

Abstract
Biodegradable nanoparticles have emerged as a versatile platform for the design and implementation of new intranasal vaccines against respiratory infectious diseases. Specifically, polyanhydride nanoparticles composed of the aliphatic sebacic acid (SA), the aromatic 1,6-bis(p-carboxyphenoxy)hexane (CPH), or the amphiphilic 1,8-bis(p-carboxyphenoxy)-3,6-dioxaoctane (CPTEG) display unique bulk and surface erosion kinetics1,2 and can be exploited to slowly release functional biomolecules (e.g., protein antigens, immunoglobulins, etc.) in vivo3,4,5. These nanoparticles also possess intrinsic adjuvant activity, making them an excellent choice for a vaccine delivery platform6,7,8.
In order to elucidate the mechanisms governing the activation of innate immunity following intranasal mucosal vaccination, one must evaluate the molecular and cellular responses of the antigen presenting cells (APCs) responsible for initiating immune responses. Dendritic cells are the principal APCs found in conducting airways, while alveolar macrophages (AMɸ) predominate in the lung parenchyma9,10,11. AMɸ are highly efficient in clearing the lungs of microbial pathogens and cell debris12,13. In addition, this cell type plays a valuable role in the transport of microbial antigens to the draining lymph nodes, which is an important first step in the initiation of an adaptive immune response9. AMɸ also express elevated levels of innate pattern recognition and scavenger receptors, secrete pro-inflammatory mediators, and prime naïve T cells12,14. A relatively pure population of AMɸ (e.g., greater than 80%) can easily be obtained via lung lavage for study in the laboratory. Resident AMɸ harvested from immune competent animals provide a representative phenotype of the macrophages that will encounter the particle-based vaccine in vivo. Herein, we describe the protocols used to harvest and culture AMɸ from mice and examine the activation phenotype of the macrophages following treatment with polyanhydride nanoparticles in vitro.
Keywords: Bioengineering, Issue 64, Microbiology, alveolar macrophages, AMɸ, lung lavage, polyanhydride, nanoparticles, harvesting, activation
Protocol
1. Harvesting Alveolar Macrophages (AMɸ) from the Mouse using Lung Lavage
Wear appropriate personal protective equipment (PPE) such as a lab coat, disposable gloves, and proper eye protection.
Prepare complete AMɸ (cAMɸ) medium before initiation of harvest. Add 2.5 mL of penicillin/streptomycin (pen/strep) solution, 250 μL of β-mercaptoethanol (2-mercaptoethanol), and 25 mL of heat inactivated fetal bovine serum (FBS) to 222.25 mL of Dulbelcco's Modified Eagle Medium (DMEM). Filter-sterilize cAMɸ medium using a 0.2 μm pore size bottle top filter under vacuum.
Euthanize a healthy mouse by CO2 asphyxiation in an euthanization chamber. Although a C57BL/6 mouse (6-8 weeks old) was used for the purposes of this project, this lung lavage technique can be performed on any strain of mouse. Using a compressed gas cylinder, let gas flow into the chamber for 1 min prior to placing the mouse in the chamber to assure that the chamber is fully charged with CO2. Place the mouse in the chamber for at least one minute to euthanize the mouse. In addition, exsanguinate each mouse via cardiac puncture as a physical method to confirm mouse death as described in 1.4.
Lay mouse on its back (dorsal side), spray with 70% ethanol, locate the sternum and, at a 30 degree angle from the abdomen, insert a 23 gauge needle (attached to a 1 mL syringe) about 50 mm into the thoracic cavity. With proper placement of the needle, it should be possible to collect between 0.4 to 1 mL of blood depending on the size of the mouse. Examine the animal for cessation of vital signs.
Place mouse on lab bench so that it is lying on its abdomen (ventral side). Spray mouse with 70% ethanol to sterilize the working environment. Pinch skin about halfway down the spine of the mouse. Using dissection scissors that have been sterilized with 70% ethanol, make a small incision through the skin.
Grab the skin on both sides of the incision and pull the skin in opposite directions along the cranial-caudal axis of the mouse. When done correctly, the skin will be removed from the underlying carcass.
The neck of the mouse should now show exposed muscles and glands. Carefully cut that tissue and pull it towards the anterior of the mouse using forceps, thereby exposing the larynx, trachea, and esophagus of the mouse. Avoid cutting the jugular vein or carotid artery.
The trachea is now visible and is identifiable as the tissue with cartilaginous rings around it. Delicately hold the trachea with forceps and gently lift and separate the trachea from the esophagus, which is located on the dorsal side.
Make a small incision anterior to where one holds the trachea but posterior to the larynx. The incision should be made at approximately a 45° degree angle. Also, do not completely transect the trachea, as this will prevent it from retracting into the body cavity.
Fill a 1 cc syringe fitted with a Tom Cat catheter with sterile PBS in preparation to insert the catheter into the lumen of the trachea. Hold the trachea using forceps and with your hand hold the catheter close to the tip to increase control and gently insert it into the tracheal incision site. Gently insert catheter into trachea about 20 mm. Grip the trachea that is around the catheter tube, freeing the other hand.
Using a 1 mL syringe fitted with the catheter, gently infuse 0.75 mL of room temperature, sterile PBS into the lungs and then gently aspirate the PBS back into syringe. Repeat this process three times while externally massaging the chest. Disconnect the syringe from the catheter and dispense the lavage fluid into a 15 mL centrifuge tube. After filling the syringe with 0.75 mL of fresh PBS and reconnecting the syringe to the catheter, repeat steps 1.9 and 1.10 twice for each mouse. Fluid recovery may be partial, obtaining less than the volume infused. If collecting cells from more than one mouse, place the 15 mL tube with the harvested cells on ice until all lung lavages have been performed.
2. Processing of Lung Lavage AMɸ Harvest
Centrifuge lung lavage fluid at 250 x g for 10 min at 4 °C.
Using sterile technique, decant supernatant into appropriate waste container inside a biosafety cabinet. Resuspend the cell pellet by gently raking the centrifuge tube across the top of a test tube rack. Add 1 mL of cAMɸ medium to resuspend the cells.
Enumerate the cells using a Coulter Particle Counter Z1 by pipetting 20 μL of cells into 10 mL of Isoton II Diluent in Coulter counter vial. Add 3 to 4 drops of Zap-oglobin II lytic reagent into vial, gently invert 3 to 4 times and then load the sample vial onto the counter to obtain a cell concentration. Be sure to program the counter with a dilution factor of 2 x 104 to display the cell concentration as the number of cells per mL.
Dilute cells to 2.5 x 105 per mL using cAMɸ medium.
Dispense 2 mL of 2.5 x 105 cells/mL into each well of a 6-well tissue culture dish.
To enrich for AMɸ, incubate culture dishes inside a 37 °C humidified incubator with a 5% CO2 atmosphere for 6 h to allow cells to adhere to the bottom of the well. Gently aspirate the supernatant as to not disrupt adherent cells.
Rinse the wells using sterile PBS (calcium and magnesium free) to remove non-adherent cells and debris. Add 2 mL of cAMɸ medium to each well.
Incubate cells overnight inside a 37 °C humidified incubator with a 5% CO2 atmosphere prior to addition of stimulants. After this enrichment step, approximately 87% of the cells are positive for the macrophage markers CD11b and F4/80 (Figure 2c).
3. Addition of Polyanhydride Nanoparticles and Control Treatments
Fabricate nanoparticles via polyanhydride anti-solvent nanoencapsulation15. In this process, the polymer is dissolved in methylene chloride (4 °C at a concentration of 25 mg/mL) and precipitated in pentane (-20 °C at a ratio 1:200 methylene chloride:pentane).
Weigh out polyanhydride nanoparticles using sterilized weigh paper on a balance that can accurately measure microgram amounts. Add particles to a 1.5 mL microcentrifuge tube.
Resuspend the nanoparticles in ice cold cAMɸ. Calculations are made such that no more than 0.3 mL of medium is added to each well to obtain the desired concentration of nanoparticles. Keep the resuspended particles on ice until the nanoparticles are added to the culture well.
Prior to adding the nanoparticles to the AMɸ cultures, sonicate particles using a bench top sonicator with a microtip. Sterilize microtip by spraying with 70% ethanol and wiping with a sterile Kimwipe prior to sonication. Sonicate at 10-20 joules for 30 to 45 s while keeping the tube on ice. Observe nanoparticle suspension macroscopically. If particles are not sufficiently dispersed, allow the suspension to set on ice for 1 to 2 min to ensure that the nanoparticles are below the glass transition temperature of the polymer before sonicating again for 30 to 45 sec. The intent of this sonication step is to adequately disperse the nanoparticles, as the 50:50 CPTEG:CPH nanoparticles tend to aggregate when exposed to an aqueous environment. However, some small aggregates of nanoparticles may still remain in the suspension.
Nanoparticle suspension (adequately dispersed) should be kept on ice and be used immediately to prevent premature surface erosion or aggregation.
Remove the culture dishes containing the AMɸ from incubator and place in the biosafety cabinet. Tilt plate at approximately a 45 degree angle and pipette off the amount of medium that will be added to the nanoparticle suspension (it should not exceed 0.3 mL). Do not disturb the adherent cells when removing the medium.
Vortex the polyanhydride nanoparticle suspension briefly prior to adding it to the appropriate wells. Pipette the amount of nanoparticle suspension (no more than 0.3 mL) into the specific well(s); however, do not pipette up and down to mix. This action could detach the adherent AMɸ from the well. The particles will settle to the bottom of the well and contact the cells. For the studies outlined in this report, the final concentration of nanoparticles co-cultured with the AMɸ was 125 μg/mL. Proceed with subsequent wells and desired treatment groups. Include positive and negative control groups, such as medium only and 200 ng/mL lipopolysaccharide (LPS) to designated wells.
Return the culture dishes to the 37 °C humidified incubator with a 5% CO2 atmosphere for 48 h. Kinetic studies in our laboratory have demonstrated 48 hours to be the optimal culture period for evaluating both cell-surface marker expression and cytokine secretion as it permits adequate time for transcription and protein synthesis to occur. Although greater levels of cytokines accumulate during an extended culture period (i.e., 72 hr), evaluating cell surface marker expression at this time point can be confounded by the contribution of the elevated levels of cytokines. The changes in surface marker expression observed may not be directly attributable to stimulation with the nanoparticles themselves.
4. Evaluating AMɸ Activation using Flow Cytometry
Prepare fluorescence-activated cell sorting (FACS) buffer (0.1 % sodium azide and 0.1 % bovine serum albumin (BSA)) in 0.1 M phosphate buffered saline (pH 7.4) prior to harvesting the plated macrophages that have been co-cultured with nanoparticles or stimulants.
Prepare working solutions of the blocking buffer and the monoclonal antibodies. To prepare the block buffer, dilute rat IgG (Sigma) and anti-CD16/32 (eBioscience) to 1 mg/mL and 10 μg/mL, respectively, in FACS buffer. Dilute surface marker antibody panel in FACS buffer to either the manufacturer recommended concentration or the user optimized concentration. For the results presented in this work, the surface marker panel consists of antibodies against MHC II, CD86, CD40 and CIRE (CD209). Anti-mouse antibodies against the macrophage markers F4/80 and CD11b are also included in the panel to positively identify the macrophage phenotype during data analysis. Care must be taken when selecting antibodies against MHC II molecules, as inbred mouse strains often differ in their MHC II haplotypes. The antibodies selected must react with the specific MHC alloantigens expressed by your mouse strain of interest.
After 48 h of incubation with nanoparticles and/or stimulants, place cell culture dishes on ice for 20 min. To remove the adherent macrophages from the plates, gently scrape wells with a 24 cm cell scraper.
Using a 5 mL serological pipette, gently aspirate the cells and medium up and down five times to generate a single cell suspension. Pipette the contents of individual wells into separate 5 mL polystyrene tubes so as not to mix together cells from different experimental treatments.
Add 2 mL of FACS buffer to each tube.
Centrifuge tubes of cells at 250 x g at 4 °C for 10 min.
Decant supernatant and gently resuspend cell pellet in residual fluid by raking the tubes across the top of a test tube rack.
In order to prevent non-specific binding of the monoclonal antibodies, add 10 μL of blocking buffer (prepared in 4.2) to each tube of cells. Vortex tubes and incubate on ice for 30 min.
Add appropriate volume of diluted monoclonal antibodies to each tube and incubate in the dark on ice for 15 min. Tubes that contain samples to be analyzed should receive the combination of antibodies of the polycromatic panel (previous optimization of combination of antibodies should be performed by individual labs). Additional control groups should be included to correctly set the acquisition parameters on the flow cytometer, including a tube of cells that were not labeled, and compensation tubes that correspond to labeled cells using each single color separately. To correctly set the gates for flow cytometric analysis, the Fluorescence Minus One (FMO) controls should be included during acquisition. FMO controls are cells labeled with all the fluorochrome-conjugated antibodies that are used on the panel except one that is replaced by its respective isotype control. Positive and negative populations (i.e., separate aliquots of cells treated with a positive control stimulant and medium alone) should also be included in FMO and compensation control tubes.
Add 2 mL of FACS buffer to each tube, vortex and centrifuge at 250 x g at 4 °C for 10 min.
Decant supernatant and resuspend cell pellet in residual fluid by raking the tubes across the top of a test tube rack.
If the antibody panel consists of an antibody not directly conjugated to a fluorochrome but is instead conjugated to biotin, a fluorochrome-conjugated streptavidin is needed to visualize the surface marker. In this step, add an appropriate volume of the diluted fluorochrome-conjugated streptavidin and incubate in the dark for 15 min on ice. As in 4.2, the streptavidin will be diluted to a concentration recommended by the manufacturer or to one optimized by the user. This step is only needed for panels containing biotinylated primary antibodies.
Perform a wash step as described in 4.10.
Dilute an appropriate volume of BD stabilizing fixative 1:3 in deionized water. Add 100 μL of diluted fixative to each tube while gently vortexing to prevent clumping of cells. Labeled and fixed cells can be stably stored in the dark at 4 °C for approximately one week prior to acquisition on a flow cytometer.
5. Representative Data
Nanoparticles fabricated in step 3.1 had an average diameter of 163 ± 24 nm and a morphology consistent with that obtained in previous studies5,16. Nanoparticles in solution prior to and after sonication are shown in Figure 1 to demonstrate the need for sonication to ensure adequate dispersal of the particles. Flow cytometric analysis of harvested AMɸs obtained via lung lavage is shown in Figure 2. Labeling cells with a combination of anti-mouse CD11b and anti-mouse F4/80 antibodies as well as the corresponding FMO controls allows for establishing background labeling, identifying AMɸs and gating for further analysis. Treatment of alveolar macrophages with polyanhydride nanoparticles enhances activation as shown by the increased mean fluorescence intensity of MHC II, CD40, CD86, and CIRE (Figure 3).
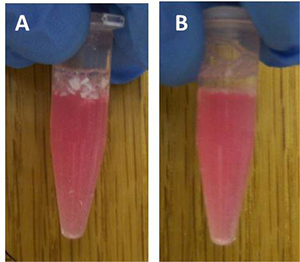 Figure 1. 50:50 CPTEG:CPH nanoparticles prior to (A) and after (B) 30 s of sonication.
Figure 1. 50:50 CPTEG:CPH nanoparticles prior to (A) and after (B) 30 s of sonication.
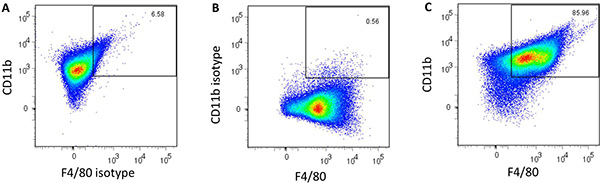 Figure 2. Flow cytometric analysis of harvested alveolar macrophages labeled with (A) Alexa Fluor 700 anti-CD11b and the PE-Cy7 FMO Control, (B) Alexa Fluor 700 FMO Control and the PE-Cy7 anti-F4/80 and (C) Alexa Fluor 700 anti-CD11b and PE-Cy7 anti-F4/80. The number in the top right corner represents the percentage of double-positive cells.
Figure 2. Flow cytometric analysis of harvested alveolar macrophages labeled with (A) Alexa Fluor 700 anti-CD11b and the PE-Cy7 FMO Control, (B) Alexa Fluor 700 FMO Control and the PE-Cy7 anti-F4/80 and (C) Alexa Fluor 700 anti-CD11b and PE-Cy7 anti-F4/80. The number in the top right corner represents the percentage of double-positive cells.
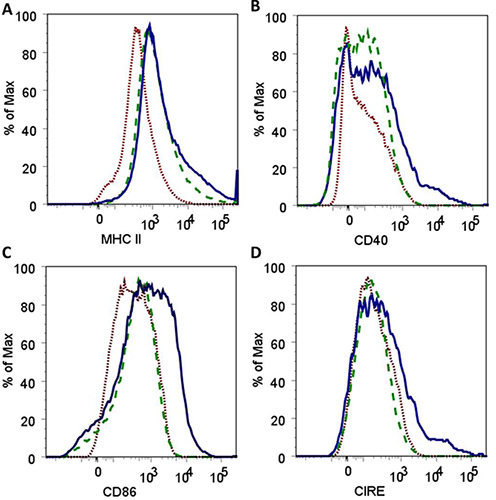 Figure 3. Histograms demonstrating an increase in the fluorescence intensity for surface expression of MHC II (A), CD40 (B), CD86 (C) and CIRE (D) after co-culture of alveolar macrophages with 50:50 CPTEG:CPH polyanhydride nanoparticles for 48 hr. Histograms depict results for AMɸ labeled as FMO controls (
Figure 3. Histograms demonstrating an increase in the fluorescence intensity for surface expression of MHC II (A), CD40 (B), CD86 (C) and CIRE (D) after co-culture of alveolar macrophages with 50:50 CPTEG:CPH polyanhydride nanoparticles for 48 hr. Histograms depict results for AMɸ labeled as FMO controls (![]() ), untreated AMɸ labeled with antibodies against all cell surface markers (
), untreated AMɸ labeled with antibodies against all cell surface markers (![]() ) and AMɸ cultured with nanoparticles and labeled with antibodies against all cell surface markers (
) and AMɸ cultured with nanoparticles and labeled with antibodies against all cell surface markers (![]() ).
).
Discussion
Polyanhydride nanoparticle vaccine platforms have shown efficacy when administered intranasally in single dose regimens5. Measuring the activation of the resident phagocytic cell populations in the lungs induced by this vaccine delivery platform permits evaluation of its potential capability to ultimately promote adaptive immune responses.
Specifically, harvesting alveolar macrophages from lung lavage fluid and treating them with different formulations of the nanoparticles provides insights into the abilities of different particle chemistries to activate macrophages, leading to antigen presentation6,8. In addition, these in vitro studies are useful for assessing the capacity of these particulate adjuvant formulations to activate alveolar macrophages prior to embarking on larger and more complex in vivo studies. Experiments should always contain a positive control treatment for surface marker up-regulation, such as LPS, an agonist of Toll-like receptor 4. Care should be taken in planning experiments prior to harvesting alveolar macrophages as this protocol may not produce adequate numbers of cells needed for multiple treatments in one experiment. Lung lavages may therefore need to be performed on multiple mice to ensure adequate cell numbers (~ 5.0 x 105 cells per mouse) for larger experiments with more treatment groups. Lung lavage techniques have also been reported for other species, and the amount of fluid utilized is proportional to the species being studied (i.e., mouse lung capacity is 1 mL, rat lung capacity is 10 mL and human lung capacity is 6 L17.
Polyanhydride nanoparticles are formulated and stored in a dry powder form to prevent premature surface erosion. Because of static interactions that result in clumping of the particles, sonication is necessary before addition to cell cultures. This step allows for uniform distribution and leads to more reproducible results. The quantification of cell surface markers on alveolar macrophages using flow cytometry is complicated by strong autofluorescence signals. This obstacle can be overcome by optimizing FACS antibody concentrations, polychromatic fluorochrome combinations, and flow cytometry capture parameters (i.e., use of compensation controls). FMO controls allow the change of one fluorescent parameter at a time, are useful for setting gates for each antibody and enable correct quantification of cell surface marker expression. Differences in the strains of mice should also be considered in the selection of antibodies, particularly haplotype specificity of MHC II.
It is important to have standardized methods to determine activation of antigen presenting cells in the lung, as this will greatly facilitate comparisons of novel biomaterials between different laboratories and institutions. Generating consistent populations of alveolar macrophages through the methods presented here, provide optimal conditions for obtaining useful data regarding different formulations of nanoparticles and their immune enhancing potential.
Disclosures
No conflicts of interest declared.
Acknowledgments
The authors would like to thank the U.S. Army Medical Research and Materiel Command (Grant Numbers W81XWH-09-1-0386 and W81XWH-10-1-0806) for financial support and Dr. Shawn Rigby from the Iowa State University Flow Cytometry Facility for his expert technical assistance.
References
- Mallapragada SK, Narasimhan B. Immunomodulatory biomaterials. Int. J. Pharm. 2008;364:265–271. doi: 10.1016/j.ijpharm.2008.06.030. [DOI] [PubMed] [Google Scholar]
- Torres MP, Vogel BM, Narasimhan B, Mallapragada SK. Synthesis and characterization of novel polyanhydrides with tailored erosion mechanisms. J. Biomed. Mater. Res. A. 2006;76:102–110. doi: 10.1002/jbm.a.30510. [DOI] [PubMed] [Google Scholar]
- Lopac SK, Torres MP, Wilson-Welder JH, Wannemuehler MJ, Narasimhan B. Effect of polymer chemistry and fabrication method on protein release and stability from polyanhydride microspheres. Journal of Biomedical Materials Research Part B: Applied Biomaterials. 2009;91:938–947. doi: 10.1002/jbm.b.31478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres MP, Determan AS, Anderson GL, Mallapragada SK, Narasimhan B. Amphiphilic polyanhydrides for protein stabilization and release. Biomaterials. 2007;28:108–116. doi: 10.1016/j.biomaterials.2006.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulery BD. Design of a Protective Single-Dose Intranasal Nanoparticle-Based Vaccine Platform for Respiratory Infectious Diseases. PLoS ONE. 2011;6:e17642. doi: 10.1371/journal.pone.0017642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen LK, Xue L, Wannemuehler MJ, Rajan K, Narasimhan B. The simultaneous effect of polymer chemistry and device geometry on the in vitro activation of murine dendritic cells. Biomaterials. 2009;30:5131–5142. doi: 10.1016/j.biomaterials.2009.05.069. [DOI] [PubMed] [Google Scholar]
- Petersen LK. Activation of innate immune responses in a pathogen-mimicking manner by amphiphilic polyanhydride nanoparticle adjuvants. Biomaterials. 2011;32:6815–6822. doi: 10.1016/j.biomaterials.2011.05.063. [DOI] [PubMed] [Google Scholar]
- Torres MP. Polyanhydride microparticles enhance dendritic cell antigen presentation and activation. Acta. Biomater. 2011;7:2857–2864. doi: 10.1016/j.actbio.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby AC, Coles MC, Kaye PM. Alveolar macrophages transport pathogens to lung draining lymph nodes. J. Immunol. 2009;183:1983–1989. doi: 10.4049/jimmunol.0901089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barletta KE. Leukocyte compartments in the mouse lung: Distinguishing between marginated, interstitial, and alveolar cells in response to injury. J. Immunol. Methods. 2011. [DOI] [PMC free article] [PubMed]
- Rubins JB. Alveolar macrophages: wielding the double-edged sword of inflammation. Am. J. Respir. Crit. Care Med. 2003;167:103–104. doi: 10.1164/rccm.2210007. [DOI] [PubMed] [Google Scholar]
- Sun K, Gan Y, Metzger DW. Analysis of Murine Genetic Predisposition to Pneumococcal Infection Reveals a Critical Role of Alveolar Macrophages in Maintaining the Sterility of the Lower Respiratory Tract. Infect. Immun. 2011;79:1842–1847. doi: 10.1128/IAI.01143-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto K. Alveolar Macrophages that Phagocytose Apoptotic Neutrophils Produce Hepatocyte Growth Factor during Bacterial Pneumonia in Mice. Am. J. Respir. Cell Mol. Biol. 2001;24:608–615. doi: 10.1165/ajrcmb.24.5.4292. [DOI] [PubMed] [Google Scholar]
- Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- Ulery BD. Polymer chemistry influences monocytic uptake of polyanhydride nanospheres. Pharm. Res. 2009;26:683–690. doi: 10.1007/s11095-008-9760-7. [DOI] [PubMed] [Google Scholar]
- Petersen LK, Sackett CK, Narasimhan B. High-throughput analysis of protein stability in polyanhydride nanoparticles. Acta Biomater. 2010;6:3873–3881. doi: 10.1016/j.actbio.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Irvin CG, Bates JH. Measuring the lung function in the mouse: the challenge of size. Respir. Res. 2003;4:4. doi: 10.1186/rr199. [DOI] [PMC free article] [PubMed] [Google Scholar]