

Abstract
The embryonic neural crest (NC) is a multipotent progenitor population that originates at the dorsal aspect of the neural tube, undergoes an epithelial to mesenchymal transition (EMT) and migrates throughout the embryo, giving rise to diverse cell types 1-3. NC also has the unique ability to influence the differentiation and maturation of target organs4-6. When explanted in vitro, NC progenitors undergo self-renewal, migrate and differentiate into a variety of tissue types including neurons, glia, smooth muscle cells, cartilage and bone.
NC multipotency was first described from explants of the avian neural tube7-9. In vitro isolation of NC cells facilitates the study of NC dynamics including proliferation, migration, and multipotency. Further work in the avian and rat systems demonstrated that explanted NC cells retain their NC potential when transplanted back into the embryo10-13. Because these inherent cellular properties are preserved in explanted NC progenitors, the neural tube explant assay provides an attractive option for studying the NC in vitro.
To attain a better understanding of the mammalian NC, many methods have been employed to isolate NC populations. NC-derived progenitors can be cultured from post-migratory locations in both the embryo and adult to study the dynamics of post-migratory NC progenitors11,14-20, however isolation of NC progenitors as they emigrate from the neural tube provides optimal preservation of NC cell potential and migratory properties13,21,22. Some protocols employ fluorescence activated cell sorting (FACS) to isolate a NC population enriched for particular progenitors11,13,14,17. However, when starting with early stage embryos, cell numbers adequate for analyses are difficult to obtain with FACS, complicating the isolation of early NC populations from individual embryos. Here, we describe an approach that does not rely on FACS and results in an approximately 96% pure NC population based on a Wnt1-Cre activated lineage reporter23.
The method presented here is adapted from protocols optimized for the culture of rat NC11,13. The advantages of this protocol compared to previous methods are that 1) the cells are not grown on a feeder layer, 2) FACS is not required to obtain a relatively pure NC population, 3) premigratory NC cells are isolated and 4) results are easily quantified. Furthermore, this protocol can be used for isolation of NC from any mutant mouse model, facilitating the study of NC characteristics with different genetic manipulations. The limitation of this approach is that the NC is removed from the context of the embryo, which is known to influence the survival, migration and differentiation of the NC2,24-28.
Keywords: Neuroscience, Issue 64, Developmental Biology, neural crest, explant, cell culture, mouse, embryo
Protocol
1. Preparing Plates
Use sterile technique at all times.
Prepare fibronectin (FN) by diluting 100 μL of human plasma FN stock into a final volume of 3.3 mL in Dulbecco's PBS (dPBS). Final concentration is 30 μg/mL and this can be stored at 4 °C for 1 week.
Cover bottom of each well of a sterile tissue culture four well plate with FN solution and let sit for 15 minutes. Make sure the entire surface is covered. Make media during this time (steps 2 and 3).
Remove FN solution and allow plates to dry. Gently rinse wells with 500 μL DMEM, remove DMEM, and add 500 μL of Self-Renewal (SR) medium (see below). Incubate at 37 °C in a humidified incubator containing 5 percent CO2. Complete this approximately one hour before dissection so that the plates will be dry before use.
2. Preparing SR Medium
For the culture of vagal and trunk NC from ten embryos (approximately one litter, depending on genetic background of the mouse line), prepare 25 mL of SR medium. Combine 12.5 mL low glucose DMEM, 7.5 mL Neurobasal Medium, 25 μL retinoic acid (117 μM final concentration), and 25 μL 2-mercaptoethanol (50 mM final concentration). Mix well.
Add 3.75 mL Chick Embryo Extract, 250 μL N2 salt supplement, 500 μL B27 supplement and 250 μL penicillin-streptomycin (1% final concentration). Filter the medium through a 0.22 μm filter.
Add 10 μL sterile IGF1 (20 μg/mL final concentration) and 20 μL sterile bFGF (20 μg/mL final concentration). Mix by inverting. Store at 4 °C.
3. Preparing Wash Medium
For approximately 10 embryos prepare 50 mL of wash medium. Combine 50 mg BSA with 35 mL low glucose DMEM, 15 mL Neurobasal Medium, and 500 μL penicillin-streptomycin (1% final concentration). Sterile filter with a 0.22 μm filter.
4. Preparing Collagenase/dispase
Add 50 μL of 100 mg/mL collagenase/dispase to 5 mL dPBS. Mix well.
Syringe filter with a 0.2 μm filter and add 1.5 mL into each of three wells of a twelve well plate. Pipette approximately 1 mL wash medium in the remaining wells. Store entire plate on ice until ready to digest dissected tissue.
Cut tips of a p20 and p1000 pipette filter tip with a sterile razor blade. Cut just below the beveled edge of the tip. The cut off p1000 will be used to transfer the whole embryo while the p20 will be used to transfer pieces of isolated tissue.
5. Isolating Vagal and Trunk Neural Tube from 9.5 dpc Embryos
For timed pregnancies, dams with a vaginal plug are considered 0.5 dpc at noon the morning the plug is observed. Sacrifice and remove the uterus at 9.5 dpc.
Remove the decidua from uterus and gently remove the embryo from the decidua. Once experienced with this protocol, isolate 3-4 9.5 dpc embryos at a time in sterile dPBS. Use sterile technique and sterilized dissection instruments. Instruments can be sterilized by autoclaving, heat sterilization or by incubation in ethanol.
For anterior vagal NC: using insulin needles, cut the neural tube at the mid otic placode. Cut again at the posterior edge of the 4th somite. Trim tissue ventral to the neural tube to remove the pharyngeal arches and heart.
For trunk NC: using insulin needles, remove the portion of the neural tube between somites 16-22 (or the last somite if embryos are developmentally earlier than the 22 somite stage).
Keep the yolk sac and any remaining embryonic tissue for genotyping.
6. Removal of Non-neural Ectoderm and Mesoderm
Place neural tube containing segments into collagenase/dispase at room temperature for 10 minutes. Immediately wash in wash medium.
Return tissue to sterile dPBS. Using sterile insulin needles, gently remove the non-neural ectoderm from the tissue and separate the somites away from the neural tube. The last remaining parts of the mesoderm from somite tissue can be removed by triturating gently using a cut-down p20 tip. Be careful to not damage the neural tube. Observe this closely during trituration.
Place the isolated neural tube through a second and third 30 second wash of wash medium.
Wash once in SR medium, and place the isolated neural tube into the center of a FN-coated well that was prepared previously (Step 1.3). Humidify the hypoxia chamber with a dish of sterile water. Place the dish into the hypoxia chamber and flush the chamber with mixed gas to 3% O2 (use a tank containing a mixture of 1% O2, 6% CO2, 93% N2).
Always handle the chamber extremely carefully to ensure that explants are undisturbed and remain in the center of the wells.
Incubate at 37 °C. Summary of steps 5-6 is shown in Figure 1.
7. Removal of Neural Tube
After 24 hours of incubation, remove the neural tube by gently teasing the edge of the neural tube away from the migrating cells using a sterile insulin needle. Remove and discard the neural tube from the medium using a sterile p20 cut as in step 6.2 and replace the medium with fresh SR medium (Figure 2). This is most easily done using an inverted microscope.
8. Representative Results
Following 24 hours of incubation at 37 °C in hypoxic conditions, NC cells have migrated away from the neural tube in a nearly pure population (Figure 3a). Sometimes, less than ideal cultures will not yield robust outgrowths. For example, it is possible that after 24 hours the neural tube will have curled up upon itself and the NC will not migrate away from the neural tube (Figure 3b). Occasionally the neural tube will not attach to the fibronectin coated plates.
In our experience, sub-optimal NC migration or problems with attachment of the neural tube can be adversely affected by normoxia conditions or concentration of the fibronectin, respectively. Enzymatic activity of the collagenase/dispase varies slightly by batch and digestion time must be adjusted appropriately, however, do not digest the tissue longer than fifteen minutes. Overdigestion of the neural tube containing tissue in collagenase/dispase will also result in deficient outgrowths. If somite tissue is not easily removed from the neural tube after incubation in collagenase/dispase, the neural tube can be incubated for longer than ten minutes. Sometimes the neural tube will not attach to the substrate. If this is the case, double-check the fibronectin concentration and the hypoxia conditions.
While normoxic conditions can be used to culture wild-type NC, hypoxic conditions more closely mimic the in vivo environment29,30. In our experience, hypoxic conditions became critical when culturing mutant NC. For example, when Foxd3 mutant NC were cultured in normoxic conditions, trunk NC had a greatly reduced cell outgrowth compared to controls. This disparity in outgrowth size was removed when the explants were cultured in hypoxic conditions (Figure 4). Furthermore, when wild-type neural tube explants were cultured in normoxia, the number of caspase-positive cells was greater then that of similar explants cultured in hypoxia (data not shown). By maintaining all NC culture in hypoxia, comparisons can more easily be made between dynamics of control and mutant cultures.
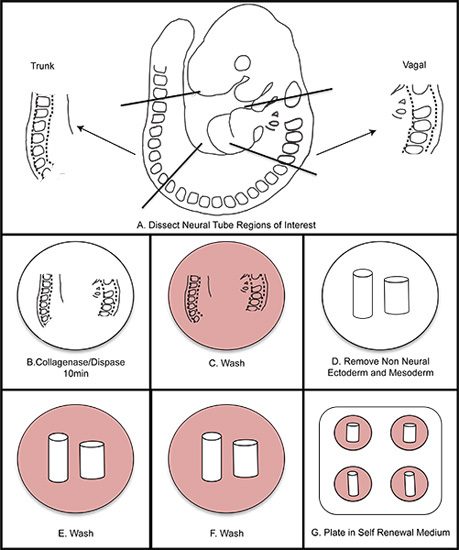 Figure 1. Overall schematic of NC isolation. A) Dissect regions of interest from the embryo. B) Digest neural tube in collagenase/dispase for ten minutes (do not exceed fifteen minutes). C) Wash in wash medium. D) Dissect away the non-neural ectoderm and mesoderm. E-F) Wash twice in wash medium. G) Plate in self renewal medium. Incubate at 37 °C in 3% O2 hypoxic conditions.
Figure 1. Overall schematic of NC isolation. A) Dissect regions of interest from the embryo. B) Digest neural tube in collagenase/dispase for ten minutes (do not exceed fifteen minutes). C) Wash in wash medium. D) Dissect away the non-neural ectoderm and mesoderm. E-F) Wash twice in wash medium. G) Plate in self renewal medium. Incubate at 37 °C in 3% O2 hypoxic conditions.
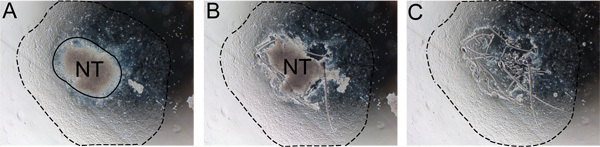 Figure 2. Stepwise removal of neural tube from explant. The neural tube must be removed after 24-48 hours to prevent contamination with non-NC cells. A) Notice the boundary between the neural tube and the NC outgrowth (solid line). B) Cut along the edge of the neural tube with an insulin needle. C) Discard the neural tube and replace medium with fresh self renewal medium. Dashed line indicates extent of the outgrowth. Abbreviation: NT, neural tube.
Figure 2. Stepwise removal of neural tube from explant. The neural tube must be removed after 24-48 hours to prevent contamination with non-NC cells. A) Notice the boundary between the neural tube and the NC outgrowth (solid line). B) Cut along the edge of the neural tube with an insulin needle. C) Discard the neural tube and replace medium with fresh self renewal medium. Dashed line indicates extent of the outgrowth. Abbreviation: NT, neural tube.
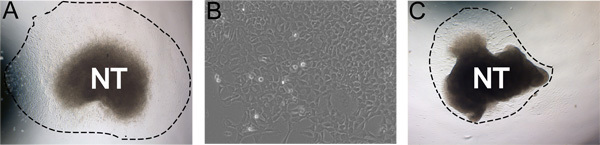 Figure 3. Examples of representative results. A) Typical explant outgrowth after 24 hours incubation in self renewal medium in hypoxic conditions (dashed line indicates extent of the outgrowth). B) Magnified view of NC outgrowth after 48 hours in culture. C) Less ideal culture with low outgrowth yield. A and C were cultured under the same conditions. Images demonstrate the natural range in culture robustness. This can be affected by efficiency of neural tube isolation, concentration of FN, hypoxic conditions, and time in collagenase/dispase digestions.
Figure 3. Examples of representative results. A) Typical explant outgrowth after 24 hours incubation in self renewal medium in hypoxic conditions (dashed line indicates extent of the outgrowth). B) Magnified view of NC outgrowth after 48 hours in culture. C) Less ideal culture with low outgrowth yield. A and C were cultured under the same conditions. Images demonstrate the natural range in culture robustness. This can be affected by efficiency of neural tube isolation, concentration of FN, hypoxic conditions, and time in collagenase/dispase digestions.
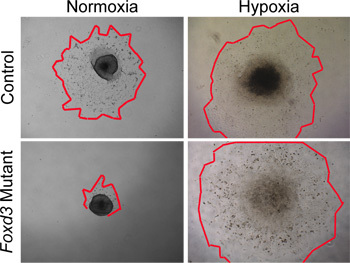 Figure 4.
In vitro analyses of NC explant cultures in normoxia versus hypoxia. Control (wild type) NC cells migrated from neural tube explants after 48 hours in normoxic culture conditions. In contrast, Foxd3 mutant NC had greatly reduced cell outgrowths in normoxia (red outlines mark edges of the NC outgrowths). When comparable explants were grown under hypoxic conditions, Foxd3 mutant NC explants grew comparably to controls, allowing subsequent analyses. Note, this behavior correlated well with the behavior of Foxd3 mutant NC in vivo.
Figure 4.
In vitro analyses of NC explant cultures in normoxia versus hypoxia. Control (wild type) NC cells migrated from neural tube explants after 48 hours in normoxic culture conditions. In contrast, Foxd3 mutant NC had greatly reduced cell outgrowths in normoxia (red outlines mark edges of the NC outgrowths). When comparable explants were grown under hypoxic conditions, Foxd3 mutant NC explants grew comparably to controls, allowing subsequent analyses. Note, this behavior correlated well with the behavior of Foxd3 mutant NC in vivo.
Discussion
Careful attention should be paid to the developmental stage of embryo to ensure the success of this approach. Counting somites of early mouse embryos is critical both for stage matching embryos within a litter and determining the correct regions of neural tube for isolation. A variation of one or two somites between embryos is within a reasonable range of developmental timing, depending upon the resolution of the experiment conducted. An embryo between 9 and 9.5 dpc will have between 17 and 25 somites. If the embryo has more than 25 somites, the NC is further advanced in developmental time, and NC outgrowths will be less robust in culture. Staging embryos based on somite number facilitates precision in experiments when developmental stage of the embryo, and therefore NC, can influence the outcome of the experiments. In a 9.5 dpc embryo, the vagal NC migrates from the dorsal neural tube in defined anterior-posterior limits: from the mid-otic placode to somite 7. Similarly, trunk NC migrates from the neural tube at levels somite 8 to somite 24. To isolate distinct populations of NC progenitors as described in this protocol, discrete sub-regions within each of those domains are isolated. The region of the neural tube used will vary based upon the design of the experiment and the anterior-posterior region of the NC being studied.
The culture conditions described above, including the medium and incubation conditions, are specifically adapted for culture of murine NC. Similar SR medium was first used for the culture of rat NC11. In our hands, this rat medium, with the addition of Neurobasal Medium, produces a robust outgrowth of murine NC progenitors. Furthermore, unlike previous work culturing avian NC cells, a feeder layer is unnecessary for culture of the mammalian NC using this method11,13,21,22. While there are numerous protocols detailing both avian and mammalian NC culture in normoxic conditions9,22, we23 (Figure 4), along with others in the field20, have found that culturing murine NC cells in hypoxic conditions as described here greatly aids survival and self-renewal, presumably because hypoxia more closely mimics physiological oxygen levels within the embryo20,29,30.
Evaluation of the expression of molecular markers greatly facilitates the identification of NC cells and their differentiated derivatives. These include expression of Foxd3, p75, and Sox10 for NC stem cells. Markers of differentiated NC include smooth muscle alpha actin (SMA) for myofibroblasts, glial fibrillary acidic protein (GFAP) for glia, microphthalmia-associated transcription factor (MITF) for melanocytes, and beta-III tubulin, protein gene product 9.5 (PGP9.5), and peripherin for neurons. These markers can be used to determine explant efficiency and differentiation of NC progenitors.
Once this technique is mastered, cultured NC can be used in a variety of assays including quantification of proliferation, cell death, migration dynamics, and/or differentiation status. NC can also be clonally cultured to investigate self-renewal or multipotency of individual NC progenitors23. After the neural tube is removed from the cultures, dissociate the NC cells in 100 μl Trypsin-EDTA (0.25%) for exactly 5 minutes at 37 °C. Quench Trypsin-EDTA in excess wash medium (8-10 mLs) by adding 800 μl per well and transferring to individual 15 mL tubes containing wash medium. Gently centrifuge cells at 150 x g for 3 minutes, remove supernatant and resuspend cell pellet in 1 mL SR medium. Count cells using a hemocytometer and plate them at a density of 25 cells/cm2. These colonies can be serially passaged to assay self-renewal. To assay multipotency, maintain the NC cells at clonal density in SR medium for 6 days and then switch to differentiation medium (10 ng/mL bFGF and 1% chick embryo extract). Culture the cells in differentiation medium under hypoxia for 8 days before analyzing colony composition with molecular markers as above11.
In conclusion, this method for isolating NC cells from mouse embryos produces a feeder free adherent culture of premigratory NC cells without the use of FACS. Analysis of experiments using these cells can easily be quantified. While this technique is straightforward, its applications for the study of NC characteristics of migration, self-renewal, and differentiation are vast. Furthermore, isolating NC from genetically modified mouse embryos allows for the direct study of particular genes and pathways in the context of NC migration, survival, and/or differentiation.
Disclosures
We have nothing to disclose.
Acknowledgments
We would like to acknowledge Marc Wozniak for video assistance. We would also like to acknowledge Sean Morrison at UT Southwestern for the original protocol for culturing rat NC cells. This work was supported Vanderbilt University Medical Center Academic Program Support and by grants from the NIH (HD36720 and HD036720-11S109) and the AHA 11GRNT7690040 to PAL, predoctoral fellowships from the AHA (0615209B) and NIH (NS065604) to NAM, and ERP was supported by an NIH training grant T32HD007502.
References
- Le Douarin N, Kalcheim C. The neural crest. 2nd edn. Cambridge University Press; 1999. [Google Scholar]
- Kulesa PM, Gammill LS. Neural crest migration: patterns, phases and signals. Developmental biology. 2010;344:566–568. doi: 10.1016/j.ydbio.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-Jeannet J-P. Neural crest induction and differentiation. Springer Science+Business Media, Landes Bioscience/Eurekah.com; 2006. [Google Scholar]
- Plank JL. Influence and timing of arrival of murine neural crest on pancreatic beta cell development and maturation. Developmental biology. 2011;349:321–330. doi: 10.1016/j.ydbio.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekrep N, Wang J, Miyatsuka T, German MS. Signals from the neural crest regulate beta-cell mass in the pancreas. Development. 2008;135:2151–2160. doi: 10.1242/dev.015859. [DOI] [PubMed] [Google Scholar]
- Freem LJ. The intrinsic innervation of the lung is derived from neural crest cells as shown by optical projection tomography in Wnt1-Cre;YFP reporter mice. J. Anat. 2010;217:651–664. doi: 10.1111/j.1469-7580.2010.01295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AM, Konigsberg IR. A clonal approach to the problem of neural crest determination. Developmental biology. 1975;46:262–280. doi: 10.1016/0012-1606(75)90104-9. [DOI] [PubMed] [Google Scholar]
- Sieber-Blum M, Cohen AM. Clonal analysis of quail neural crest cells: they are pluripotent and differentiate in vitro in the absence of noncrest cells. Developmental biology. 1980;80:96–106. doi: 10.1016/0012-1606(80)90501-1. [DOI] [PubMed] [Google Scholar]
- Baroffio A, Dupin E, Douarin NMLe. Common precursors for neural and mesectodermal derivatives in the cephalic neural crest. Development. 1991;112:301–305. doi: 10.1242/dev.112.1.301. [DOI] [PubMed] [Google Scholar]
- White PM. Neural crest stem cells undergo cell-intrinsic developmental changes in sensitivity to instructive differentiation signals. Neuron. 2001;29:57–71. doi: 10.1016/s0896-6273(01)00180-5. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, White PM, Zock C, Anderson DJ. Prospective identification, isolation by flow cytometry, and in vivo self-renewal of multipotent mammalian neural crest stem cells. Cell. 1999;96:737–749. doi: 10.1016/s0092-8674(00)80583-8. [DOI] [PubMed] [Google Scholar]
- Bronner-Fraser M, Sieber-Blum M, Cohen AM. Clonal analysis of the avian neural crest: migration and maturation of mixed neural crest clones injected into host chicken embryos. J. Comp. Neurol. 1980;193:423–434. doi: 10.1002/cne.901930209. [DOI] [PubMed] [Google Scholar]
- Stemple DL, Anderson DJ. Isolation of a stem cell for neurons and glia from the mammalian neural crest. Cell. 1992;71:973–985. doi: 10.1016/0092-8674(92)90393-q. [DOI] [PubMed] [Google Scholar]
- Corpening JC. Isolation and live imaging of enteric progenitors based on Sox10-Histone2BVenus transgene expression. Genesis. 2011;49:599–618. doi: 10.1002/dvg.20748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biernaskie JA, McKenzie IA, Toma JG, Miller FD. Isolation of skin-derived precursors (SKPs) and differentiation and enrichment of their Schwann cell progeny. Nat. Protoc. 2006;1:2803–2812. doi: 10.1038/nprot.2006.422. [DOI] [PubMed] [Google Scholar]
- Chung IH. Stem cell property of postmigratory cranial neural crest cells and their utility in alveolar bone regeneration and tooth development. Stem Cells. 2009;27:866–877. doi: 10.1002/stem.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biernaskie J. SKPs derive from hair follicle precursors and exhibit properties of adult dermal stem cells. Cell Stem Cell. 2009;5:610–623. doi: 10.1016/j.stem.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagedorn L, Suter U, Sommer L. P0 and PMP22 mark a multipotent neural crest-derived cell type that displays community effects in response to TGF-beta family factors. Development. 1999. pp. 126–3781. [DOI] [PubMed]
- Heanue TA, Pachnis V. Prospective identification and isolation of enteric nervous system progenitors using Sox2. Stem Cells. 2011;29:128–140. doi: 10.1002/stem.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SJ. Culture in reduced levels of oxygen promotes clonogenic sympathoadrenal differentiation by isolated neural crest stem cells. J. Neurosci. 2000;20:7370–7376. doi: 10.1523/JNEUROSCI.20-19-07370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Morita T, Sieber-Blum M. In vitro clonal analysis of mouse neural crest development. Developmental biology. 1993;157:517–525. doi: 10.1006/dbio.1993.1154. [DOI] [PubMed] [Google Scholar]
- Etchevers H. Primary culture of chick, mouse or human neural crest cells. Nat. Protoc. 2011;6:1568–1577. doi: 10.1038/nprot.2011.398. [DOI] [PubMed] [Google Scholar]
- Mundell NA, Labosky PA. Neural crest stem cell multipotency requires Foxd3 to maintain neural potential and repress mesenchymal fates. Development. 2011;138:641–652. doi: 10.1242/dev.054718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammill LS, Gonzalez C, Gu C, Bronner-Fraser M. Guidance of trunk neural crest migration requires neuropilin 2/semaphorin 3F signaling. Development. 2006;133:99–106. doi: 10.1242/dev.02187. [DOI] [PubMed] [Google Scholar]
- Gammill LS, Gonzalez C, Bronner-Fraser M. Neuropilin 2/semaphorin 3F signaling is essential for cranial neural crest migration and trigeminal ganglion condensation. Dev. Neurobiol. 2007;67:47–56. doi: 10.1002/dneu.20326. [DOI] [PubMed] [Google Scholar]
- Kasemeier-Kulesa JC, Bradley R, Pasquale EB, Lefcort F, Kulesa PM. Eph/ephrins and N-cadherin coordinate to control the pattern of sympathetic ganglia. Development. 2006;133:4839–4847. doi: 10.1242/dev.02662. [DOI] [PubMed] [Google Scholar]
- Osborne NJ, Begbie J, Chilton JK, Schmidt H, Eickholt BJ. Semaphorin/neuropilin signaling influences the positioning of migratory neural crest cells within the hindbrain region of the the chick. Dev. Dyn. 2005;232:939–949. doi: 10.1002/dvdy.20258. [DOI] [PubMed] [Google Scholar]
- Schwarz Q, Maden CH, Vieira JM, Ruhrberg C. Neuropilin 1 signaling guides neural crest cells to coordinate pathway choice with cell specification. Proc. Natl. Acad. Sci. U.S.A. 2009;106:6164–6169. doi: 10.1073/pnas.0811521106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon MC, Keith B. The role of oxygen availability in embryonic development and stem cell function. Nat. Rev. Mol. Cell Biol. 2008;9:285–296. doi: 10.1038/nrm2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanovic Z. Hypoxia or in situ normoxia: The stem cell paradigm. J. Cell Physiol. 2009;219:271–275. doi: 10.1002/jcp.21690. [DOI] [PubMed] [Google Scholar]