

Abstract
Nanoparticulate systems have emerged as valuable tools in vaccine delivery through their ability to efficiently deliver cargo, including proteins, to antigen presenting cells1-5. Internalization of nanoparticles (NP) by antigen presenting cells is a critical step in generating an effective immune response to the encapsulated antigen. To determine how changes in nanoparticle formulation impact function, we sought to develop a high throughput, quantitative experimental protocol that was compatible with detecting internalized nanoparticles as well as bacteria. To date, two independent techniques, microscopy and flow cytometry, have been the methods used to study the phagocytosis of nanoparticles. The high throughput nature of flow cytometry generates robust statistical data. However, due to low resolution, it fails to accurately quantify internalized versus cell bound nanoparticles. Microscopy generates images with high spatial resolution; however, it is time consuming and involves small sample sizes6-8. Multi-spectral imaging flow cytometry (MIFC) is a new technology that incorporates aspects of both microscopy and flow cytometry that performs multi-color spectral fluorescence and bright field imaging simultaneously through a laminar core. This capability provides an accurate analysis of fluorescent signal intensities and spatial relationships between different structures and cellular features at high speed.
Herein, we describe a method utilizing MIFC to characterize the cell populations that have internalized polyanhydride nanoparticles or Salmonella enterica serovar Typhimurium. We also describe the preparation of nanoparticle suspensions, cell labeling, acquisition on an ImageStreamX system and analysis of the data using the IDEAS application. We also demonstrate the application of a technique that can be used to differentiate the internalization pathways for nanoparticles and bacteria by using cytochalasin-D as an inhibitor of actin-mediated phagocytosis.
Keywords: Bioengineering, Issue 64, Microbiology, ImageStream, phagocytosis, nanoparticles, pathogen, bacteria, Salmonella, imaging, multi-spectral imaging, flow cytometry
Protocol
1. RAW 264.7 Cell Culture
Harvest RAW 264.7 cells from their flasks when they reach confluency by scraping them gently with a cell scraper. Count and plate them into a 24-well cell culture dish at a density of 5 x 105 cells/well in 0.5 mL complete Dulbecco's Modified Eagle Medium (cDMEM; 10% heat-inactivated fetal bovine serum (FBS), 2 mM Glutamax, and 10 mM HEPES) and incubate overnight at 37 °C in a 5% CO2 incubator.
2. Pathogenic Salmonella enterica Serovar Typhimurium 14028 Transformation and Culture
Introduce recombinant plasmids expressing green fluorescent protein (GFP) to S. Typhimurium by electroporation. First, prepare culture of Salmonella enterica serovar Typhimurium (ATCC 14028) in LB broth and incubate overnight at 37 °C with aeration.
Next day, pellet 1 mL of bacteria at maximum speed in a microcentrifuge. Wash the cells four times with 1 mL of cold MOPS/glycerol solution (1 mM 3-(N-morpholino)propanesulfonic acid (MOPS) buffer in 20% glycerol) by gently resuspending the pellet with a pipette between each wash9. After the final wash, resuspend the pellet in 50 μL of MOPS/glycerol into which ~0.5 μg of pAKgfp1 or pAKgfplux1 plasmid DNA has been added10.
Transfer the cell suspension to a pre-chilled 1 mm gap electroporation cuvette and electroporate using the settings: 1800 V, 200 W, 25 mF. Immediately after pulse delivery, add 1 mL of LB broth and transfer the cell suspension to a new 15 mL tube. Allow the cells to recover at 37 °C with aeration. After one hour, concentrate the cells by centrifugation and resuspend in ~100 μL of remaining supernatant.
Finally, plate the cells on LB plates containing 100 μg/mL ampicillin and incubate overnight at 37 °C. Single colonies are purified by restreaking with antibiotic selection and expression of GFP is confirmed by illumination with a standard fluorescein isothiocyanate (FITC) filter set.
Inoculate 10 mL of LB broth containing 25 μg/mL ampicillin (or appropriate antibiotic) with a loopful of Salmonella that has been transformed to stably express GFP.
Grow the bacteria overnight at 37 °C.
Dilute Salmonella 1:10 into a fresh culture tube containing 10 mL LB supplemented with ampicillin and incubate for 5 h at 37 °C.
Measure the absorbance of the culture at 600 nm and calculate the concentration of the Salmonella per mL using a previously establish growth curve.
Dilute the Salmonella in cDMEM (0.5 mL/well) to obtain a multiplicity of infection (MOI) of 100 per RAW 264.7 cell.
3. Preparation of Nanoparticle Suspension
Fabricate 1% FITC-loaded nanoparticles as described previously11. Briefly, particles are fabricated by polyanhydride anti-solvent nanoencapsulation, in which the polymer is dissolved in methylene chloride (4 °C at a concentration of 25 mg/mL) and precipitated in pentane (-30 °C at a ratio 1:200 methylene chloride: pentane). Recover nanoparticles by vacuum filtration. After evaporating any residual solvent, weigh the dried polyanhydride nanoparticles using sterilized weigh paper.
Add 5 mg of nanoparticles to 0.5 mL of cold phosphate buffered saline (PBS, calcium and magnesium free, pH 7.4) in a 1.5 mL microcentrifuge tube and keep it on ice until the nanoparticles are added to the RAW 264.7 cells.
Sonicate the nanoparticle suspension (while keeping on ice) using an ultrasonic liquid processor fitted with a microtip for approximately 25 s at 4 to 6 joules.
4. Phagocytosis Assay
Pretreat a subset of RAW 264.7 cells with 5 μg/mL cytochalasin-D 1 hr prior to introduction of NP or Salmonella by aspirating medium and replacing with fresh cDMEM supplemented with the inhibitor. Return the cultures to 37 °C incubator.
After the 1 h incubation with the inhibitor, remove the plates from the incubator, vortex the nanoparticle suspension, and add 10 μL to the appropriate wells.
Vortex the Salmonella and infect the RAW 264.7 cells with an MOI of 100 by adding the bacteria to the appropriate wells.
Tap the plate few times to mix and incubate at 37 °C or 4 °C (control) for 45 min.
Remove the plates from the incubator or refrigerator and place on ice. Wash the cells twice with ice cold PBS (without Ca2+ and Mg2+) by aspirating and discarding the old medium to remove unbound particles, Salmonella, and dead or detached RAW 264.7 cells.
To harvest the RAW 264.7 cells after the second wash, add 250 μL of ice-cold PBS and gently scrape the wells.
Pipet the harvested cells into clear-view snap cap microcentrifuge tubes and keep them on ice.
Wash the cells by adding 1 mL of cold wash buffer (2% heat inactivated FBS, 0.1% sodium azide in PBS) and centrifuge at 250 x g for 10 min at 4 °C.
Discard the supernatant and remove residual buffer by tapping the inverted microcentrifuge tube on paper towel. Resuspend the cell pellet by gently raking the microcentrifuge tube across a test tube rack.
Fix the RAW 264.7 cells by adding 100 μL of 4% paraformaldehyde (PFA) in PBS and allow the cells to stand for 15 min at room temperature (RT).
Wash the RAW 264.7 cells by adding 1 mL of Perm/Wash buffer (BD Biosciences) and centrifuge at 250 x g for 10 min at 4 °C.
Repeat step 4.9.
Stain the RAW 264.7 cells for actin by adding 100 μl of Perm/Wash buffer containing Alexa Fluor phalloidin 660 (AF 660; 1:150 dilution) for 15 min at RT. Repeat steps 4.11 and 4.12.
Resuspend RAW 264.7 cells in 50 μL of PBS containing 1% PFA and store them in the dark at 4 °C until acquisition.
Tips & notes:
Single color fluorescence samples should be prepared at this step to be used as compensation controls while setting up the ImageStreamX instrument. The compensation controls included in this experiment were: RAW 264.7 cells (not labeled for actin) incubated with nanoparticles; RAW 264.7 cells (not labeled for actin) incubated with Salmonella; actin only labeled RAW 264.7 cells.
For the inhibitor studies, it is important to include the vehicle control in which the inhibitor was dissolved as a control. In this experiment, cytochalasin-D was dissolved in 100% DMSO. We, therefore, incubated cells in cDMEM containing DMSO and then evaluated the internalization of nanoparticles or Salmonella. We did not detect any significant difference between the medium group and DMSO control (data not shown).
The sample preparation guide available on the Amnis website (https://www.amnis.com/images/1__Sample_Prep_Guide_3L_-_0418081.pdf) is a good resource for checking the compatibility of fluorophores with the ImageStreamX. To access this document, users must first register (free-of-charge) with Amis.
5. Sample Acquisition on the ImageStreamX
Power up ImageStreamX and launch INSPIRE.
Initialize Fluidics. At the end of this script SpeedBeads should be running.
In the file menu, choose Load Default Template.
In the Image Gallery view menu, select ALL and Press Run Setup to start imaging the beads.
Adjust Core Tracking to center images laterally (if necessary).
Select the Brightfield (BF) channel and click Set Intensity.
Wait until the Flow Speed CV is consistently less than 0.2%.
Calibrate the instrument daily. In the ASSIST tab, click Start All to run calibrations and tests and verify that all have passed.
Press Flush Lock and Load (FLL) to load the first sample. Load the brightest sample in the experiment that fluoresces with each fluorochrome used. It's critical that you run this sample first to establish the instrument settings and then DO NOT change them for the entire experiment.
Turn on each laser used in the experiment and set the Laser Power so each fluorochrome has max pixel values between 100 and 4000 counts, as measured in the scatter plots.
Set Cell Classification criteria, to eliminate collection of unwanted objects. For collecting data from only the cells, select the Area Lower Limit in BF channel to 50 μm. Objects with area less than 50 μm will be considered debris and will not be acquired. Select channels to be collected.
Enter the File Name, Destination Folder, set Sequence # to 1 and the Number of Events to acquire.
Click Run Acquire to collect and save the first experiment data file.
Click FLL and run the next experimental sample. Repeat until all experimental samples have been collected.
Click Comp Settings (turns off brightfield and scatter laser and enables collection of all channels) and collect 500 positive cells from each single stained sample for each fluorophore in the experiment to develop a compensation matrix.
To summarize, samples can be run in the following order:
Brightest sample first.
Remaining test samples.
Compensation controls containing single color fluorescence.
Tips & notes:
An INSPIRE template can be saved and reloaded to set the instrument settings. Once templates are saved, the autosampler can be used for unattended operation to run the experimental samples with one template and the compensation controls with a template for each single color control.
The ImageStreamX configuration for these experiments: 488 and 658 nm excitation lasers; 785 nm scatter laser; 40X (0.75 NA) objective, 6 channel system with a standard ISX filter stack. The autosampler option was used to allow unattended acquisition of the samples.
The following settings were used for the experiment. Nanoparticles were imaged with the 488 nm laser set to 10mW, the 658 nm laser set to 50 mW, the 785 nm laser set to 2 mW and BF in channel 1. Salmonella infected cells were imaged with the 488 nm laser set to 20 mW, 658 nm laser set to 50 mW, the 785 nm laser set to 2 mW, and BF in channel 1. Compensation controls were collected in the absence of BF and 785. At least 5000 events (i.e., cells) were imaged for each of the test samples.
6. Image Analysis
Launch IDEAS and double-click on the Internalization wizard and load one of the test sample .rif files.
Create a compensation matrix by clicking on 'New Matrix' in step 2. The compensation wizard is launched. Add files for the single color controls in the experiment. Click next through the wizard following directions until the compensation matrix file is saved and loaded into the box in step 2 of the Internalization wizard.
Click Next and follow directions until a .daf file is generated.
Set image display properties by selecting the image channels used during acquisition. Click on Ch 2 (FITC) and Ch 5 (AF 660). (BF and SSC are default selected).
Select the image channel for making the cell boundary (CH01) and the channel in which nanoparticles or bacteria were collected (CH02).
A scatter plot of Brightfield Area versus Brightfield Aspect Ratio of all of the cells is generated. Define the single cell population by clicking on individual dots and gating around singlets. Single cells have an aspect ratio of around 1 and doublets around 0.5 (Figure 1A).
A histogram of the Brightfield Gradient Root Mean Square (RMS) of the brightfield image is generated and the population view in the image gallery is set to selected bin (Figure 1B). Click on the bins to determine where cells in best focus begin and draw a line region to gate focused cells. The higher the Gradient RMS, the better focused. Skip the next step unless there are other stains you want to gate on.
A new scatter plot of Intensity of Channel 2 on the x-axis versus Max Pixel of Ch2 on the y-axis is generated (Figure 1C). Click on the dots and view the images to help you draw the region around the cells that are positive for nanoparticles or bacteria.
A histogram of the Internalization feature is generated with a region that begins at 0, which should be adjusted by observing images (Figure 1D). The Internalization feature is a ratio of the intensity inside the cell to the intensity of the whole cell. It is scaled such that at a value of 0 about half of the intensity is inside. The wizard has created a region to designate the inside by making a mask that has used the cell image input from step 5 to find the cell surface and eroded this by 4 pixels. Note that this mask can be manually adjusted for different cell types when necessary. In this experiment, we manually adjusted the feature by creating an Object mask on the brightfield image first and eroded this by 4 pixels. The Internalization feature was then calculated based on this 4 pixel eroded Object mask. This feature allows us to distinguish internalized particles and bacteria, which have the majority of their fluorescence signal within the mask boundary, from surface bound particles and bacteria, which have the majority of their fluorescence signal outside the mask boundary (Figure 2).
Create a new histogram with the new Internalization feature based on the eroded Object mask. Draw a region to gate on internalized cells by viewing the imagery in selected bin mode. In our experiments, we set this gate at 0.3. Cells with a score lower than 0.3 were considered surface bound particle positive cells.
Finally, to eliminate the cells with background labeling and to identify specific internalized nanoparticles or bacteria, the IDEAS Spot Count feature was used. Spot count is a feature that counts the number of connected components or small masks in an image. The mask functions, Spot, Peak and Intensity were used to define the spots. The spot mask finds the bright details in an image that have a user specified radius and threshold above the local background, the mask is refined by breaking apart high intensities into individual spots using the Peak function and then spots above an intensity of 200 counts were included. See the Amnis spot masking guide for further information (Figure 1E). A statistics report template was generated by including various features in the Reports menu.
This file was saved as template file to be used for batch analysis of all experimental files.
In the IDEAS software, click Tools→ Batch Data Files and input all the .rif files. Add the compensation matrix file (.ctm) and the template file (.ast) in the corresponding sections. Submit the batch for processing. After the processing step, all the .rif files are analyzed and .daf files are generated for each of the individual raw files. A final report file is generated with statistics for all the samples.
Tips & notes:
Image analysis was performed using the IDEAS software version 4.0 and the Internalization wizard with some modifications. The wizard is self-instructive and the ouptput .daf file can be generated by following the instructions of the wizard.
7. Representative Results
The representative images in Figure 2 demonstrate that MIFC can be used to successfully distinguish between internalized (left panel) versus surface bound (right panel) NP (Figure 2A) or Salmonella (Figure 2B). Internalization of NP and Salmonella were reduced by both inhibiting actin and lowering the temperature to 4 °C (Fig 3A). The percentage of cells positive for surface bound NP increased from approximately 8% at 37 °C to greater than 35% after either cytochalasin-D or 4 °C treatment (Fig 3B). In contrast, the percentage of cells with surface bound Salmonella was reduced from 35% to 15% following cytochalasin-D treatment. Incubation of RAW 264.7 cells with Salmonella at 4 °C decreased internalization without an apparent increase in the amount of surface bound bacteria as compared to the 37 °C control. Together, these data demonstrate that Salmonella and NP are internalized by a similar cellular process that requires actin and is temperature dependent. Moreover, the data indicate that sustained attachment of Salmonella to macrophages requires actin polymerization.
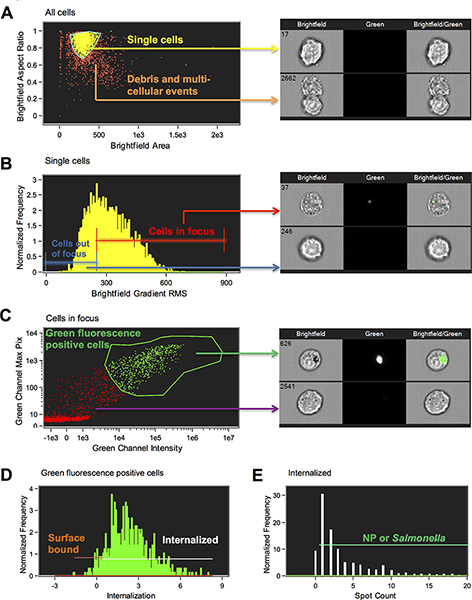 Figure 1. Schematic of the gating strategy utilized to determine internalized versus surface bound nanoparticles and Salmonella. (A) To limit the analysis to single cells, it is important to eliminate debris and multi-cellular events. Single cells and doublets were separated from multicellular aggregates using the IDEAS features area and aspect ratio of the brightfield image (M01). Area is the size of the image in square microns and aspect ratio is the minor axis divided by the major axis and therefore a measure of circularity (a perfect circle will have an aspect ratio of 1; doublets typically have aspect ratios of around 0.5 and multicellular aggregates are typically less than 0.5). A region was drawn to gate on single cell events (Step 6.6). (B) To gate on cells in-focus, the IDEAS feature Gradient RMS of the brightfield image is plotted in a histogram. The Gradient RMS feature measures the sharpness quality of an image by detecting changes of pixel values in the image. A higher Gradient RMS value indicates a more focused image (Step 6.7). (C) Green fluorescence positive cells were selected by gating on the cells with high Max Pixel values and Intensity in the green fluorescence channel (Step 6.8). (D) Cells with internalized NP or Salmonella were selected by choosing the cell population with an internalization score equal to or greater than 0.3. Cells receiving a score less than 0.3 were considered to be surface bound (Step 6.9). (E) Cells in the "internalized" gate were further characterized based on the number of spots (NP or STM) because there were some cells with background staining that were counted as internalized but had a spot value of zero (Step 6.11). Representative images are presented for each gate. Click here to view larger figure.
Figure 1. Schematic of the gating strategy utilized to determine internalized versus surface bound nanoparticles and Salmonella. (A) To limit the analysis to single cells, it is important to eliminate debris and multi-cellular events. Single cells and doublets were separated from multicellular aggregates using the IDEAS features area and aspect ratio of the brightfield image (M01). Area is the size of the image in square microns and aspect ratio is the minor axis divided by the major axis and therefore a measure of circularity (a perfect circle will have an aspect ratio of 1; doublets typically have aspect ratios of around 0.5 and multicellular aggregates are typically less than 0.5). A region was drawn to gate on single cell events (Step 6.6). (B) To gate on cells in-focus, the IDEAS feature Gradient RMS of the brightfield image is plotted in a histogram. The Gradient RMS feature measures the sharpness quality of an image by detecting changes of pixel values in the image. A higher Gradient RMS value indicates a more focused image (Step 6.7). (C) Green fluorescence positive cells were selected by gating on the cells with high Max Pixel values and Intensity in the green fluorescence channel (Step 6.8). (D) Cells with internalized NP or Salmonella were selected by choosing the cell population with an internalization score equal to or greater than 0.3. Cells receiving a score less than 0.3 were considered to be surface bound (Step 6.9). (E) Cells in the "internalized" gate were further characterized based on the number of spots (NP or STM) because there were some cells with background staining that were counted as internalized but had a spot value of zero (Step 6.11). Representative images are presented for each gate. Click here to view larger figure.
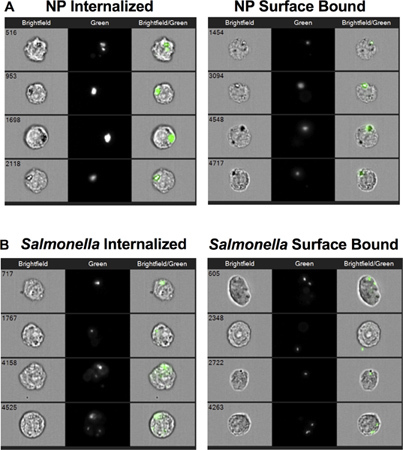 Figure 2. Cell images of internalized and surface bound nanoparticles (NP) or Salmonella. (A) Representative images of RAW 264.7 cells that internalized NP (left panel) and cells to which NP were bound to their surface but not internalized (right panel). (B) Representative images of RAW 264.7 cells that internalized Salmonella (left panel) and cells to which Salmonella were bound to their surface but not internalized (right panel).
Figure 2. Cell images of internalized and surface bound nanoparticles (NP) or Salmonella. (A) Representative images of RAW 264.7 cells that internalized NP (left panel) and cells to which NP were bound to their surface but not internalized (right panel). (B) Representative images of RAW 264.7 cells that internalized Salmonella (left panel) and cells to which Salmonella were bound to their surface but not internalized (right panel).
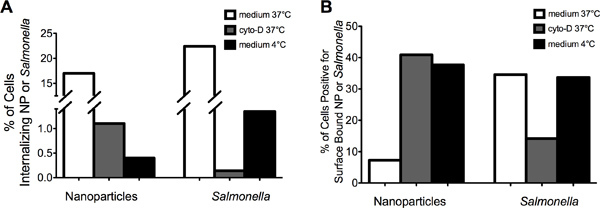 Figure 3. Cytochalasin-D treatment of cells inhibited internalization of NP and Salmonella. (A) Pretreatment of RAW 264.7 cells with cytochalasin-Dor incubation at 4 °C reduced the incidence of NP or Salmonella internalization as compared to RAW 264.7 cells incubated at 37 °C in medium. RAW 264.7 cells incubated in medium containing DMSO (i.e., vehicle control) showed similar internalization levels for NP and Salmonella compared to medium alone (data not shown). (B) Pretreatment of RAW 264.7 cells with cytochalasin-D increased the percent of cells with surface bound NP while decreasing the percent of cells with surface bound Salmonella.
Figure 3. Cytochalasin-D treatment of cells inhibited internalization of NP and Salmonella. (A) Pretreatment of RAW 264.7 cells with cytochalasin-Dor incubation at 4 °C reduced the incidence of NP or Salmonella internalization as compared to RAW 264.7 cells incubated at 37 °C in medium. RAW 264.7 cells incubated in medium containing DMSO (i.e., vehicle control) showed similar internalization levels for NP and Salmonella compared to medium alone (data not shown). (B) Pretreatment of RAW 264.7 cells with cytochalasin-D increased the percent of cells with surface bound NP while decreasing the percent of cells with surface bound Salmonella.
Discussion
Studies have shown that biodegradable nanoparticles based on poly(lactic-co-glycolic acid (PLGA) or polyanhydrides can be used to deliver encapsulated antigens or drugs to target cells. Uptake of these nanoparticles by phagocytic cells is important for their effectiveness, thus making quantitative analysis of internalization critical in designing novel nanoparticle delivery systems. By using this method, differential uptake of nanoparticles by various cell types can be analyzed with ease. To date, conventional microscopy and flow cytometry have been used for quantifying particle uptake; however, their respective limitations with high throughput and resolution call for alternative approaches to study internalization. In this article, we perform MIFC analysis to characterize and compare how the biological process of phagocytosis differs between two types of targets- a bacterial pathogen and synthetic nanoparticles.
The MIFC phagocytosis assay was used to delineate differences in the mechanism underlying the uptake of Salmonella compared to nanoparticles. It should be noted that inhibitors can be cytotoxic depending on their incubation time and concentration; hence a prior cytotoxicity profiling (i.e., exposure time and concentration) is necessary before their use13,14. Depending on their chemical formulation, nanoparticles can have a glass transition temperature (Tg = 13 °C) below RT, consequently, they form aggregates at RT15. To overcome the aggregation, we sonicated the particles and kept them on ice prior to addition to the cell cultures. Kinetic studies of nanoparticle uptake provide important information on the efficiency of these particles to deliver the encapsulated payload to antigen presenting cells. Care must be taken to keep cells on ice at the end of a given time point to inhibit further cellular processes. To this end, we have noted variability in cellular uptake when cells were not placed on ice. One limitation of the method described above is that the experiment is performed on adherent cells that require scraping techniques to harvest the phagocytic cells. This procedure could result in cell death, thus introducing some error in the data analysis. The procedure we describe herein could also be performed using cells that grow in suspension.
The incubation of macrophages with reactive dyes or immunolabeling reagents was optimized by reducing the amount of residual buffer present following fixation. The presence of residual buffer creates a dilution effect and can produce sample to sample variation among the multiple samples, thereby resulting in inconsistent mean fluorescence intensity values. Sample concentration (i.e., cells/mL) is also an important factor to consider during the acquisition step, as dilute samples result in longer run times. For a multi-parametric comparison between different experimental samples, it is important to keep the laser intensity constant.
Disclosures
Sherree L. Friend is employed by Amnis Corporation, which manufactures the ImageStreamX system.
Acknowledgments
The authors would like to thank the ONR-MURI Award (NN00014-06-1-1176) and the U.S. Army Medical Research and Materiel Command (Grant Numbers W81XWH-09-1-0386 and W81XWH-10-1-0806) for financial support.
References
- Ulery BD, Kumar D, Ramer-Tait AE, Metzger DW, Wannemuehler MJ, Narasimhan B. Design of a protective single-dose intranasal nanoparticle-based vaccine platform for respiratory infectious diseases. PLoS One. 2011;6:e17642. doi: 10.1371/journal.pone.0017642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasturi SP, Skountzou I, Albrecht RA, Koutsonanos D, Hua T, Nakaya HI, Ravindran R, Stewart S, Alam M, Kwissa M, Villinger F, Murthy N, Steel J, Jacob J, Hogan RJ, García-Sastre A, Compans R, Pulendran B. Programming the magnitude and persistence of antibody responses with innate immunity. Nature. 2011;470:543–547. doi: 10.1038/nature09737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice-Ficht AC, Arenas-Gamboa AM, Kahl-McDonagh MM, Ficht TA. Polymeric particles in vaccine delivery. Curr. Opin. Microbiol. 2010;13:106–112. doi: 10.1016/j.mib.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Jain JP, Chitkara D, Kumar N. Polyanhydrides as localized drug delivery carrier: an update. Expert. Opin. Drug. Deliv. 2008;5:889–907. doi: 10.1517/17425247.5.8.889. [DOI] [PubMed] [Google Scholar]
- Pfeifer BA, Burdick JA, Little SR, Langer R. Poly(ester-anhydride):poly(beta-amino ester) micro- and nanospheres: DNA encapsulation and cellular transfection. Int. J. Pharm. 2005;304:210–219. doi: 10.1016/j.ijpharm.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Ahmed F, Friend S, George TC, Barteneva N, Lieberman J. Numbers matter: quantitative and dynamic analysis of the formation of an immunological synapse using imaging flow cytometry. J. Immunol. Methods. 2009;347:79–86. doi: 10.1016/j.jim.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton MB, Winterbourn CC. Methods for quantifying phagocytosis and bacterial killing by human neutrophils. J. Immunol. Methods. 1999;232:15–22. doi: 10.1016/s0022-1759(99)00147-7. [DOI] [PubMed] [Google Scholar]
- Rieger AM, Hall BE, Barreda DR. Macrophage activation differentially modulates particle binding, phagocytosis and downstream antimicrobial mechanisms. Dev. Comp. Immunol. 2010;34:1144–1159. doi: 10.1016/j.dci.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Murphy KC, Campellone KG. Lambda Red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC. Mol. Biol. 2003;4:11. doi: 10.1186/1471-2199-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsi A, Lawrence ML. Broad host range fluorescence and bioluminescence expression vectors for Gram-negative bacteria. Plasmid. 2007;57:286–295. doi: 10.1016/j.plasmid.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Ulery BD, Phanse Y, Sinha A, Wannemuehler MJ, Narasimhan B, Bellaire BH. Polymer chemistry influences monocytic uptake of polyanhydride nanospheres. Pharm. Res. 2009;26:683–690. doi: 10.1007/s11095-008-9760-7. [DOI] [PubMed] [Google Scholar]
- Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009;78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- Vercauteren D, Vandenbroucke RE, Jones AT, Rejman J, Demeester J, De Smedt SC, Sanders NN, Braeckmans K. The use of inhibitors to study endocytic pathways of gene carriers: optimization and pitfalls. Mol. Ther. 2010;18:561–569. doi: 10.1038/mt.2009.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzio L, Marianecci C, Cinque B, Nazzarri M, Cimini AM, Cristiano L, Cifone MG, Alhaique F, Carafa M. pH-sensitive non-phospholipid vesicle and macrophage-like cells: binding, uptake and endocytotic pathway. Biochim. Biophys. Acta. 2008;1778:2749–2756. doi: 10.1016/j.bbamem.2008.07.029. [DOI] [PubMed] [Google Scholar]
- Torres MP, Vogel BM, Narasimhan B, Mallapragada SK. Synthesis and characterization of novel polyanhydrides with tailored erosion mechanisms. J. Biomed. Mater. Res. A. 2006;76:102–110. doi: 10.1002/jbm.a.30510. [DOI] [PubMed] [Google Scholar]