

Abstract
The opportunistic pathogen Legionella pneumophila is an amoeba-resistant bacterium, which also replicates in alveolar macrophages thus causing the severe pneumonia "Legionnaires' disease"1. In protozoan and mammalian phagocytes, L. pneumophila employs a conserved mechanism to form a specific, replication-permissive compartment, the "Legionella-containing vacuole" (LCV). LCV formation requires the bacterial Icm/Dot type IV secretion system (T4SS), which translocates as many as 275 "effector" proteins into host cells. The effectors manipulate host proteins as well as lipids and communicate with secretory, endosomal and mitochondrial organelles2-4.
The formation of LCVs represents a complex, robust and redundant process, which is difficult to grasp in a reductionist manner. An integrative approach is required to comprehensively understand LCV formation, including a global analysis of pathogen-host factor interactions and their temporal and spatial dynamics. As a first step towards this goal, intact LCVs are purified and analyzed by proteomics and lipidomics.
The composition and formation of pathogen-containing vacuoles has been investigated by proteomic analysis using liquid chromatography or 2-D gel electrophoresis coupled to mass-spectrometry. Vacuoles isolated from either the social soil amoeba Dictyostelium discoideum or mammalian phagocytes harboured Leishmania5, Listeria6, Mycobacterium7, Rhodococcus8, Salmonella9 or Legionella spp.10. However, the purification protocols employed in these studies are time-consuming and tedious, as they require e.g. electron microscopy to analyse LCV morphology, integrity and purity. Additionally, these protocols do not exploit specific features of the pathogen vacuole for enrichment.
The method presented here overcomes these limitations by employing D. discoideum producing a fluorescent LCV marker and by targeting the bacterial effector protein SidC, which selectively anchors to the LCV membrane by binding to phosphatidylinositol 4-phosphate (PtdIns(4)P)3,11 . LCVs are enriched in a first step by immuno-magnetic separation using an affinity-purified primary antibody against SidC and a secondary antibody coupled to magnetic beads, followed in a second step by a classical Histodenz density gradient centrifugation12,13 (Fig. 1).
A proteome study of isolated LCVs from D. discoideum revealed more than 560 host cell proteins, including proteins associated with phagocytic vesicles, mitochondria, ER and Golgi, as well as several GTPases, which have not been implicated in LCV formation before13. LCVs enriched and purified with the protocol outlined here can be further analyzed by microscopy (immunofluorescence, electron microscopy), biochemical methods (Western blot) and proteomic or lipidomic approaches.
Keywords: Infection, Issue 64, Immunology, amoeba, Dictyostelium discoideum, density gradient centrifugation, effector protein, Icm/Dot type IV secretion system, immuno-magnetic separation, Legionella pneumophila, macrophage, pathogen vacuole
Protocol
1. Preparations for LCV Isolation
Streak out Legionella pneumophila producing DsRed-Express13,14 from glycerol stocks on CYE agar plates with 5 μg/mL chloramphenicol (Cam) four days before LCV isolation. Incubate the bacteria at 37 °C.
Seed out 1 x 107D. discoideum producing GFP-calnexin or RAW264.7 murine macrophages in 75 cm2 tissue culture flasks one day prior infection. Use 10 ml HL5 medium with 20 μg/mL G418 for D. discoideum and incubate the amoebae at 23 °C. For RAW264.7 macrophages use 10 ml RPMI 1640 medium supplemented with 10% FCS (heat inactivated) and 1% glutamine, and grow the cells at 37 °C and 5% CO2. Use at least three 75 cm2 flasks per infection and sample (minimum 6 x 107 cells).
Inoculate an overnight culture with L. pneumophila from the CYE plate. Take a 15 mL test tube with 3 mL AYE medium and 5 μg/mL Cam. Inoculate with 100 μl of a bacterial suspension to yield an OD600nm of 0.1. Incubate the overnight culture on an overhead rotation wheel at 37 °C for 21-22 hr.
2. LCV Isolation
Change the medium of D. discoideum cells to remove the antibiotic, which would interfere with the following infection.
Measure the OD of the overnight culture. Bacteria should have reached their peak infectivity at an OD600nm of ≥3, which corresponds to 2 x 109 bacteria/mL.
Infect the cells by adding approximately 500 μl of the L. pneumophila overnight culture to the cells growing in 10 ml HL5 medium (D. discoideum) or 10 ml RPMI 1640 (macrophages). This corresponds to a multiplicity of infection (MOI) of 50. Subsequently, the infection is synchronized by centrifugation of the bacteria onto the cells at 500 × g for 10 min. Centrifugation is followed by an incubation of the D. discoideum cells at 25 °C and the RAW264.7 macrophages at 37 °C and 5% CO2, respectively. The infection time of 1 hr includes the centrifugation step.
After infection remove the medium and wash the cells once to remove extracellular bacteria. Use ice-cold SorC buffer for D. discoideum and ice-cold PBS for RAW264.7 macrophages. Add 3 mL HS buffer, supplemented with protease inhibitors (Roche) to each flask and collect the cells by using a cell scraper. Pool the corresponding samples in a 15 mL test tube.
For homogenization of the sample use 3 mL plastic Luer-Lock syringes and the stainless steel ball homogenizer. Make sure that you work on ice. Before starting, wash the ball homogenizer with distilled water, to avoid any detergent contamination and flush it with ice cold HS buffer to get rid of air bubbles. Use the 8 μm clearance ball.
Fill the first 3 mL into the syringe und mount it on the homogenizer. Press the sample back and forth nine times through the homogenizer. Exchange the syringes afterwards to avoid contamination with not-homogenized material. Collect and pool the homogenized sample in a 15 mL test tube and take a 150 μl sample for microscopic analysis. Before proceeding with a different sample dismantle and wash the ball homogenizer.
Block the homogenate with 2% CS or FCS for 30 min on a shaker on ice or on an overhead-spinning wheel (10-20 rpm) at 4 °C.
After blocking use an affinity-purified primary antibody against SidC (dilution 1:3000) or against any other bacterial marker exclusively binding to the LCV membrane. Vortex the antibody solution before adding it to the homogenate. Incubate the sample for 1 hr on a shaker on ice or on an overhead-spinning wheel (10-20 rpm) at 4 °C.
In the meantime, prepare the Histodenz gradient. Use a 15 mL test tube per gradient and three 75 cm2 flasks of one sample. First, add 5.75 mL of 35% Histodenz to the tube, and second, carefully add 5.75 mL of 10% Histodenz solution on top. Afterwards carefully lay the tubes down horizontally for 1 hr.
After incubation of the homogenate with the blocking reagent and the primary antibody, centrifuge at 600 × g for 15 min at 4°C to pellet the sample. Discard the supernatant and resuspend the pellet in 1.5 mL HS buffer. Transfer the samples to a fresh 15 mL test tube and take a 40 μl sample for microscopic analysis later on.
Incubate the pellet with the secondary antibody - MACS goat anti-rabbit IgG micro beads (dilution 1:25) - for 30 min on a shaker at 4°C. In the meantime, put the columns on the MACS magnetic holder and equilibrate them with 0.5 mL HS buffer.
Subsequently, apply the samples to the column. Collect 150 μl of the flow-through for subsequent microscopic analysis. Wash the column three times with 0.5 mL HS buffer.
Remove the columns from the magnet and eluate the LCVs with 0.5 mL HS buffer by immediate squirting. Take a 20 μl sample for microscopic analysis. The purification step by immuno-magnetic separation is expected to yield per 1 × 107 infected D. discoideum cells approximately 1 × 106 intact LCVs, which are about sevenfold enriched in the eluate compared with the flow-through 13.
Put the Histodenz gradient tubes back to a vertical position and carefully load the eluate on top. Centrifuge the tubes at 3350 × g for 1 hr at 4 °C.
After centrifugation take 1.5 mL fractions starting from the bottom of the test tube by using a long glass Pasteur pipette. Altogether collect eight fractions, each from the bottom of the tube. No visible layers are observed in the continuous Histodenz gradient. The highest yield of LCVs is typically expected in fraction 4 at a ratio of ~2 × 105 intact LCVs per 1 × 107 infected amoebae 13. Minor quantities of LCVs are also present in fractions 3 and 5, respectively. Thus, the overall yield of purified LCVs is ~1 × 106 intact LCVs per 6 × 107 infected D. discoideum cells 13. The recovery of LCVs can be performed at room temperature (RT), and the LCVs appear stable for several hours.
3. Microscopic Analysis of the Collected Samples
Prepare a 24-well flat-bottom tissue culture plate with poly-L-lysine coated coverslips.
Pipette every sample taken during the LCV purification plus 150 μl of each density gradient fraction into the 24-well plate. Add 0.5 mL HS buffer to each well to dilute the high Histodenz concentration. Afterwards, centrifuge the plate at 600 × g for 10 min at 4 °C. Carefully remove the supernatant and fix the samples with 4% paraformaldehyde (PFA) for 20 min at RT. After fixation wash the samples twice. Use SorC for D. discoideum and PBS for RAW264.7 macrophages.
- Mount the samples from GFP-calnexin expressing D. discoideum, taken at different steps during LCV isolation, directly on glass-slides using e.g. Vectashield.
- Incubate the samples from RAW264.7 macrophages, taken at different steps during LCV isolation, with blocking solution (1% BSA in PBS) for 10 min at RT. Use an anti-SidC primary antibody to stain the LCVs and incubate for 1h in a wet chamber at RT (affinity-purified rabbit anti-SidC; 1:1000 in blocking buffer). Subsequently, wash the samples three times with PBS. Incubate with a secondary antibody (e.g. anti-rabbit Cy5; 1:200 in blocking solution) for 30 min in a wet and dark chamber at RT. Finally, wash the samples three times with PBS and mount them on glass-slides using e.g. Vectashield.
Morphology, integrity and quantity of the LCVs isolated from GFP-calnexin expressing D. discoideum or from RAW264.7 macrophages are analyzed by a fluorescence microscope equipped with the adequate filters.
4. Representative Results
The quality and yield of the LCV purification by immuno-affinity separation and density gradient centrifugation can be followed by fluorescence microscopy (Fig. 1). An overview of samples collected during the LCV isolation from D. discoideum or macrophages is shown (Fig. 2). The homogenate of L. pneumophila-infected phagocytes shows intact LCVs, but also a lot of cell debris and extracellular bacteria. After pelleting the sample, the image is similar to the homogenate, sometimes a bit denser. Since intact LCVs should stick to the column, the flow-through contains mostly extracellular bacteria and cell debris. After eluting the sample from the column, the eluate contains intact LCVs in large amounts. In our hands, the LCV purification appears to be more effective for D. discoideum than for macrophages. In D. discoideum the pathogen vacuoles appear rounder and the yield of isolated LCVs is more than 10 times higher compared with macrophages (Fig. 3). In intact macrophages LVCs stained with different antibodies also appear more irregularly shaped.
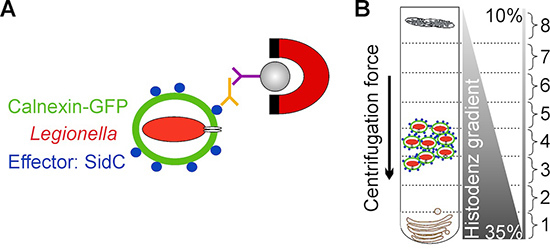 Figure 1. Schematic overview of LCV isolation. A) Isolation of LCVs by immuno-magnetic separation using an affinity-purified antibody against the bacterial effector protein SidC, localizing exclusively to the LCV membrane. A secondary MACS micro bead-coupled antibody and a magnet are used to isolate LCVs from cell debris. B) After immuno-magnetic separation LCVs are further purified by Histodenz density gradient centrifugation. LCVs are enriched in fraction 4.
Figure 1. Schematic overview of LCV isolation. A) Isolation of LCVs by immuno-magnetic separation using an affinity-purified antibody against the bacterial effector protein SidC, localizing exclusively to the LCV membrane. A secondary MACS micro bead-coupled antibody and a magnet are used to isolate LCVs from cell debris. B) After immuno-magnetic separation LCVs are further purified by Histodenz density gradient centrifugation. LCVs are enriched in fraction 4.
 Figure 2. Samples collected during LCV purification. Images from homogenate, pellet, flow-through and eluate from D. discoideum or macrophages are depicted. The samples show L. pneumophila producing DsRed-Express (red) in D. discoideum producing GFP-calnexin signal (green, upper panel) or in macrophages stained with an anti-SidC antibody (cyan, lower panel). Bars, 10 μm.
Figure 2. Samples collected during LCV purification. Images from homogenate, pellet, flow-through and eluate from D. discoideum or macrophages are depicted. The samples show L. pneumophila producing DsRed-Express (red) in D. discoideum producing GFP-calnexin signal (green, upper panel) or in macrophages stained with an anti-SidC antibody (cyan, lower panel). Bars, 10 μm.
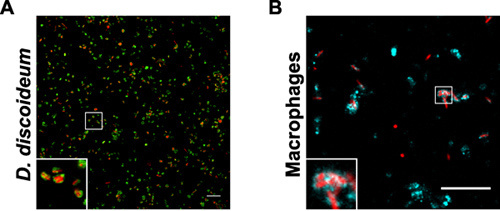 Figure 3. Purified LCVs from D. discoideum or macrophages. The samples show fraction 4 after Histodenz density gradient centrifugation of L. pneumophila producing DsRed-Express (red) in (A) D. discoideum producing GFP-calnexin signal (green) or (B) in macrophages stained with an anti-SidC antibody (cyan). Bars, 10 μm.
Figure 3. Purified LCVs from D. discoideum or macrophages. The samples show fraction 4 after Histodenz density gradient centrifugation of L. pneumophila producing DsRed-Express (red) in (A) D. discoideum producing GFP-calnexin signal (green) or (B) in macrophages stained with an anti-SidC antibody (cyan). Bars, 10 μm.
Discussion
In contrast to previously published methods, this protocol is based on two steps, first the separation of LCVs by an immuno-magnetic approach, and second, the purification of LCVs by density gradient centrifugation. The LCV isolation can be easily followed by fluorescence microscopy, when either fluorescently labeled bacteria and GFP-producing cells or antibody staining is used. The protocol described here is a simple, straight-forward method, which results in the enrichment of LCVs with high purity. Importantly, the protease inhibitor in the homogenization buffer is required for RAW264.7 macrophages, but optional for D.discoideum13.
In principle, the protocol can be adopted for other pathogens or host cells, as long as a specific, selectively localizing marker is present, which is needed for the separation step. Ideally, the distinct marker is of bacterial origin, transported to the vacuole membrane, and selectively localizing there. Targeting a host cell marker likely results in the undesired co-purification of the pathogen vacuole with other host cell compartments.
Once a distinctive marker of the pathogen vacuole is identified, a suitable polyclonal or monoclonal antibody has to be raised. For every primary antibody the concentration and the blocking reagent should be optimized, as blocking reagents other than FCS could be more appropriate (e.g. BSA, de-lipidated milk, other commercially available blocking reagents).
Another critical point is the infectivity of the pathogen. The bacteria used for infection should be in their most infective stage. L. pneumophila, e.g., needs to be grown to the post-exponential/early stationary growth phase in liquid culture. Under these conditions, the bacteria are also morphologically uniform (~2 x 0.5 μm), as opposed to bacteria grown on CYE agar plates. Furthermore, antibiotics possibly used in the cell culture medium must be removed prior to an infection to avoid low infectivity and harm to the bacteria. To avoid a negative effect on the uptake efficiency, the phagocytes should have reached a confluency of approximately 80% (not more) at the time of infection. Finally, although an incubation period of 1 hr is the standard infection time for L. pneumophila, yielding a rather homogenous and robust population of LCVs, other time points can be chosen for proteomic or biochemical studies on LCV formation13.
Once the above method is established for a given pathogen-host cell pair, it is also possible to test bacterial mutant strains and/or different host cells, including D. discoideum defined deletion mutants. In general, pathogen vacuoles purified with this protocol can be analyzed by different methods, including microscopy (fluorescence and electron microscopy), biochemical assays (Western blot), as well as proteomics and lipidomics. For the latter methods, it is advisable to coordinate with the person in charge of the proteomics/ lipidomics analysis the buffer conditions, sample amounts and downstream processing of purified pathogen vacuoles.
Disclosures
No conflicts of interest declared.
Acknowledgments
Research in our lab was supported by the Max von Pettenkofer Institute, Ludwig-Maximilians University Munich, the German Research Foundation (DFG, HI 1511/3-1) and the Bundesministerium für Bildung und Forschung (BMBF) "Medical Infection Genomics" initiative (0315834C).
References
- Hilbi H, Hoffmann C, Harrison CF. Legionella spp. outdoors: colonization, communication and persistence. Environ. Microbiol. Rep. 2011;3:286–296. doi: 10.1111/j.1758-2229.2011.00247.x. [DOI] [PubMed] [Google Scholar]
- Zhu W. Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS ONE. 2011;6:e17638. doi: 10.1371/journal.pone.0017638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilbi H, Weber S, Finsel I. Anchors for effectors: subversion of phospho-inositide lipids by Legionella. Front Microbiol. 2011;2:91. doi: 10.3389/fmicb.2011.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubber A, Roy CR. Modulation of host cell function by Legionella pneumophila type IV effectors. Annu. Rev. Cell Dev. Biol. 2010;26:261–283. doi: 10.1146/annurev-cellbio-100109-104034. [DOI] [PubMed] [Google Scholar]
- Kima PE, Dunn W. Exploiting calnexin expression on phagosomes to isolate Leishmania parasitophorous vacuoles. Microb. Pathog. 2005;38:139–145. doi: 10.1016/j.micpath.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Lührmann A, Haas A. A method to purify bacteria-containing phagosomes from infected macrophages. Methods Cell Sci. 2000;22:329–341. doi: 10.1023/a:1017963401560. [DOI] [PubMed] [Google Scholar]
- Sturgill-Koszycki S, Haddix PL, Russell DG. The interaction between Mycobacterium and the macrophage analyzed by two-dimensional polyacrylamide gel electrophoresis. Electrophoresis. 1997;18:2558–2565. doi: 10.1002/elps.1150181411. [DOI] [PubMed] [Google Scholar]
- Fernandez-Mora E, Polidori M, Lührmann A, Schaible UE, Haas A. Maturation of Rhodococcus equi-containing vacuoles is arrested after completion of the early endosome stage. Traffic. 2005;6:635–653. doi: 10.1111/j.1600-0854.2005.00304.x. [DOI] [PubMed] [Google Scholar]
- Mills SD, Finlay BB. Isolation and characterization of Salmonella typhimurium and Yersinia pseudotuberculosis-containing phagosomes from infected mouse macrophages: Y. pseudotuberculosis traffics to terminal lysosomes where they are degraded. Eur. J. Cell Biol. 1998;77:35–47. doi: 10.1016/S0171-9335(98)80100-3. [DOI] [PubMed] [Google Scholar]
- Shevchuk O. Proteomic analysis of Legionella-containing phagosomes isolated from Dictyostelium. Int. J. Med. Microbiol. 2009;299:489–508. doi: 10.1016/j.ijmm.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Brombacher E. Rab1 guanine nucleotide exchange factor SidM is a major phosphatidylinositol 4-phosphate-binding effector protein of Legionella pneumophila. J. Biol. Chem. 2009;284:4846–4856. doi: 10.1074/jbc.M807505200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urwyler S, Finsel I, Ragaz C, Hilbi H. Isolation of Legionella-containing vacuoles by immuno-magnetic separation. Curr. Protoc. Cell Biol. 2010;Chapter 3:Unit 3. doi: 10.1002/0471143030.cb0334s46. [DOI] [PubMed] [Google Scholar]
- Urwyler S. Proteome analysis of Legionella vacuoles purified by magnetic immunoseparation reveals secretory and endosomal GTPases. Traffic. 2009;10:76–87. doi: 10.1111/j.1600-0854.2008.00851.x. [DOI] [PubMed] [Google Scholar]
- Mampel J. Planktonic replication is essential for biofilm formation by Legionella pneumophila in a complex medium under static and dynamic flow conditions. Appl. Environ. Microbiol. 2006;72:2885–2895. doi: 10.1128/AEM.72.4.2885-2895.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]