

Abstract
The steroid hydroxylases CYP17A1 (P450c17, 17-hydroxylase/17,20-lyase) and CYP21A2 (P450c21, 21-hydroxylase) catalyze progesterone hydroxylation at one or more sites within a 2 Å radius. We probed their hydrogen atom abstraction mechanisms and regiochemical plasticity with deuterium-labeled substrates: 17-[2H]-pregnenolone; 17-[2H]-, 16α-[2H]-, 21,21,21-[2H3]-, and 21-[2H]-progesterone; and 21,21,21-[2H3]-17-hydroxyprogesterone. Product distribution and formation rates with recombinant human P450-oxidoreductase and wild-type human CYP17A1 or mutation A105L (reduced progesterone 16α-hydroxylation) and wild-type human CYP21A2 or mutation V359A (substantial progesterone 16α-hydroxylation) were used to calculate intramolecular and intermolecular kinetic isotope effects (KIEs). The intramolecular KIEs for CYP17A1 and mutation A105L were 4.1 and 3.8, respectively, at H-17 and 2.9 and 5.1, respectively, at H-16α. Mutation A105L 21-hydroxylates progesterone (5% of products), and wild-type CYP17A1 also catalyzes a trace of 21-hydroxylation, which increases with 16α-[2H]- and 17-[2H]-progesterone. The intramolecular KIEs with CYP21A2 mutation V359A and progesterone were 6.2 and 3.8 at H-21 and H-16α, respectively. Wild-type CYP21A2 also forms a trace of 16α-hydroxyprogesterone, which increased with 21,21,21-[2H3]-progesterone substrate. Competitive intermolecular KIEs paralleled the intramolecular KIE values, with DV values of 1.4–5.1 and DV/K values of 1.8–5.1 for these reactions. CYP17A1 and CYP21A2 mutation V359A both 16α-hydroxylate 16α-[2H]-progesterone with 33–44% deuterium retention, indicating stereochemical inversion. We conclude that human CYP17A1 has progesterone 21-hydroxylase activity and human CYP21A2 has progesterone 16α-hydroxylase activity, both of which are enhanced with deuterated substrates. The transition states for C-H bond cleavage in these hydroxylation reactions are either significantly non-linear and/or asymmetric, and C-H bond breakage is partially rate-limiting for all reactions.
Steroid 21-hydroxylation is the biochemical reaction that led to the discovery of the cytochrome P450 enzymes (1). Studies over the subsequent decades revealed that, in human beings, six cytochromes P450 participate in steroid biosynthesis, three mitochondrial/type 1 (CYP11A1, CYP11B1, and CYP11B2) and three microsomal/type 2 (CYP17A1, CYP21A2, CYP19A1). Together with the hydroxysteroid dehydrogenases and 5α-reductases, this limited set of enzymes generates from the same cholesterol scaffold a remarkable repertoire of steroid hormones with diverse functional properties including androgens, estrogens, progestins, mineralocorticoids, and glucocorticoids (2). The biologic functions of these enzymes derive from their substrate specificity and regiochemical selectivity, delivering oxygenation chemistry to carbon atoms necessary to generate ligands for their cognate nuclear hormone receptors. Steroid 21-hydroxylation via CYP21A2, for example, is required for the biosynthesis of glucocorticoids and mineralocorticoids (Figure 1). Deficiency of CYP21A2 (P450c21) causes the most common form of congenital adrenal hyperplasia (CAH) (3), which occurs in 1:15,000 live births (4) and in an attenuated or nonclassic form 10–100 times more commonly. Conversely, CYP17A1 (P450c17) is required for androgen biosynthesis, and this enzyme is the target of ketoconazole and abiraterone acetate, drugs employed for the treatment of prostate cancer (5, 6). Because these enzymes are central to normal human physiology and relevant to common diseases, an understanding of their mechanisms and biochemistry is of considerable importance.
Figure 1.

Major steroid hydroxylase activities of human CYP17A1 and CYP21A2 with principal substrates. The 17,20-lyase reactions catalyzed by CYP17A1 are omitted for simplicity.
Although these steroidogenic P450s have been known and studied for many years, several unsolved mysteries about their catalytic mechanisms remain, despite the recent x-ray crystal structures of modified bovine CYP21A2 (7) and human CYP17A1 (8). First, CYP21A2 oxygenates a methyl group adjacent to other more easily oxidized carbon atoms. Second, CYP17A1 performs not only the 17α-hydroxylase reaction but also the 16α-hydroxylase reaction with progesterone as substrate (Figure 1) in a 3:1 ratio (9), and the small side chain of A105 allows 16α-hydroxylation (10). Furthermore, CYP17A1 performs the 17,20-lyase reaction, involving the oxidative cleavage of a carbon-carbon bond. Only a few P450 enzymes incorporate carbon-carbon cleavages in their physiologic functions, including the steroidogenic enzymes CYP11A1 (P450scc, the cholesterol side chain cleavage enzyme), CYP17A1, and CYP19A1 (P450aro, aromatase) as well as CYP51A1 (lanosterol demethylase) (2, 11). Common catalytic mechanisms or themes for these enzymes have not emerged from the literature, and debate continues for the mechanisms of individual reactions. The participation of cytochrome b5 in the 17,20-lyase reaction has been extensively documented (12–15) and physiologically validated by the description of patients with isolated 17,20-lyase deficiency due to CYB5 mutations (16, 17), yet the mechanism of this stimulation is not yet resolved (18). Finally, the steroidogenic P450s are very slow catalysts, with turnover numbers <10 min−1, compared to related members of the superfamily such as to CYP7A1 (cholesterol 7α-hydroxylase), with a turnover number of 200 min−1 (19), or the soluble bacterial enzymes P450cam and P450BM3, which catalyze thousands of turnovers per minute (20). Consequently, the fundamental assumptions regarding the catalytic cycle and rate-determining steps gleaned from prokaryotic P450 enzymes might apply differently to the steroid hydroxylases.
The available evidence supports a model in which the first chemical step for cytochrome P450 hydroxylations involving substrate is hydrogen atom abstraction from a C-H bond using a highly reactive oxygenated heme species resembling a ferryl oxene with radical (odd-electron) character (21). For several P450 enzymes, the C-H abstraction step has been studied in detail by measuring the kinetic isotope effects (KIEs) in order to determine the contribution of this step to the reaction rate relative to the other steps associated with hydroxylation (22–27). Depending on the specific cytochrome P450, the contribution of C-H bond cleavage to the overall rate varies, and it is not clear what properties of the proteins determine these kinetic features. Furthermore, for P450-catalyzed reactions yielding two or more products from a common E•S complex, intramolecular KIE experiments have been employed to deduce the contribution of C-H bond breakage to product partitioning (23, 25). For some P450 enzymes, anomalously large intramolecular KIEs >10 have been observed (22), suggesting that proton-coupled electron transfer or hydrogen atom tunneling rather than classical reaction mechanics best describes these reactions (28). Comprehensive KIE studies for CYP3A4 and CYP7A1 with steroid (26) and sterol (19) substrates, respectively, have appeared; however, no such studies have been reported for the human biosynthetic steroid hydroxylases. Therefore, we conducted a series of intramolecular and intermolecular KIE experiments to probe the mechanisms of human CYP17A1 and CYP21A2.
Experimental Procedures
General Methods
NMR spectra were obtained using Varian instruments at frequencies for 1H and 13C as specified in the experimental detail. Chemical shifts were referenced to the chloroform peak in the 1H NMR assigned at 7.26 ppm and in the 13C NMR assigned at 77.16 ppm. NMR spectra are provided in the Supporting Information. Reaction progress was determined either by TLC monitoring, or an aliquot was taken and analyzed by NMR. Pregnenolone was purchased from Waterstone Technology (St. Carmel, IN), and all other reagents and solvents were purchased from Sigma Aldrich (St. Louis, MO), Steraloids (Newport, RI), ThermoFisher Scientific (Pittsburgh, PA), or as specified. Oligonucleotides were obtained from Integrated DNA Technologies (Coralville, IA). Cholesterol oxidase was purchased from Sigma Aldrich (Brevibacterium sp., product C8868-100UN). Site-directed mutagenesis employed the primers for CYP17A1-A105L as reported (10) hCYP17A105L_S: 5′-TCA AAT GGC AAC TCT AGA CAT CCT GTC CAA CAA C-3′ and hCYP17A105L_AS: 5′-GAT GTC TAG AGT TGC CAT TTG AGG CCG CC-3′; primers for CYP21A2 V359A (29) were C21V359A_S: 5′-CCC GTT GCG CCC TTA GCC TTG-3′ and C21V359A_AS: 5′-AAG GCT AAG GGC GCA ACG GGC C-3′. Constructs were sequenced to assure accurate mutagenesis. Stock solutions of the deuterated substrates were prepared with the polar aprotic solvent acetonitrile to minimize exchange of labile deuterium atoms with protic solvents. Protein determinations used the Coomassie Plus Reagent (Pierce, Rockford, IL).
Steroid syntheses
16α-bromopregnenolone-3-acetate (2)
To a solution of tetrabutylammonium bromide (2.0 g, 6.2 mmol) and 16,17-dehydropregnenolone-3-acetate (1.21 g, 3.4 mmol) in CH2Cl2 (20 mL) was added concentrated sulfuric acid (1.0 mL). The reaction was stirred for 2 h, and the reaction mixture was directly loaded onto a silica gel column and purified (100% hexanes to 50% hexanes in ethyl acetate, v/v) to afford bromide 2 (0.5 g, 1.1 mmol, 32%). If the 1H NMR indicated starting material remaining (vinyl 16-proton), then the mixture of 16α-bromopregnenolone-3-acetate and 16,17-dehydropregnenolone-3-acetate could be subjected to the same reaction conditions to afford the 16α-bromopregnenolone-3-acetate 2. 1H NMR of 2 (400 MHz, CDCl3) δ 5.38 (broad s, 1H), 4.82 (apparent t, J = 9.0 Hz, 1H), 4.66-4.55 (m, 1H), 3.11 (d, J = 7.4 Hz, 1H), 2.33-2.30 (m, 2H), 2.18 (s, 3H), 2.15-2.10 (m, 1H), 2.03 (s, 3H), 2.04-1.85 (m, 4H), 1.80-1.42 (m, 7H), 1.24-1.05 (m, 2H), 1.01 (s, 3H), 0.61 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 206.0, 170.7, 139.8, 122.1, 75.6, 73.8, 54.7, 49.7, 46.4, 46.2, 38.6, 38.1, 37.7, 37.0, 36.7, 31.9, 31.6, 31.2, 27.8, 21.6, 20.8, 19.4, 13.7.
1H NMR of 5,6,16-tribromopregnenolone-3-acetate (400 MHz, CDCl3) δ 5.54-5.46 (m, 1H), 4.81-4.76 (m, 1H), 3.13 (d, J = 7.5 Hz, 1H), 2.32 (dd, J1 = 13.0 Hz, J2 = 4.9 Hz, 1H), 2.16 (s, 3H), 2.14-1.94 (m, 4H), 2.02 (s, 3H), 1.89-1.48 (m, 5H), 1.08 (s, 3H), 0.57 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 206.4, 170.5, 87.4, 75.5, 71.3, 54.1, 47.6, 46.5, 46.2, 42.4, 41.3, 38.6, 37.4, 34.0, 33.0, 31.9, 31.7, 27.3, 26.5, 22.8, 21.4, 15.4, 14.0. This side product is efficiently converted to 2 with sodium iodide in acetone.
16α-[2H]-pregnenolone-3-acetate (3)
To a stirring solution of acetic anhydride (0.2 mL) and [2H2]-H2O (0.2 mL) was added 16α-bromopregnenolone-3-acetate (77 mg, 0.177 mmol) in diethyl ether (2 mL). Zinc dust (500 mg, 7.8 mmol) was added, and the reaction mixture was stirred for 1 h under a N2 atmosphere, then purified directly on a silica gel column (100% hexanes to 50% hexanes in ethyl acetate, v/v) to afford 16α-[2H]-pregnenolone-3-acetate 3 (34 mg, 0.095 mmol, 54%). 1H NMR (400 MHz, CDCl3) δ 5.37 (broad s, 1H), 4.68-4.54 (m, 1H), 2.53 (d, J = 8.8 Hz, 1H), 2.38-2.41 (m, 1H), 2.21-2.15 (m, 1H), 2.13 (s, 3H), 2.05-1.95 (m, 1H), 2.04 (s, 3H), 1.73-1.55 (m, 2H), 1.50-1.43 (m, 2H), 1.91-1.83 (m, 2H), 1.28-1.12 (m, 2H), 1.02 (s, 3H), 0.63 (s, 3H).
16α-[2H]-pregnenolone (4)
To a solution of 16α-[2H]-pregnenolone-3-acetate (34 mg, 0.09 mmol) in CH2Cl2 (5 mL) and methanol (2 mL) was added 0.1 mL of 12 M HCl. The reaction was stirred for 10 h and washed with saturated aqueous NaHCO3 solution (2 × 10 mL). The aqueous layer was extracted with ethyl acetate (2 × 15 mL), and the combined organic extracts were concentrated via reduced pressure. The crude material was purified via flash column chromatography (100% hexanes to 50% hexanes in ethyl acetate, v/v) to afford 16α-[2H]-pregnenolone 4 (10 mg, 0.05 mmol, 56%). 1H NMR (400 MHz, CDCl3) δ 5.36-5.35 (m, 1H), 3.57-3.49 (m, 1H), 2.52 (d, J = 9.0 Hz, 1H), 2.33-2.25 (m, 2H), 2.22-2.14 (m, 1H), 2.12 (s, 3H), 2.07-1.97 (m, 2H), 1.75-1.43 (m, 7H), 1.27-1.03 (m, 5H), 1.01 (s, 3H), 0.99-0.98 (m, 1H), 0.63 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 209.8, 140.9, 121.6, 71.9, 63.8, 57.1, 50.1, 44.2, 42.4, 39.0, 37.4, 36.7, 32.0, 31.9, 31.8, 31.7, 24.5, 21.2, 19.5, 13.4.
16α-[2H]-progesterone (5)
Dess-Martin periodinane (12 mg) was added to 16α-[2H]-pregnenolone (10 mg) in CH2Cl2 (3 mL), and the reaction was stirred for 1 h. The reaction was washed with saturated NaHCO3 aqueous solution (2 × 15 mL) and extracted with ethyl acetate (2 × 15 mL). The combined organic extracts were concentrated via reduced pressure and purified via flash column chromatography (100% hexanes to 50% hexanes in ethyl acetate, v/v) to afford 16-deutero-Δ5,6-progesterone (5 mg, 50%). The 16α-[2H]-Δ5,6-progesterone was dissolved in MeOH (2 mL) and CH2Cl2 (1 mL), treated with 10 μL of 12 M HCl, stirred for 30 min at RT and purified directly on a silica gel column (100% hexanes to 40% hexanes in ethyl acetate, v/v) to afford 16α-[2H]-progesterone 5 (2 mg, 40%). 1H NMR (400 MHz, CDCl3) δ 5.73 (s, 1H), 2.52 (d, J = 9.1 Hz, 1H), 2.47-2.34 (m, 3H), 2.34-2.25 (m, 1H), 2.19-2.15 (m, 1H), 2.12 (s, 3H), 2.07-1.98 (m, 2H), 1.88-1.83 (m,1H), 1.75-1.61 (m, 3H), 1.57-1.51 (m, 1H), 1.47-1.39 (m, 2H), 1.30-1.21 (m, 2H), 1.20-1.15 (m, 1H), 1.18 (s, 3H), 1.15-0.95 (m, 3H), 0.66 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 209.5, 199.6, 171.1, 124.1, 63.6, 56.1, 53.8, 44.1, 38.8, 38.7, 35.8, 35.7, 34.1, 32.9, 32.0, 31.7, 24.4, 21.1, 17.5, 13.5. The 1H NMR resonance at 2.52 ppm is H-17, which is normally a triplet but is a doublet in the spectrum of compound 5 due to mono-deuteration at C-16. From nuclear Overhauser effect spectra, the resonance at 2.19-2.15 ppm, overlapping with the methyl singlet at 2.12 ppm, was previously identified as H-16β (30). The presence of this feature in the spectrum of compound 5 confirms that the 16α-position has been selectively deuterated. The H-16β resonance is seen more clearly in the 1H NMR spectrum of compound 11, which lacks the methyl singlet at 2.12 ppm.
17-[2H]-pregnenolone (7)
A solution of 17-bromopregnenolone (6, 61 mg, 0.16 mmol) in diethyl ether (1.0 mL) was added with stirring to a solution of [2H2]-H2O (0.3 mL, 15.0 mmol) and acetic anhydride (0.3 mL, 3.2 mmol) in diethyl ether (0.5 mL), which was first pre-stirred for 5 min. Zinc dust (160 mg, 2.5 mmol) was added, and the reaction was stirred at RT for 1 h under a N2 atmosphere. The reaction was diluted with diethyl ether (10 mL) and washed with [2H2]-H2O (1 mL). The organic layer was concentrated via reduced pressure and quickly purified to avoid any deuterium-hydrogen exchange on the silica gel column via flash column chromatography (100% hexanes to 50% hexanes/ethyl acetate) to afford 17-[2H]-pregnenolone (40 mg, 0.13 mmol, 81%). 1H NMR (400 MHz, CDCl3) δ 5.35-5.34 (m, 1H), 3.55-3.49 (m, 1H), 2.30 (ddd, J1 = 7.2 Hz, J2 = 5.2 Hz, J3 = 2.30 Hz, 1H), 2.27-2.19 (m, 1H), 2.19-2.14 (m, 1H), 2.12 (s, 3H), 2.08-1.97 (m, 2H), 1.88-1.81 (m, 2H), 1.74-1.42 (m, 4H), 1.28-1.06 (m, 3H), 1.00 (s, 3H), 0.98 (dd, J1 = 11.5 Hz, J2 = 4.8 Hz, 1H), 0.63 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 209.8, 140.9, 121.5, 71.8, 57.1, 50.1, 44.1, 42.4, 38.9, 37.4, 36.7, 32.0, 31.9, 31.8, 31.7, 24.6, 22.9, 21.2, 19.5, 13.4.
17-[2H]-progesterone (9)
Acetic anhydride (0.30 mL, 3.18 mmol) was added to a vial containing [2H2]-H2O (0.30 mL, 6.0 mmol) and diethyl ether (1.0 mL) under a N2 atmosphere. After stirring for 3 min at RT, 17-bromoprogesterone (8, 62 mg, 0.16 mmol) was added as a solid, and the reaction was stirred for 1 min under inert atmosphere. Zinc dust (0.16 g, 2.45 mmol) was added, and the reaction was stirred for 1 h. The reaction mixture was purified via flash column chromatography (100% hexanes to 50% hexanes in ethyl acetate) to afford 17-[2H]-progesterone (40 mg, 0.13 mmol, 81%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 5.72 (s, 1H), 2.47-2.35 (m, 2H), 2.20-2.12 (m, 1H), 2.11 (s, 3H), 2.06-2.01 (m, 2H), 1.86-1.83 (m, 1H), 1.76-1.62 (m, 4H), 1.58-1.50 (m, 1H), 1.50-1.41 (m, 2H), 1.31-1.24 (m, 2H), 1.18 (s, 3H), 1.21-1.14 (m, 1H), 1.09-0.95 (m, 2H), 0.66 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 209.5, 199.6, 171.1, 124.0, 56.1, 53.8, 44.0, 38.75, 38.70, 35.9, 35.7, 34.1, 32.9, 32.0, 31.6, 24.5, 22.9, 21.1, 17.5, 13.5.
21,21,21-[2H3]-progesterone (11)
To a stirring solution of [2H2]-H2O (0.2 mL, 10.0 mmol) and acetic anhydride (0.20 mL, 2.12 mmol) was added 21,21,21-tribromoprogesterone (10, 63 mg, 0.11 mmol) in diethyl ether (5 mL). Zinc dust (0.20 g, 3.06 mmol) was added, and the reaction was stirred at RT under a N2 atmosphere for 1 h, then directly purified on a silica gel column (100% hexanes to 50% hexanes in ethyl acetate) to afford 21,21,21-[2H3]-progesterone (17 mg, 0.05 mmol, 47%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 5.73 (s, 1H), 2.53 (apparent t, J1 = 9.0 Hz, 1H), 2.46-2.35 (m, 2H), 2.33-2.25 (m, 1H), 2.21-2.15 (m, 1H), 2.08-2.01 (m, 2H), 1.85-1.83 (m, 1H), 1.76-1.62 (m, 4H), 1.57-1.52 (m, 1H), 1.50-1.42 (m, 2H), 1.28-1.22 (m, 2H), 1.18 (s, 3H), 1.16-1.13 (m, 1H), 1.10-0.95 (m, 2H), 0.66 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 199.6, 171.1, 124.1, 63.6, 56.2, 53.8, 44.1, 38.8, 38.7, 35.9, 35.7, 34.1, 32.9, 32.0, 24.5, 22.9, 21.1, 17.5, 13.5. The C-21 and C-20 resonances were obscured in the 13C NMR spectra by one- and two-bond 13C-2H couplings.
21,21,21-[2H3]-17-hydroxyprogesterone (14)
To a stirring solution of 17-hydroxypregnenolone (248 mg, 0.747 mmol) in 1-[2H]-CH3OH (5 mL) and THF (1 mL) was added [2H]-KOH (40% wt/v, 0.5 mL) and the reaction was stirred for 7 days. The reaction was stopped and purified directly on a silica gel column to afford 21,21,21-[2H3]-17-hydroxypregnenolone (compound 13) (160 mg, 0.478 mmol, 64%). Deuterium incorporation was confirmed through the loss of the C21-methyl singlet in 1H NMR. Dess-Martin periodinane (200 mg, 0.472 mmol) was added to a stirring solution of 21,21,21-[2H3]-17-hydroxypregnenolone (160 mg, 0.478 mmol) and the reaction was stirred for 1.5 h. The reaction was purified directly on a silica gel column (100% hexanes to 50% hexanes in ethyl acetate) to afford 21,21,21-[2H3]-Δ5,6-17-hydroxyprogesterone (40 mg, 0.120 mmol, 26%) and 21,21,21-[2H3]-17-hydroxyprogesterone (50 mg, 0.150 mmol, 32%).
1H NMR of 21,21,21-[2H3]-17-hydroxy-Δ5,6-progesterone: (500 MHz, CDCl3) δ 5.33 (broad s, 1H), 3.27 (d, J = 16.4 Hz, 1H), 2.81 (d, J = 16.4 Hz, 1H), 2.70-2.64 (m, 1H), 2.45 (ddd, J1 = 19.7 Hz, J2 = 14.1 Hz, J3 = 5.8 Hz, 1H), 2.31-2.22 (m, 1H), 2.06-2.01 (m, 3H), 1.85-1.41 (m, 9H), 1.36-1.29 (m, 2H), 1.17 (s, 3H), 1.07 (ddd, J1 = 16.0 Hz, J2 = 12.2 Hz, J3 = 4.7 Hz, 1H), 0.73 (s, 3H).
1H NMR of 21,21,21-[2H3]-17-hydroxyprogesterone (14): (500 MHz, CDCl3) δ 5.73 (s, 1H), 2.81 (s, 1H), 2.67 (apparent t, J = 12.0 Hz, 1H), 2.45-2.25 (m, 4H), 1.90-1.79 (m, 2H), 1.75-1.56 (m, 7H), 1.45-1.35 (m, 3H), 1.18 (s, 3H), 1.11 (ddd, J1 = 16.8 Hz, J2 = 13.1 Hz, J3 = 3.6 Hz, 1H), 0.97 (ddd, J1 = 15.7 Hz, J2 = 11.4 Hz, J3 = 3.5 Hz, 1H), 0.75 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 199.7, 171.1, 124.1, 89.9, 53.4, 50.1, 48.4, 38.7, 35.8, 35.6, 34.1, 33.6, 32.9, 32.1, 30.1, 24.1, 20.6, 17.5, 15.6.
General enzyme assay methods
The enzyme source for most experiments was microsomes from yeast cells expressing native human P450-oxidoreductase (POR) plus the cytochrome P450 (31): CYP17A1 wild-type or mutation A105L, and CYP21A2 wild-type or mutation V359A. Control microsomes lacking CYP17A1 or CYP21A2 were prepared from yeast transformed with empty V60 expression vector as described (31). Experiments were repeated with purified CYP17A1 and POR reconstituted using 1,2-didodecanoyl-sn-glycero-3-phosphocholine as phospholipid, and these incubations gave an anomalously low proportion of 16α-hydroxyprogesterone (~10% of products, not shown), compared to 20–25% from experiments with yeast microsomes or transfected cells (32). In contrast, experiments with purified proteins using control yeast microsomes as the source of phospholipid gave 20–25% 16α-hydroxyprogesterone; therefore, control yeast microsomal lipids were used for obtaining KIE data with reconstituted, purified CYP17A1 and POR. For incubations with pregnenolone substrates, whose extinction coefficients are too low to quantitate by integration of UV absorbance, the reaction products were converted from the 3β-hydroxy-Δ5-ene steroids to the corresponding 3-keto-Δ4-ene steroids (ε240 ~16,000 M−1•cm−1), using cholesterol oxidase. CYP17A1 experiments were performed in the absence of cytochrome b5 and under partial substrate consumption, to minimize 19-carbon steroid products.
Microsomal enzyme incubations
The primary intramolecular KIEs were measured by incubation of the enzyme (1–10 pmol P450, 20–200 μg protein) in 1 mL of 50 mM potassium phosphate (pH 7.4) with 20–40 μM deuterium-labeled or unlabeled steroid. For intermolecular KIEs, incubations included deuterium-labeled steroid plus tracer amounts of steroid with tritium label distant from the reaction sites (1,2,6,7-[3H4]-pro gesterone, 90 Ci/mmol 7-[3H]-pregnenolone, 25 Ci/mmol [both PerkinElmer NEN, Waltham, MA] or 1,2-[3H2]-17-hydroxyprogesterone, 50 Ci/mmol [American Radiolabeled Chemicals, St. Louis, MO], 0.2–0.6 μCi, 2–6 pmol, 2–6 nM). One set of incubations replaced the 1,2,6,7-[3H4]-progesterone with 4-[14C]-progesterone (55 mCi/mmol, PerkinElmer NEN, 120 nCi, 2.2 μM). The reaction was started by addition of 1 mM NADPH at 37 °C and continued for 20–60 min, then terminated by extracting the steroids with 1 mL methylene chloride.
Reconstituted CYP17A1 assays
Modified human CYP17A1 was expressed in E. coli JM109 cells and purified to homogeneity as described (33). GroEL/ES chaperones (pGro7 plasmid) were co-expressed with the P450 to increase expression of active enzyme. Modified human POR was expressed in E. coli C41(DE3) cells and purified according to the previously published procedure (34), except that the bound protein was eluted from the nickel-agarose resin in buffer containing 200 mM imidazole rather than histidine.
CYP17A1 (30 pmol), POR (120 pmol) and control yeast microsomes (20 μg protein) were added to a 2 mL polypropylene tube in <10 μL, and the contents were gently swirled and set at room temperature for 5 min. The mixture was diluted to 0.2 mL containing 40 mM HEPES buffer (pH = 7.4), 30 mM MgCl2, 2.4 mM glutathione, 25 μM progesterone or [2H]-labeled progesterone, 1,2,6,7-[3H4]-progesterone (~0.4 μCi, 4 nM), and 20% glycerol (34). The resulting mixture was mixed gently and set at room temperature for 3 min. NADPH (1 mM) was added, and the incubation was started at 37 °C for 30 min. The incubation was extracted with 1 mL methylene chloride, and the organic phase was dried under nitrogen flow.
Cholesterol oxidase transformation
For incubations involving the pregnenolone substrates, the dried incubation extracts were dissolved in methanol (70 μL) and suspended in the same 0.1 M potassium phosphate buffer (100 μL). Water (100 μL) and cholesterol oxidase (70 μL, 28 units/mL) were added, and the resulting solution was shaken at 30 °C at 200 rpm for 6 h. The reaction was stopped by the addition of 1 mL methylene chloride. The mixture was extracted, and the organic layer was dried under nitrogen flow.
Chromatography and data acquisition
Reaction products were analyzed using either a Breeze 1525 high-performance liquid chromatography (HPLC) system equipped with in-line UV detector set to 254 nm (Waters, Woburn, MA) and β-RAM3 in-line scintillation counter or an Agilent 1260 Infinity HPLC system with UV detector and β-RAM4 in-line scintillation counter (LabLogic, Brandon, FL). Extracted steroid products were dissolved in 20 μL of methanol, and 5 μL injections were resolved with a 50 × 2.1 mm, 2.6 μm, C18 or C8 Kinetex column (Phenomenex, Torrance, CA), equipped with a guard column, at a flow rate of 0.4 mL/min and a methanol/water linear gradient: 27% methanol from 0 to 0.5 min, 39% to 16 min, 44% to 20 min, 60% to 22 min, 71% to 30 min, 75% to 30.5 min, 27% to 33 min. Products were identified by retention times of external standards chromatographed at the beginning and ends of the experiments. The flow rate of the scintillation cocktail (Bio-SafeII, Research Products International, Mount Prospect, IL) was 1.2 mL/min, and the data were processed with Laura4 software (LabLogic). The method settings were adjusted under the channel parameters to detect either 14C or 3H radioisotopes.
Mass spectrometry
The products from a subset of reactions were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) after extraction with methylene chloride, drying under nitrogen, and dissolving in aqueous methanol. The samples were injected into an Agilent 1290 HPLC coupled to a 6490 triple-quadrupole mass spectrometer, equipped with a Kinetex C8 column with methanol-water gradients to resolve the products. Replicate injections from duplicate incubations were analyzed in multiple reaction monitoring (MRM) mode with differential gating of precursor ions to discriminate and quantify deuterium-retained and deuterium-removed products. For example, starting with 21-[2H]-progesterone, the MRM parameters for the 21-hydroxylation product (11-deoxycorticosterone) are m/z 331.2/97.0 for natural-abundance (deuterium-removed) and m/z 332.2/97.0 for deuterium-retained species. Results were deconvoluted algebraically from [M+H]+, [M+H+1]+, and [M+H+2]+ precursor ions to account for the [M+H+1]+ isotopomers (contributing 15–20% of [M+H]+ peak based on standards run with the same instrumentation settings) in the natural-abundance and deuterated products and corrected for the deuterium incorporation in the synthetic substrates (90–95%).
KIE calculations
The first set of experiments involved the use of one substrate in each incubation to measure the intramolecular KIEs (35). The KIE equals the ratio of hydroxylation products in the deuterated vs. the non-deuterated substrates, and these ratios can be given by integration of the peaks in the output of the UV detector. For example, the intramolecular KIE of the 17-position for CYP17A1 can be calculated from data obtained from two separate incubations (Equation 1).
| (1) |
where [17OHProg] is the amount of 17α-hydroxyprogesterone formed, and Σ([21OHProg]+[16OHProg]) is the sum of all other hydroxylated products, which includes 16α-hydroxyprogesterone and 21-hydroxyprogesterone (if any). The values in the numerator derive from experiments with natural-abundance progesterone, and those in the denominator derive from experiments with 17α-[2H]-progesterone. An example calculation using Equation 1, extracted from the raw data, is given in the Supporting Information.
In the second set of experiments, the incubations contained both the substrate selectively deuterated at the site of reaction (i.e. 17-[2H]-progesterone) and substrate bearing hydrogen at the site of reaction but incorporating a tritium label distant from the site of reaction (i.e. 1,2,6,7-[3H4]-progesterone) to measure the intermolecular KIE. The products are analyzed via HPLC coupled to both an UV detector and in-line scintillation counter. The radioactivity detector only measures the tritiated compound, reflecting product partitioning of the natural-abundance substrate; this product distribution was verified using separate incubations with unlabeled steroid and UV detection of products. The UV detector measures only the deuterium-labeled steroid, because the concentration of the tritiated compound is ~1000 times lower than the non-radioactive compound, and its UV absorbance is thus negligible. In order to calculate the competitive intermolecular KIE, one begins with the relationship derived by Northrop (36) between initial velocity and substrate concentration (Equation 2).
| (2) |
As shown in equation 2, the KIE DV/K is dependent on f, the fractional conversion of the substrate to the product. The equation to calculate the competitive KIE for the 17-position with CYP17A1 and 17-[2H]-progesterone following Northrop’s derivation is given by Equation 3.
| (3) |
where the “*” denotes non-deuterated compound tracked with the radioactivity measurements. Under this experimental paradigm, we calculated the competitive intermolecular KIE values DV at Vmax conditions from the ratio of product formation rates derived from the non-deuterated substrate compared to the product formation rate derived from the deuterated substrate. For example, using 17-[2H]-pregnenolone plus 7-[3H]-pregnenolone incubated with CYP17A1, in which products other than 17-hydroxylation are negligible, DV is given by simply (after cholesterol oxidase conversion) the fractional conversion to 17-hydroxyprogesterone from radiochemical detection divided by the fractional conversion to 17-hydroxyprogesterone from UV detection, as shown in Equation 4.
| (4) |
For other reactions with multiple products, the denominators in equation 4 are replaced by the sum of remaining substrate plus all products, which equates the ratio of percent conversion to the product hydroxylated at the site of deuterium labeling for the labeled versus unlabeled substrates. Example calculations using Equations 3 and 4, extracted from the raw data, are given in the Supporting Information with a discussion of Northrop’s equation.
Results
Synthesis of isotopically-labeled steroids
The key feature of synthesizing deuterium-labeled substrates was the use of a Reformatzky-type reduction of halogenated precursors, which involved subjecting the halosteroids to zinc dust in the presence of 1-[2H]-acetic acid (generated in situ from [2H2]-H2O and acetic anhydride) (Scheme 1). We used brominated steroid precursors because iodides were relatively labile and chlorides or fluorides were less reactive than bromides in the zinc reduction, and deuterium incorporation was high. The brominated precursors 17-bromopregnenolone (6), 17-bromoprogesterone (8) and 21,21,21-tribromoprogesterone (10) were obtained via the procedures we previously reported (37). To access 21,21,21-[2H3]-17-hydroxyprogesterone, we exhaustively deuterated the 21-position of 17-hydroxypregnenolone under basic conditions (1-[2H]-CH3OH/[2H]-KOH) and oxidized the 3-hydroxy group using a mild Dess-Martin periodinane protocol, and some of this product spontaneously isomerized to the Δ4,5-enone during the reaction workup and purification (Scheme 2).
Scheme 1.

Synthesis of 16α-[2H]-progesterone (5).
Scheme 2.

Synthesis of 17-[2H]-pregnenolone (7), 17-[2H]-progesterone (9), and 21,21,21-[2H3]-progesterone (11).
The most difficult synthesis was the 16α-bromoprogesterone precursor of 16α-[2H]-progesterone. Reduction of 16,17-dehydropregnenolone with [2H2]-H2 using palladium on BaSO4 catalyst selectively deuterated the Δ16,17-double bond, but following Oppenauer oxidation with washout of the 17-[2H]-label, this route yielded <80% deuterium incorporation on the 16α-position by 1H NMR. We next brominated the 16α-position of 16,17-dehydropregnenolone-3-acetate using an acid-catalyzed Michael addition with tetrabutylammonium bromide and sulfuric acid (Scheme 3). By limiting tetrabutylammonium bromide to 2 molar equivalents, keeping reaction times short (2 h), and purifying the crude reaction mixture on a silica gel column without workup, we avoided dibromination of the Δ5,6-olefin and HBr elimination (Michael elimination reverting to starting material) and obtained sufficient product.
Scheme 3.
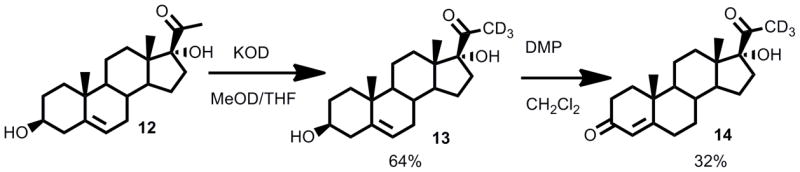
Synthesis of 21,21,21-[2H3]-17-hydroxyprogesterone (14).
KIE For CYP17A1 at H-17α and H-16α, and H-21
When incubated with pregnenolone, human CYP17A1 essentially only hydroxylates the 17α-position, whereas for progesterone substrate, 16α-hydroxyprogesterone accounts for 20–25% of the products (9). Swart and colleagues used phylogenetic analysis to identify A105 as a key residue enabling 16α-hydroxylase activity, and mutation A105L in human CYP17A1 reduces 16α-hydroxyprogesterone to 10% of the products (10). Consequently, we measured intramolecular and intermolecular KIEs at the 17α- and 16α-position for progesterone and pregnenolone substrates for both wild-type CYP17A1 and mutation A105L.
Intramolecular (intrinsic) KIEs
When yeast microsomes with CYP17A1 and POR are incubated with pregnenolone, 17-hydroxypregnenolone is the exclusive hydroxylation product identified, but with 17-[2H]-pregnenolone, a trace amount of 16α-hydroxypregnenolone appeared to be formed in amounts too low to calculate an intramolecular KIE (Figure S1 of the Supporting Information). Given the known product partitioning for progesterone substrate during CYP17A1 catalysis, we studied progesterone hydroxylation in detail, first measuring the intramolecular KIEs at the 17α- and 16α-positions. Incubations with 17-[2H]- or 16α-[2H]-progesterone both demonstrated suppression of hydroxylation at the site of deuteration, affording intramolecular KIE values of kH/kD = 4.1 ± 0.2 or 2.9 ± 0.2, respectively (Figure 2A, Table 1). Experiments with reconstituted, purified CYP17A1 and POR gave comparable results (Figure S1 and Table S1 of Supporting Information).
Figure 2.

Intramolecular KIEs and metabolic switching with wild-type human CYP17A1 (A) and mutation A105L (B). HPLC tracings show products obtained by incubation with microsomal enzyme system with (top to bottom) 16α-[2H]-, 17-[2H]-, 21,21,21-[2H3]-, and natural abundance progesterone (P4) substrates. Products were identified by retention times of standards chromatographed before and after samples: 1, 16α-hydroxyprogesterone; 2, 21-hydroxyprogesterone (DOC); 3, 17-hydroxyprogesterone; 4, progesterone. Ordinate scales are 0.15–0.25 and 0.08–0.25 AU full scale for Panels A and B, respectively.
Table 1.
Kinetic isotope effects for human CYP17A1 and mutation A105L
| Enzyme and substrate | C-H Bond | Intramolecular kH/kD (n)a | Intermolecular | |
|---|---|---|---|---|
| DV (n)b | DV/K (n) | |||
| CYP17A1 | ||||
| 16α-[2H]-progesterone | C16 | 2.9 ± 0.2 (3) | 3.0 ± 0.1 (3) | 3.2 ± 0.1 (3) |
| 17-[2H]-progesterone | C17 | 4.1 ± 0.2 (3) | 1.7 ± 0.2 (11) | 2.1 ± 0.4 (11) |
| 17-[2H]-progesterone [using 14C] | C17 | not determined | 2.2 ± 0.3 (3) | 1.8 ± 0.9 (3) |
| 17-[2H]-progesterone [using 10x 3H] | C17 | not determined | 2.0 ± 0.4 (3) | 2.3 ± 0.1 (3) |
| 17-[2H]-pregnenolone | C17 | not determined | 2.0 ± 0.4 (6) | 2.3 ± 0.5 (6) |
| 21,21,21-[2H3]-progesterone | C21 | 4.0 ± 0.6 (3) | 5.8 ± 0.8 (3) | 5.8 ± 0.6 (3) |
| CYP17A1-A105L | ||||
| 16α-[2H]-progesterone | C16 | 5.1 ± 0.2 (3) | 5.1 ± 0.3 (3) | 5.1 ± 0.3 (3) |
| 17-[2H]-progesterone | C17 | 3.8 ± 0.2 (3) | 1.4 ± 0.0 (3) | 2.0 ± 0.0 (3) |
| 17-[2H]-pregnenolone | C17 | not determined | 2.3 ± 0.4 (3) | 2.7 ± 0.6 (3) |
| 21,21,21-[2H3]-progesterone | C21 | 3.3 ± 0.7 (3) | 3.9 ± 0.9 (3) | 3.9 ± 0.9 (3) |
Data are given as means ± standard deviations for n experiments.
Ratio of relative yield for probed hydroxylated product to the sum of other hydroxylated products.
Ratio of measured hydroxylated products.
To characterize metabolic switching for human CYP17A1 in greater detail, we performed intramolecular KIE experiments with mutation A105L, for which 16α-hydroxylation constitutes only 10% of turnover. Experiments with recombinant enzyme in yeast microsomes confirmed the altered product distribution reported using transfected COS-7 cells (10), with one exception. These incubations yielded an additional product, which co-chromatographs with 21-hydroxyprogesterone (11-deoxycorticosterone, DOC) in a ~1:1 ratio to 16α-hydroxyprogesterone (Figure 2B). Incubations of CYP17A1 mutation A105L with 17-[2H]-progesterone afforded an intramolecular KIE value of kH/kD = 3.8 ± 0.2, similar to that observed with wild-type CYP17A1 (Table 1). By comparison, the intramolecular KIE observed with mutation A105L using 16α-[2H]-progesterone substrate was 5.1 ± 0.2, considerably higher than observed with wild-type CYP17A1 (Figure 2B, Table 1). Because the 21-hydroxylation product for mutation A105L is also observed, we conducted experiments with 21,21,21-[2H3]-progesterone and found an intramolecular KIE of kH/kD = 3.3 ± 0.7, similar to values observed at H-17 for both wild-type CYP17A1 and mutation A105L (Figure 2A, Table 1). A trace of 16α-hydroxypregnenolone also appeared in incubations with mutation A105L and 17-[2H]-pregnenolone (Figure S2 of Supporting Information).
After observing that mutation A105L reproducibly 21-hydroxylates progesterone, review of chromatograms from exhaustive incubations with wild-type CYP17A1 and progesterone suggested DOC formation as well. DOC comprises ~1% of the total products, but the percentage increases for incubations with 17-[2H]- or 16α-[2H]-progesterone, due to metabolic switching (Figure 2A). The identification of DOC was confirmed by mass spectrometry, and pre-incubation with abiraterone or ketoconazole eliminated the formation of all these metabolites in parallel (Figure S3 of Supporting Information). We pursued this observation by conducting incubations with 21,21,21-[2H3]-progesterone and wild-type CYP17A1, and the deuterium labels suppressed 21-hydroxylation to <1% of products (Figure 2A). The intramolecular KIE kH/kD = 4.0 ± 0.6 was similar to the KIE at H-21 for the A105L mutation; however, the calculated KIE values at H-21 might be distorted by the very small peak areas for this metabolite, consistent with the large standard deviations. These results demonstrate modest intrinsic primary KIEs on C-H abstraction for CYP17A1 catalysis at H-17α, H-16α, and H-21. Finally, we characterized the composition of 16α-hydroxyprogesterone product derived from incubations with 16α-[2H]-progesterone. For wild-type CYP17A1, 33–40% of the deuterium was retained in the 16α-hydroxyprogesterone product (range of three experiments, Figure S4 of Supporting Information), indicating that the 16β-hydrogen atom is abstracted during more than one in three oxygenations at C-16.
Intermolecular KIEs
Co-incubations with deuterium-labeled and tracer tritium-labeled steroids were employed to calculate intermolecular competitive KIEs DV and DV/K using Northrop’s equations for CYP17A1 and mutation A105L at H-17α, H-16α, and H-21 of progesterone and at H-17α of pregnenolone. For 17-[2H]-pregnenolone, which has negligible product partitioning to complicate analysis, competitive KIE values at H-17α of DV = 2.0 ± 0.4 and DV/K = 2.3 ± 0.5 was observed for wild-type CYP17A1 (Figure 3A) and DV = 2.3 ± 0.4 and DV/K = 2.7 ± 0.6 for mutation A105L (Figure 3B). Experiments using 17-[2H]-progesterone gave competitive KIE values at H-17α of DV = 1.7 ± 0.2 and DV/K = 2.1 ± 0.4 for wild-type CYP17A1 and DV = 1.4 ± 0.0 and DV/K = 2.0 ± 0.0 for mutation A105L (Figure 3, Table 1). Experiments using 16α-[2H]-progesterone gave KIE values of DV = 3.0 ± 0.1 and DV/K = 3.2 ± 0.1 at H-16α for wild-type CYP17A1 and DV = 5.1 ± 0.3 and DV/K = 5.1 ± 0.3 for mutation A105L, respectively (Figure 3, Table 1). These values are considerably higher than the intermolecular KIE values at H-17α. Experiments using 21,21,21-[2H3]-progesterone gave intermolecular KIE values at H-21 of DV = 5.8 ± 0.8 and DV/K = 5.8 ± 0.6 for wild-type CYP17A1 and DV = 3.9 ± 0.9 and DV/K = 3.9 ± 0.9 for mutation A105L (Table 1). These values, which are also higher than the intramolecular KIE values at H-17α, are prone to greater error due to low fractional conversion but are internally consistent and reproducible.
Figure 3.

Intermolecular KIE data with wild-type human CYP17A1 (A) and mutation A105L (B). Chromatograms show radioactivity (top, 400–2500 CPM full scale) and absorbance at 254 nm (bottom, 0.08–1.8 AU full scale) derived from co-incubations with deuterium-labeled steroid and tracer tritium-labeled steroid as indicated. Products were identified by retention times of standards chromatographed before and after samples, same key as Figure 2: 1, 16α-hydroxyprogesterone; 2, DOC; 3, 17-hydroxyprogesterone; 4, progesterone (P4). Experiments with pregnenolone were followed by cholesterol oxidase treatment prior to analysis.
The concentrations of the deuterated and non-deuterated substrates in these incubations were very dissimilar, which might introduce error; however, increasing the [3H]-labeled, non-deuterated progesterone substrate concentration by a factor of 10 or substituting [14C]-labeled, non-deuterated progesterone at 1000-fold higher concentration gave equivalent results (Table 1). In addition, similar results were obtained with purified, reconstituted CYP17A1 and POR (Table S1 of Supporting Information). Our data indicate an intermolecular KIE significantly >1 at all sites of oxygenation, suggesting that C-H bond breaking is partially rate-limiting in the CYP17A1 reaction cycle, and least contributory for the dominant oxygenation at H-17α.
KIE For CYP21A2 at H-21 and H-16α
Intramolecular (intrinsic) KIEs
Upon incubation of yeast microsomes containing CYP21A2 and POR with either progesterone or 17-hydroxyprogesterone, the 21-hydroxylation products DOC and 11-deoxycortisol, respectively, are observed. Incubations with 21,21,21-[2H3]-progesterone, however, consistently afforded an additional product corresponding to 16α-hydroxyprogesterone (Figure 4A). This product was not observed in control experiments omitting enzyme and reduced in parallel with the 21-hydroxylated products using enzyme inhibited with ketoconazole, and the identity of the 16α-hydroxyprogesterone was confirmed by mass spectrometry (Figure S5 of Supporting Information). Further inspection of chromatograms from incubations with unlabeled progesterone and CYP21A2 also suggested a trace of 16α-hydroxyprogesterone (Figure 4A), but the amount is too low to determine an intramolecular KIE value.
Figure 4.

Intramolecular KIEs and metabolic switching with wild-type human CYP21A2 (A) and mutation V359A (B). HPLC tracings show products obtained by incubation with microsomal enzyme system with (top to bottom) 16α-[2H]-, 21,21,21-[2H3]-, and natural abundance progesterone (P4) substrates. Products were identified by retention times of standards chromatographed before and after samples same key as Figure 2: 1, 16α-hydroxyprogesterone; 2, DOC; 4, progesterone (P4). Ordinate scales are 0.02–0.08 AU full scale. Peaks near 16α-hydroxyprogesterone (1) derived from incubations with 16α-[2H]-progesterone in A are trace contaminants in substrate with different retention times than 1.
Consequently, to better quantify the intramolecular KIEs for CYP21A2 at H-21 and H-16α, we used mutation V359A, which yields 60% 21-hydroxylation and 40% 16α-hydroxylation (29) and thus leads to more accurately measured changes in product distributions. Experiments using 21,21,21-[2H3]-progesterone substrate afforded an intramolecular KIE at H-21 of kH/kD = 6.2 ± 1.0, and those using 16α-[2H]-progesterone substrate afforded an intramolecular KIE at H-16α of kH/kD = 3.8 ± 0.8 (Figure 4B, Table 2). These intramolecular KIE values are similar to values for CYP17A1 at all sites of oxygenation.
Table 2.
Kinetic isotope effects for human CYP21A2 and mutation V359A
| Enzyme and substrate | C-H Bond | Intramolecular kH/kD (n)a | Intermolecular | |
|---|---|---|---|---|
| DV (n)b | DV/K (n) | |||
| CYP21A2 | ||||
| 21-[2H]-progesteronec | C21 | 2.5 ± 0.0 (2) | not determined | not determined |
| 21,21,21-[2H3]-progesterone | C21 | not determined | 1.9 ± 0.2 (10) | 2.0 ± 0.2 (10) |
| 21,21,21-[2H3]-17-hydroxyprogesterone | C21 | not determined | 2.3 ± 0.3 (7) | 2.4 ± 0.3 (7) |
| CYP21A2-V359A | ||||
| 16α-[2H]-progesterone | C16 | 3.8 ± 0.8 (4) | 2.8 ± 0.3 (7) | 3.0 ± 0.4 (7) |
| 21,21,21-[2H3]-progesterone | C21 | 6.2 ± 1.0 (4) | 3.0 ± 0.6 (4) | 3.1 ± 0.7 (4) |
| 21,21,21-[2H3]-17-hydroxyprogesterone | C21 | not determined | 2.4 ± 0.1 (4) | 3.8 ± 0.7 (4) |
Data are given as means ± standard deviations for n experiments.
Ratio of relative yield for probed hydroxylated product to the sum of other hydroxylated products.
Ratio of measured hydroxylated products.
Determined by LC-MS/MS.
To complete the intramolecular KIE analyses for CYP21A2, we determined a competitive KIE at H-21 using 21-[2H]-progesterone and measuring the fractional deuterium retention in the DOC product by LC-MS/MS. These experiments gave a kH/kD = 2.5 ± 0.0, similar to values from experiments with 21,21,21-[2H3]-progesterone and CYP17A1 (Tables 1 and 2). Finally, we determined the isotopic composition of the 16α-hydroxyprogesterone formed by mutation V359A from 16α-[2H]-progesterone. Mass spectrometry with deconvolution of isotopomer abundance showed 42–44% deuterium retention in the 16α-hydroxyprogesterone product (range of 2 experiments, Figure S6 of Supporting Information). The greater deuterium retention for the 16α-hydroxylation of 16α-[2H]-progesterone when catalyzed by CYP21A2-V359A versus CYP17A1 is consistent with the predicted progesterone orientations in the active sites of these enzymes (29, 38). The 16β-hydrogen is more accessible when progesterone binds with the D-ring perpendicular to the plane of the heme ring, as in CYP21A2, than when the D-ring is parallel to the heme with α-hydrogens facing the iron-oxygen complex, as in CYP17A1.
Intermolecular KIEs
Using similar conditions to those employed with CYP17A1, intermolecular competitive KIE experiments gave values at H-21 for wild-type CYP21A2 of DV = 1.9 ± 0.2 and DV/K = 2.0 ± 0.2 for 21,21,21-[2H3]-progesterone and DV = 2.3 ± 0.3 and DV/K = 2.4 ± 0.3 for 21,21,21-[2H3]-17-hydroxyprogesterone substrates (Figure 5A, Table 2). The intermolecular KIE values with mutation V359A were slightly larger at H-21, DV = 3.0 ± 0.6 and DV/K = 3.1 ± 0.7 with 21,21,21-[2H3]-progesterone and DV = 2.4 ± 0.1 and DV/K = 3.8 ± 0.7 with 21,21,21-[2H3]-17-hydroxyprogesterone. Additionally, experiments using 16α-[2H]-progesterone gave an intermolecular KIE at H-16α for mutation V359A of DV = 2.8 ± 0.3 and DV/K = 3.0 ± 0.4 (Figure 5B, Table 2). These data demonstrate that C-H bond breakage is also partially rate-limiting for the reactions catalyzed by CYP21A2.
Figure 5.

Intermolecular KIE data with wild-type human CYP21A2 (A) and mutation V359A (B). Chromatograms show radioactivity (top, 800–2000 CPM full scale) and absorbance at 254 nm (bottom, 0.2–0.6 AU full scale; inset in Panel A is 0.03 AU full scale) derived from co-incubations with deuterium-labeled steroid and tracer tritium-labeled steroid as indicated. Products were identified by retention times of standards chromatographed before and after samples, same key as Figure 2 [1, 16α-hydroxyprogesterone; 2, DOC; 3, 17-hydroxyprogesterone; 4, progesterone (P4)] plus 5, 11-deoxycortisol; 6, 16α,17α-dihydroxyprogesterone (pregn-4-ene-16α,17α-diol-3,20-dione). A trace of 6 appears to be produced by mutation V359A (lower right of B).
Discussion
For an enzyme-catalyzed C-H bond cleavage reaction following classical mechanics without proton-coupled electron transfer (hydrogen atom tunneling), the magnitude of the kinetic isotope effects is indicative of the transition state structure (36). A symmetrical, linear transition state leads to a high KIE value, whereas a bent or asymmetric transition state yields lower KIE values (25). Asymmetric transition states for C-H bond cleavage might be not only non-linear or angled but also “early” or “late,” meaning that the C-H bond resembles more the reactant (C-H bond mostly formed) or the product (C-H bond mostly broken) in the transition state. The magnitude of an intermolecular KIE is also reduced or “masked” by other steps in the catalytic cycle if C-H bond cleavage is not substantially rate-limiting. Masking might complicate intermolecular KIE experiments in which two different molecules compete for metabolism in a single incubation, limiting the information obtained. Many cytochrome P450 reactions, however, demonstrate incomplete regiochemical selectivity, executing hydroxylation at more than one site. This relaxed catalytic selectivity leads to the phenomenon of metabolic switching, where oxygenation shifts to an alternate site upon deuterium substitution at the principal site of reactivity. Metabolic switching permits the calculation of intramolecular KIEs, reflecting the intrinsic KIE for the C-H bond breakage step, independent of masking from slower steps in the reaction cycle. In the present study, we used intramolecular KIEs to compare transition state features for steroid hydroxylations at H-16α, H-17α, and H-21 catalyzed by CYP17A1 and CYP21A2.
We found relatively modest intramolecular KIEs for wild-type CYP17A1 and mutation A105L at H-17α, the principal site of steroid hydroxylation. The intramolecular KIE at H-16α was much lower than at H-17α for wild-type CYP17A1 but higher for mutation A105L. The KIEs well below 9 indicate that the transition state for hydrogen atom abstraction is either somewhat bent or asymmetrical, particularly for 16α-hydroxylation by wild-type CYP17A1 (KIE <3). Computer modeling studies of CYP17A1 predict that steroids bind with the cyclopentanophenanthrene nucleus parallel to the plane of the heme with H17α very close to the iron-oxygen complex (38), although the recent x-ray crystal structure of human CYP17A1 with bound inhibitor abiraterone suggests that other substrate orientations are possible (8). If the transition state geometry mimics that predicted from computational studies, then the C—H—O(Fe) alignment should not significantly deviate from linearity, and the most plausible explanation for low KIEs would be an early or late transition state. The bond energy of the C-H17α bond, a methine adjacent to a carbonyl, is predicted to be weaker (82 kcal/mol) than a carboxymethyl C-H21 (85 kcal/mol) or methylene C-H16α (99 kcal/mol). On the basis of this analysis, we favor an early transition state for C-H17α bond cleavage, which reduces the observed intramolecular KIE to less than half of theoretical maximum. Transition state geometry for the secondary sites of reactivity H-16α and H-21 are likely to be more distorted than for H-17α, but the similar KIE values observed at H-21 suggest that the transition state for C-H bond cleavage is more symmetrical than for H-17α, possibly due to the stronger C-H bonds. It is not obvious why the intramolecular KIE for mutation A105L at H-16α is larger than at H-17α—the largest intramolecular KIE observed in our studies with CYP17A1—whereas the converse is true for wild-type CYP17A1. Even this value at 5.1 is small compared to values of 10 or higher documented for other P450-catalyzed oxygenations (26, 39–41).
Our observation that one-third to one-half of the deuterium is retained in the 16α-hydroxyprogesterone product derived from 16α-[2H]-progesterone for both CYP17A1 and CYP21A2-V359A softens our interpretation of the KIE values at C-H16α, since the H16β proton is abstracted in a fraction of these turnovers. Ours are not the first examples of stereochemical inversion during a cytochrome P450 reaction, with abstraction of one hydrogen or deuterium atom and delivery of a hydroxyl group with the opposite stereochemistry. During P450LM2-catalyzed oxidation of exo, exo, exo, exo-2,3,5,6-[2H4]-norbornane, 25% of the exo-norborneol formed retains all four deuterium atoms, implying that one-fourth of substrate oxidation involves abstraction of an endo-hydrogen (42). More dramatically, over half of the 5-exo-hydroxycamphor formed during the P450cam-catalyzed hydroxylation of 5-exo-[2H]-camphor retains the deuterium, demonstrating endo-hydrogen abstraction in 55% of the reactions (43). The 33–44% deuterium retention we observed for the 16α-hydroxylation of 16α-[2H]-progesterone, either using CYP17A1 or CYP21A2 mutation V359A, is intermediate the values for P450LM2 and P450cam. This stereochemical inversion, which has not been previously documented for a steroid hydroxylase reaction, indicates that substrate trajectories during the hydrogen atom abstraction step are less constrained than during the oxygen rebound step. If this inference is correct, this phenomenon might explain why some progesterone analogs are inhibitors but not substrates for these steroid hydroxylases (37).
For CYP21A2, intramolecular KIEs were best obtained for mutation V359A with progesterone, which gave values of 3.8 and 6.2 at H-16α and H-21, respectively. Computer modeling studies suggested that progesterone binds to CYP21A2 with the steroid nucleus perpendicular to the plane of the heme ring with the C-21 methyl group dangling over the iron-oxygen complex (29). In mutation V359A, the larger steroid-binding pocket enables trajectories with the steroid tipping on its long axis to present the more reactive H-16α in addition to H-21, yielding both products (29). The crystal structure of bovine CYP21A2 with 17-hydroxyprogesterone bound confirms the orthogonal orientation of heme and steroid, plus a second apparently structural steroid distant from the steroid-binding pocket (7). On the basis of our results, the transition states for these two hydrogen atom abstractions must share considerable similarities. For wild-type CYP21A2, an experimental paradigm using 21-[2H]-progesterone and mass spectrometry allowed us to calculate the competitive intramolecular KIE of kH/kD = 2.5 ± 0.0, consistent with the other data for CYP21A2 hydroxylations. The coexistence of a simultaneous secondary KIE might complicate these experiments (41); however, the magnitude of the correction would not be large enough to significantly alter our conclusions.
In addition, experiments with deuterium-labeled substrates have provided compelling evidence for additional hydroxylase activities of wild-type human CYP17A1 and CYP21A2. In addition to both progesterone 17α- and 16α-hydroxylase activities, human CYP17A1 catalyzes progesterone 21-hydroxylation. This activity is augmented with 17-[2H]- or 16α-[2H]-progesterone substrate and accentuated in mutation A105L. The 21-hydroxylation comprises ~1% of the products; nonetheless, this trace of activity might be clinically significant in classical 21-hydroxylase deficiency with null CYP21A2 alleles (salt-wasting phenotype) and might explain some of the discrepancies observed between phenotype and genotype in this disease (44). We also reproducibly observed a trace of pregnenolone 16α-hydroxylation using 17α-[2H]-pregnenolone substrate (Figure S1 of Supporting Information). Analogously, CYP21A2 is a progesterone 16α-hydroxylase, accounting for <1% of the products but augmented with 21,21,21-[2H3]-progesterone and markedly increased with mutations that reduce the bulk of V359.
The phenomenon of “metabolic switching” occurs when a common E•S complex breaks down to form two (or more) products, P1 and P2, and the partitioning is altered via isotopic substitution at one reaction site. In the absence of secondary KIEs, the rate of reaction at the unlabeled site remains essentially constant, while the rate for reaction at the substituted (deuterated) site slows by an amount equal to the intramolecular KIE. For chemical reactions best described by classical mechanics, the limiting KIE at 37°C is 9, meaning that the proportion of minor product can only increase by a factor of 10 or less. Depending on the sensitivity of the assay, new products might seem to “appear” due to metabolic switching, but in fact these “new” products must be present in at least trace amounts in reactions with unlabeled substrate. This phenomenon was observed in our studies, when products were recognized from reactions with deuterium-labeled substrates and site-directed mutations. Our findings prompted us to confirm that these products were also formed in reactions with wild-type enzymes and natural abundance substrates. We could not confidently identify 16α-hydroxypregnenolone from reactions with CYP17A1 and unlabeled pregnenolone, possibly because losses from coupling the reaction with cholesterol oxidase treatment further compromised the sensitivity of our assays.
Intermolecular competitive KIE experiments gave DV and DV/K values of 1.4–5.8 for CYP17A1 and CYP21A2 hydroxylations, mostly ranging 2–3. These KIE values are unusually high for a P450 reaction, for which the second electron transfer step is traditionally assumed to be the rate-limiting step, based on studies of P450cam (45). The turnover number for the steroid hydroxylation reactions catalyzed by CYP17A1 and CYP21A2, however, are many orders of magnitude slower than those of bacterial P450s (2), and different steps might become at least partially rate-limiting in this context (19, 26, 39–41). Our data indicate that C-H bond cleavage is partially rate-limiting for all reactions catalyzed by these two enzymes in these studies. For bovine CYP21A2, pre-steady state kinetic experiments demonstrated that product release was rate-limiting for progesterone 21-hydroxylation but not for the more rapid 21-hydroxylation of 17-hydroxyprogesterone (46). The design of our experiments, which incorporates tracer tritium label at distant sites to monitor both species in the same incubation, controls for several variables that can confound intermolecular KIE determinations. Varying the ratio of deuterium-labeled to unlabeled steroid or substitution of [14C]-tracer for [3H]-tracer did not influence the results, strengthening our conclusions. Consequently, the rates of C-H bond cleavage and product release for CYP21A2-catalyzed progesterone 21-hydroxylation are probably similar.
Intramolecular non-competitive and competitive KIEs have been determined for several P450-catalyzed reactions. KIE values of 9–11 have been obtained for reactions catalyzed CYP2E1 and CYP2B4, including reactions at methyl groups (22). In contrast, the non-competitive KIE for CYP3A4-catalyzed testosterone-6β-hydroxylation of an allylic C-H bond is only 2–3 (26). Holland and colleague have reported on the intermolecular kinetic isotope effect of the 19-methyl position on testosterone with aromatase (CYP19A1) with a kH/kD of 2.3 for 19-[2H]-testosterone and 3.2 for 19,19,19-[2H3]-testosterone substrates (47). This wide range in observed KIE values suggests that, although all cytochrome P450 hydroxylation reactions might incorporate hydrogen atom abstraction in their catalytic cycles, the structural features of these transition states are considerably variable. Although some characteristics are undoubtedly common to all mammalian P450-catalzyed reactions, the steroid hydroxylases CYP17A1 and CYP21A2 are likely to require some specific properties to execute their enzymatic functions, which serve to support reproduction, response to stress, and fluid balance. Further experiments, which directly measure the rates of individual steps for these reactions, will provide insight to the mechanism of catalysis for these important enzymes in human physiology and disease.
Supplementary Material
Acknowledgments
This work was supported by grant R01-GM08659602 from the National Institutes of Health and grant I-1493 from the Robert A. Welch Foundation. F.K.Y. received support from a Chemistry and Biology Interface graduate fellowship from the University of Texas. D.S. was a UT Southwestern SUMR research fellow, and Y.Z. was a UT Southwestern SURF research fellow. Mass spectrometry used core services supported by grant DK089503 from the National Institutes of Health to the University of Michigan under the Michigan Nutrition Obesity Center (NORC).
We thank Drs. Fred Guengerich and Paul Hollenberg for critical readings of the manuscript and Drs. Chunhai Ruan and Stephen Brown for performing some of the mass spectrometry experiments.
Abbreviations
- CAH
congenital adrenal hyperplasia
- CYP17A1
cytochrome P450c17
- CYP21A2
cytochrome P450c21
- DOC
11-deoxycorticosterone
- DV
deuterium kinetic isotope effect on maximal velocity
- DV/K
deuterium kinetic isotope effect on ratio of maximal velocity to Michaelis constant
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HPLC
high-performance liquid chromatography
- KIE
kinetic isotope effect
- LC-MS/MS
liquid chromatography with tandem mass spectrometry
- MRM
multiple reaction monitoring
- NADPH
nicotinamide adenine dinucleotide phosphate (reduced form)
- NMR
nuclear magnetic resonance spectroscopy
- POR
cytochrome P450 oxidoreductase
- UV
ultraviolet light
Footnotes
Supporting Information Available
Supporting information for this publication is available. The 1H- and/or 13C-NMR spectra for compounds 2, 3, 4, 5, 7, 9, 11, 13, 16, and intermediate 21,21,21-[2H3]-17-hydroxy-Δ5,6-progesterone are provided. Figure S1 shows evidence of pregnenolone 16α-hydroxylation with CYP17A1 (yeast microsomes) and 17-[2H]-pregnenolone (A) and metabolic switching with [2H]-labeled progesterone substrates and purified, reconstituted CYP17A1 and POR (B), with derived intramolecular KIE values in Table S1. Figure S2 shows evidence of pregnenolone 16α-hydroxylation with CYP17A1 mutation A105L and 17-[2H]-pregnenolone. Figures S3 and S5 show 17α-[2H]-DOC production from incubations with CYP17A1 and 17α-[2H]-progesterone or 21,21,21-[2H3]-16α-hydroxyprogesterone formation from incubations with CYP21A2 and 21,21,21-[2H3]-progesterone, respectively, using LC-MS/MS for structure confirmation. Figures S4 and S6 show data used to calculate deuterium retention in 16α-hydroxyprogesterone product from incubations with CYP17A1 or CYP21A2 mutation V359A, respectively, and 16α-[2H]-progesterone substrate by LC-MS/MS. Figures S7 and S8 show examples of calculations used to determine intramoleccular and intermolecular KIE values, respectively, from raw data. The last page is an appendix with the equations for calculating KIEs. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cooper DY, Levin S, Narasimhulu S, Rosenthal O, Estabrook RW. Photochemical action spectrum of the terminal oxidase of mixed function oxidase systems. Science. 1965;147:400–402. doi: 10.1126/science.147.3656.400. [DOI] [PubMed] [Google Scholar]
- 2.Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev. 2011;32:81–151. doi: 10.1210/er.2010-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev. 2000;21:245–291. doi: 10.1210/edrv.21.3.0398. [DOI] [PubMed] [Google Scholar]
- 4.Therrell BLJ, Berenbaum SA, Manter-Kapanke V, Simmank J, Korman K, Prentice L, Gonzalez J, Gunn S. Results of screening 1.9 million Texas newborns for 21-hydroxylase-deficient congenital adrenal hyperplasia. Pediatrics. 1998;101:583–590. doi: 10.1542/peds.101.4.583. [DOI] [PubMed] [Google Scholar]
- 5.Auchus ML, Auchus RJ. Human steroid biosynthesis for the oncologist. J Investig Med. 2012;60:495–503. doi: 10.231/JIM.0b013e3182408567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB, Jr, Saad F, Staffurth JN, Mainwaring P, Harland S, Flaig TW, Hutson TE, Cheng T, Patterson H, Hainsworth JD, Ryan CJ, Sternberg CN, Ellard SL, Flechon A, Saleh M, Scholz M, Efstathiou E, Zivi A, Bianchini D, Loriot Y, Chieffo N, Kheoh T, Haqq CM, Scher HI. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao B, Lei L, Kagawa N, Sundaramoorthy M, Banerjee S, Nagy LD, Guengerich FP, Waterman MR. A three-dimensional structure of steroid 21-hydroxylase (Cytochrome P450 21A2) with two substrates reveals locations of disease-associated variants. J Biol Chem. 2012;287:10613–10622. doi: 10.1074/jbc.M111.323501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeVore NM, Scott EE. Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature. 2012;482:116–119. doi: 10.1038/nature10743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swart P, Swart AC, Waterman MR, Estabrook RW, Mason JI. Progesterone 16α-hydroxylase activity is catalyzed by human cytochrome P450 17α-hydroxylase. J Clin Endocrinol Metab. 1993;77:98–102. doi: 10.1210/jcem.77.1.8325965. [DOI] [PubMed] [Google Scholar]
- 10.Swart AC, Storbeck KH, Swart P. A single amino acid residue, Ala 105, confers 16α-hydroxylase activity to human cytochrome P450 17α-hydroxylase/17,20 lyase. J Steroid Biochem Mol Biol. 2010;119:112–120. doi: 10.1016/j.jsbmb.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 11.Sohl CD, Guengerich FP. Kinetic analysis of the three-step steroid aromatase reaction of human cytochrome P450 19A1. J Biol Chem. 2010;285:17734–17743. doi: 10.1074/jbc.M110.123711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Auchus RJ, Lee TC, Miller WL. Cytochrome b5 augments the 17,20 lyase activity of human P450c17 without direct electron transfer. J Biol Chem. 1998;273:3158–3165. doi: 10.1074/jbc.273.6.3158. [DOI] [PubMed] [Google Scholar]
- 13.Katagiri M, Kagawa N, Waterman MR. The role of cytochrome b5 in the biosynthesis of androgens by human P450c17. Arch Biochem Biophys. 1995;317:343–347. doi: 10.1006/abbi.1995.1173. [DOI] [PubMed] [Google Scholar]
- 14.Lee-Robichaud P, Wright JN, Akhtar ME, Akhtar M. Modulation of the activity of human 17α-hydroxylase-17,20-lyase (CYP17) by cytochrome b5: endocrinological and mechanistic implications. Biochem J. 1995;308:901–908. doi: 10.1042/bj3080901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Onoda M, Hall PF. Cytochrome b5 stimulates purified testicular microsomal cytochrome P450 (C21 side-chain cleavage) Biochem Biophys Res Commun. 1982;108:454–460. doi: 10.1016/0006-291x(82)90850-6. [DOI] [PubMed] [Google Scholar]
- 16.Idkowiak J, Randell T, Dhir V, Patel P, Shackleton CH, Taylor NF, Krone N, Arlt W. A missense mutation in the human cytochrome b5 gene causes 46,XY disorder of sex development due to true isolated 17,20 lyase deficiency. J Clin Endocrinol Metab. 2012;97:E465–E475. doi: 10.1210/jc.2011-2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kok RC, Timmerman MA, Wolffenbuttel KP, Drop SL, de Jong FH. Isolated 17,20-lyase deficiency due to the cytochrome b5 mutation W27X. J Clin Endocrinol Metab. 2010;95:994–999. doi: 10.1210/jc.2008-1745. [DOI] [PubMed] [Google Scholar]
- 18.Naffin-Olivos JL, Auchus RJ. Human cytochrome b5 requires residues E48 and E49 to stimulate the 17, 20-lyase activity of cytochrome P450c17. Biochemistry. 2006;45:755–762. doi: 10.1021/bi051623y. [DOI] [PubMed] [Google Scholar]
- 19.Shinkyo R, Guengerich FP. Cytochrome P450 7A1 cholesterol 7α-hydroxylation: individual reaction steps in the catalytic cycle and rate-limiting ferric iron reduction. J Biol Chem. 2011;286:4632–4643. doi: 10.1074/jbc.M110.193409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Whitehouse CJ, Bell SG, Yang W, Yorke JA, Blanford CF, Strong AJ, Morse EJ, Bartlam M, Rao Z, Wong LL. A highly active single-mutation variant of P450BM3 (CYP102A1) Chembiochem. 2009;10:1654–1656. doi: 10.1002/cbic.200900279. [DOI] [PubMed] [Google Scholar]
- 21.Guengerich FP. Mechanisms of cytochrome P450 substrate oxidation: MiniReview. J Biochem Mol Toxicol. 2007;21:163–168. doi: 10.1002/jbt.20174. [DOI] [PubMed] [Google Scholar]
- 22.Atkinson JK, Hollenberg PF, Ingold KU, Johnson CC, Le Tadic MH, Newcomb M, Putt DA. Cytochrome P450-catalyzed hydroxylation of hydrocarbons: kinetic deuterium isotope effects for the hydroxylation of an ultrafast radical clock. Biochemistry. 1994;33:10630–10637. doi: 10.1021/bi00201a009. [DOI] [PubMed] [Google Scholar]
- 23.Korzekwa KR, Trager WF, Gillette JR. Theory for the observed isotope effects from enzymatic systems that form multiple products via branched reaction pathways: cytochrome P-450. Biochemistry. 1989;28:9012–9018. doi: 10.1021/bi00449a009. [DOI] [PubMed] [Google Scholar]
- 24.Miwa GT, Walsh JS, Lu AY. Kinetic isotope effects on cytochrome P-450-catalyzed oxidation reactions. The oxidative O-dealkylation of 7-ethoxycoumarin. J Biol Chem. 1984;259:3000–3004. [PubMed] [Google Scholar]
- 25.Nelson SD, Trager WF. The use of deuterium isotope effects to probe the active site properties, mechanism of cytochrome P450-catalyzed reactions, and mechanisms of metabolically dependent toxicity. Drug Metab Dispos. 2003;31:1481–1498. doi: 10.1124/dmd.31.12.1481. [DOI] [PubMed] [Google Scholar]
- 26.Krauser JA, Guengerich FP. Cytochrome P450 3A4-catalyzed testosterone 6β-hydroxylation stereochemistry, kinetic deuterium isotope effects, and rate-limiting steps. J Biol Chem. 2005;280:19496–19506. doi: 10.1074/jbc.M501854200. [DOI] [PubMed] [Google Scholar]
- 27.Korzekwa KR, Gillette JR, Trager WF. Isotope effect studies on the cytochrome P450 enzymes. Drug Metab Rev. 1995;27:45–59. doi: 10.3109/03602539509029814. [DOI] [PubMed] [Google Scholar]
- 28.Knapp MJ, Rickert K, Klinman JP. Temperature-dependent isotope effects in soybean lipoxygenase-1: Correlating hydrogen tunneling with protein dynamics. J Am Chem Soc. 2002;124:3865–3874. doi: 10.1021/ja012205t. [DOI] [PubMed] [Google Scholar]
- 29.Mizrachi D, Wang Z, Sharma KK, Gupta MK, Xu K, Dwyer CR, Auchus RJ. Why human cytochrome P450c21 is a progesterone 21-hydroxylase. Biochemistry. 2011;50:3968–3974. doi: 10.1021/bi102078e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stonehouse J, Adell P, Keeler J, Shaka AJ. Ultrahigh-quality NOE spectra. J Am Chem Soc. 1994;116:6037–6038. [Google Scholar]
- 31.Sherbet DP, Tiosano D, Kwist KM, Hochberg Z, Auchus RJ. CYP17 mutation E305G causes isolated 17,20-lyase deficiency by selectively altering substrate binding. J Biol Chem. 2003;278:48563–48569. doi: 10.1074/jbc.M307586200. [DOI] [PubMed] [Google Scholar]
- 32.Costa-Santos M, Kater CE, Auchus RJ. Two prevalent CYP17 mutations and genotype-phenotype correlations in 24 Brazilian patients with 17-hydroxylase deficiency. J Clin Endocrinol Metab. 2004;89:49–60. doi: 10.1210/jc.2003-031021. [DOI] [PubMed] [Google Scholar]
- 33.Wang YH, Tee MK, Miller WL. Human cytochrome P450c17: single step purification and phosphorylation of serine 258 by protein kinase A. Endocrinology. 2010;151:1677–1684. doi: 10.1210/en.2009-1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sandee D, Miller WL. High-yield expression of a catalytically active membrane-bound protein: human P450 oxidoreductase. Endocrinology. 2011;152:2904–2908. doi: 10.1210/en.2011-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chandrasena RE, Vatsis KP, Coon MJ, Hollenberg PF, Newcomb M. Hydroxylation by the hydroperoxy-iron species in cytochrome P450 enzymes. J Am Chem Soc. 2004;126:115–126. doi: 10.1021/ja038237t. [DOI] [PubMed] [Google Scholar]
- 36.Northrop DB. Deuterium and tritium kinetic isotope effects on initial rates. Methods Enzymol. 1982;87:607–625. doi: 10.1016/s0076-6879(82)87032-8. [DOI] [PubMed] [Google Scholar]
- 37.Yoshimoto FK, Desilets MC, Auchus RJ. Synthesis of halogenated pregnanes, mechanistic probes of steroid hydroxylases CYP17A1 and CYP21A2. J Steroid Biochem Mol Biol. 2012;128:38–50. doi: 10.1016/j.jsbmb.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Auchus RJ, Miller WL. Molecular modeling of human P450c17 (17α-hydroxylase/17,20-lyase): Insights into reaction mechanisms and effects of mutations. Mol Endocrinol. 1999;13:1169–1182. doi: 10.1210/mend.13.7.0326. [DOI] [PubMed] [Google Scholar]
- 39.Guengerich FP, Krauser JA, Johnson WW. Rate-limiting steps in oxidations catalyzed by rabbit cytochrome P450 1A2. Biochemistry. 2004;43:10775–10788. doi: 10.1021/bi0491393. [DOI] [PubMed] [Google Scholar]
- 40.Guengerich FP, Miller GP, Hanna IH, Sato H, Martin MV. Oxidation of methoxyphenethylamines by cytochrome P450 2D6. Analysis of rate-limiting steps. J Biol Chem. 2002;277:33711–33719. doi: 10.1074/jbc.M205146200. [DOI] [PubMed] [Google Scholar]
- 41.Yun CH, Kim KH, Calcutt MW, Guengerich FP. Kinetic analysis of oxidation of coumarins by human cytochrome P450 2A6. J Biol Chem. 2005;280:12279–12291. doi: 10.1074/jbc.M411019200. [DOI] [PubMed] [Google Scholar]
- 42.Groves JT, McClusky GA. Aliphatic hydroxylation by highly purified liver microsomal cytochrome P-450. Evidence for a carbon radical intermediate. Biochem Biophys Res Commun. 1978;81:154–160. doi: 10.1016/0006-291x(78)91643-1. [DOI] [PubMed] [Google Scholar]
- 43.Gelb MH, Heimbrook DC, Malkonen P, Sligar SG. Stereochemistry and deuterium isotope effects in camphor hydroxylation by the cytochrome P450cam monoxygenase system. Biochemistry. 1982;21:370–377. doi: 10.1021/bi00531a026. [DOI] [PubMed] [Google Scholar]
- 44.Krone N, Braun A, Roscher AA, Knorr D, Schwarz HP. Predicting phenotype in steroid 21-hydroxylase deficiency? Comprehensive genotyping in 155 unrelated, well defined patients from southern Germany. J Clin Endocrinol Metab. 2000;85:1059–1065. doi: 10.1210/jcem.85.3.6441. [DOI] [PubMed] [Google Scholar]
- 45.Lipscomb JD, Sligar SG, Namtvedt MJ, Gunsalus IC. Autooxidation and hydroxylation reactions of oxygenated cytochrome P-450cam. J Biol Chem. 1976;251:1116–1124. [PubMed] [Google Scholar]
- 46.Kominami S, Owaki A, Iwanaga T, Tagashira-Ikushiro H, Yamazaki T. The rate-determining step in P450 C21-catalyzing reactions in a membrane-reconstituted system. J Biol Chem. 2001;276:10753–10758. doi: 10.1074/jbc.M006043200. [DOI] [PubMed] [Google Scholar]
- 47.Holland HL, Taylor GJ. Enzymic aromatization of deuterium labeled testosterone and androst-4-ene-3,17-dione. Can J Chem. 1981;59:2809–2819. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.