEG4, a Sonic hedgehog-responsive gene that potentiates Gli-dependent transcription. Genes Dev. 18:2724–2729 10.1101/gad.1221804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A.L., Oro A.E. 2012. Initial assessment of tumor regrowth after vismodegib in advanced basal cell carcinoma patients. Arch. Dermatol. In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.K., Taipale J., Young K.E., Maiti T., Beachy P.A. 2002a. Small molecule modulation of Smoothened activity. Proc. Natl. Acad. Sci. USA. 99:14071–14076 10.1073/pnas.182542899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.K., Taipale J., Cooper M.K., Beachy P.A. 2002b. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 16:2743–2748 10.1101/gad.1025302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz L.A., Jr, Williams R.T., Wu J., Kinde I., Hecht J.R., Berlin J., Allen B., Bozic I., Reiter J.G., Nowak M.A., et al. 2012. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 486:537–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkgraaf G.J.P., Alicke B., Weinmann L., Januario T., West K., Modrusan Z., Burdick D., Goldsmith R., Robarge K., Sutherlin D., et al. 2011. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 71:435–444 10.1158/0008-5472.CAN-10-2876 [DOI] [PubMed] [Google Scholar]
- Epstein E.H. 2008. Basal cell carcinomas: attack of the hedgehog. Nat. Rev. Cancer. 8:743–754 10.1038/nrc2503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Ospina N., Chang A.L.S., Qu K., Oro A.E. 2012. Translocation affecting sonic hedgehog genes in basal-cell carcinoma. N. Engl. J. Med. 366:2233–2234 10.1056/NEJMc1115123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt F., Benzing T., Katsanis N. 2011. Ciliopathies. N. Engl. J. Med. 364:1533–1543 10.1056/NEJMra1010172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D., Liu A., Rakeman A.S., Murcia N.S., Niswander L., Anderson K.V. 2003. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 426:83–87 10.1038/nature02061 [DOI] [PubMed] [Google Scholar]
- Hui C.-C., Angers S. 2011. Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 27:513–537 10.1146/annurev-cellbio-092910-154048 [DOI] [PubMed] [Google Scholar]
- Humke E.W., Dorn K.V., Milenkovic L., Scott M.P., Rohatgi R. 2010. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 24:670–682 10.1101/gad.1902910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keady B.T., Samtani R., Tobita K., Tsuchya M., San Agustin J.T., Follit J.A., Jonassen J.A., Subramanian R., Lo C.W., Pazour G.J. 2012. IFT25 links the signal-dependent movement of Hedgehog components to intraflagellar transport. Dev. Cell. 22:940–951 10.1016/j.devcel.2012.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Lee J.J., Kim J., Gardner D., Beachy P.A. 2010. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc. Natl. Acad. Sci. USA. 107:13432–13437 10.1073/pnas.1006822107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauth M., Bergström Å., Shimokawa T., Toftgård R. 2007. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA. 104:8455–8460 10.1073/pnas.0609699104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb L.A. 2011. Human cancers express mutator phenotypes: origin, consequences and targeting. Nat. Rev. Cancer. 11:450–457 10.1038/nrc3063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoRusso P.M., Rudin C.M., Reddy J.C., Tibes R., Weiss G.J., Borad M.J., Hann C.L., Brahmer J.R., Chang I., Darbonne W.C., et al. 2011. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin. Cancer Res. 17:2502–2511 10.1158/1078-0432.CCR-10-2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misale S., Yaeger R., Hobor S., Scala E., Janakiraman M., Liska D., Valtorta E., Schiavo R., Buscarino M., Siravegna G., et al. 2012. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 486:532–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Cancer Institute. 2010. Cancer Trends Progress Report. 2009/2010 Update. http://progressreport.cancer.gov (accessed May 28, 2012)
- Riobó N.A., Lu K., Ai X., Haines G.M., Emerson C.P., Jr 2006. Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA. 103:4505–4510 10.1073/pnas.0504337103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robarge K.D., Brunton S.A., Castanedo G.M., Cui Y., Dina M.S., Goldsmith R., Gould S.E., Guichert O., Gunzner J.L., Halladay J., et al. 2009. GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg. Med. Chem. Lett. 19:5576–5581 10.1016/j.bmcl.2009.08.049 [DOI] [PubMed] [Google Scholar]
- Sekulic A., Migden M.R., Oro A.E., Dirix L., Lewis K.D., Hainsworth J.D., Solomon J.A., Yoo S., Arron S.T., Friedlander P.A., et al. 2012. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 366:2171–2179 10.1056/NEJMoa1113713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J.Y., Mackay-Wiggan J.M., Aszterbaum M., Yauch R.L., Lindgren J., Chang K., Coppola C., Chanana A.M., Marji J., Bickers D.R., Epstein E.H., Jr 2012. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N. Engl. J. Med. 366:2180–2188 10.1056/NEJMoa1113538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukachinsky H., Lopez L.V., Salic A. 2010. A mechanism for vertebrate Hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J. Cell Biol. 191:415–428 10.1083/jcb.201004108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varjosalo M., Taipale J. 2008. Hedgehog: functions and mechanisms. Genes Dev. 22:2454–2472 10.1101/gad.1693608 [DOI] [PubMed] [Google Scholar]
- Von Hoff D.D., LoRusso P.M., Rudin C.M., Reddy J.C., Yauch R.L., Tibes R., Weiss G.J., Borad M.J., Hann C.L., Brahmer J.R., et al. 2009. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N. Engl. J. Med. 361:1164–1172 10.1056/NEJMoa0905360 [DOI] [PubMed] [Google Scholar]
- Wang Y., Ding Q., Yen C.-J., Xia W., Izzo J.G., Lang J.-Y., Li C.-W., Hsu J.L., Miller S.A., Wang X., et al. 2012. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell. 21:374–387 10.1016/j.ccr.2011.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss G.J., Korn R.L. 2012. Metastatic basal cell carcinoma in the era of hedgehog signaling pathway inhibitors. Cancer. In press [DOI] [PubMed] [Google Scholar]
- Yauch R.L., Dijkgraaf G.J.P., Alicke B., Januario T., Ahn C.P., Holcomb T., Pujara K., Stinson J., Callahan C.A., Tang T., et al. 2009. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science. 326:572–574 10.1126/science.1179386 [DOI] [PMC free article] [PubMed] [Google Scholar]