

Abstract
In a medical sense, biomodulation could be considered a biochemical or cellular response to a disease or therapeutic stimulus. In cancer pathophysiology, the initial oncogenic stimulus leads to cellular and biochemical changes that allow cells, tissue, and organism to accommodate and accept the oncogenic insult. In epithelial cell cancer development, the process of carcinogenesis is frequently characterized by sequential cellular and biochemical adaptations as cells transition through hyperplasia, dysplasia, atypical dysplasia, carcinoma in situ, and invasive cancer. In some cases, the adaptations may persist after the initial oncogenic stimulus is gone in a type of “hit-and-run” oncogenesis. These pathophysiological changes may interfere with cancer prevention therapies targeted solely to the initial oncogenic insult, perhaps contributing to resistance development. Characterization of these accommodating adaptations could provide insight for the development of cancer preventive regimens that might more effectively biomodulate preneoplastic cells toward a more normal state.
Keywords: cancer prevention, biomodulation, hit and run oncogenesis, viral oncogenesis, hormonal oncogenesis
Introduction
To some extent, all therapeutic interventions could be considered biomodulation, as they are all directed toward a change in the individual's pathophysiology at both cellular and systemic levels. For the purposes of discussion here, biomodulation is defined in medical terms as a change in cells or tissue in response to a pathologic or therapeutic stimulus. For example, it has been applied to specific therapies for kidney disease, that is, the impact of freeze-dried Lactobacillus acidophilus on the bacterial overgrowth syndrome occurring in end-stage kidney disease.1 In oncology, the broad concept of biomodulation is incorporated into cancer evolution and therapy and many of the approaches and techniques developed for systems biology are used within the context of oncological investigations to characterize specific “biomodulating” events at different stages of cancer evolution or therapy. In cancer therapy, this type of approach has been validated. For example, the addition of agents to previously defined standard chemotherapeutic regimens is described as biomodulating the therapeutic response.2–9 Investigations have validated the same type of approach for improving response to radiotherapy,10,11 laser irradiation,12–16 and application of Chinese medicine to cancer care.17
Evolution of cancer in epithelial tissues frequently develops as a sequence of coordinated changes in both the epithelial and stromal compartments (Fig. 1). Biomodulatory changes occur throughout the process and can be both similar and different in preneoplastic, hyperplastic, and dysplastic tissue, invasive cancer, and metastases. The field of cancer prevention seeks to interrupt cancer development before it reaches either the carcinoma in situ or invasive stages. The value of considering cancer prevention in the context of biomodulation would be to conceptually encompass the therapeutic approach as both an assault on the initiating event as targeted therapy, as well as an attack on the secondary adaptations that a preneoplastic cell, and surrounding local stromal and systemic environments, may have made to survive or adapt to that initiating stimulus (Fig. 2). The probability of epithelial cancer development can be altered by germ-line genetic predisposition such as mutations in breast cancer 1, early onset (BRCA1), breast cancer 2, early onset (BRCA2), phosphatase and tensin homolog (PTEN), and tumor protein p53 (TP53). It is possible that specific biomodulatory events accompany epithelial cancer progression in these cases. Identification of these events may lead to more targeted chemopreventive approaches for individuals carrying these mutations. Some types of sporadic cancer development are also characterized by genetic changes, sometimes in the same genes found with familial germ-line predisposition. It will be important to determine whether the types of biomodulatory changes that occur when cancer develops secondary to a germ-line risk factor are the same as those that may occur when the mutation occurs in somatic cells as part of sporadic cancer development. It is quite possible that genetic background, both germ-line and somatic, will influence the response to specific biomodulatory approaches.
Figure 1.

Biomodulation of cancer tissue occurs in both epithelial and stromal compartments throughout cancer evolution. Changes in the epithelial and stromal compartments occur as epithelial cells transition from normal through hyperplasia, dysplasia, carcinoma in situ, invasive cancer, and metastasis. The engagement of epithelial cells in crosstalk with the local and systemic environments during cancer progression results in their biomodulation. Different biomodulatory events can occur at different stages of cancer progression (reviewed in Refs. 18–20).
Figure 2.
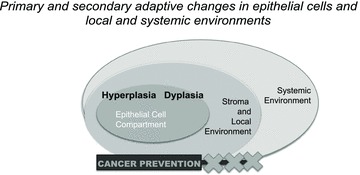
Targets for biomodulation in cancer prevention. Targets for biomodulation in epithelial cancer prevention can include the epithelial cells themselves as well as factors in the local stromal and systemic environments (reviewed in Refs. 21 and 22).
This short review concentrates on what type of molecular changes might occur in an epithelial cell as secondary cellular adaptations that occur over time following a carcinogenic stimulus, to determine if they are disease drivers, and to test if they can be recognized before therapy targeted to the initial stimulus or whether they only become apparent after the initiating stimulus has been removed. It is possible that cancer prevention approaches that target both disease-initiating stimuli and secondary adaptations would more efficiently effect disease reversal.
Identifying cellular adaptations that occur over time in cells exposed to a carcinogenic stimulus
Cancer development is not a unitary process but instead is executed by different processes. A relatively direct mechanism of cancer development and maintenance is “oncogene addiction.” In this situation the growth and survival of the cancer cell is dependent upon maintenance of a cancer causing stimulus, such as a defined oncogene,23–25 miRNA,26 or immune-mediated mechanism.27 Less direct is the multistep mechanism termed “hit-and-run” oncogenesis, where the initiating stimulus is gone but the tissue continues to progress toward, or is maintained as, cancer. Hit-and-run oncogenesis is most frequently considered in the context of virally induced cancers.28 The idea that cancer progression can persist in the absence of initiating viral oncoprotein expression is supported by laboratory models in which a transformed cellular phenotype persists despite loss of viral oncoprotein expression for adenoviruses,29,30 herpesviruses,31,32 and the polyomavirus Simian virus 40 (SV40) large T antigen (TAg).33–37 Hit-and-run–mediated pathophysiology has been suggested as a pathway for polyomavirus in human brain tumors and mesotheliomas,38–40 JC virus in colorectal cancer,41 papillomaviruses in Schneiderian inverted papillomas,42 and Hepatitis B in hepatomas.43 The challenge in accepting a hit-and-run mechanism is that, by definition, the inciting event is lost in the cancer that develops, essentially precluding fulfillment of the fourth of Koch's postulates.44 Induction of chromosomal instability45 and epigenetic reprogramming46 have been suggested as mechanisms that maintain transformed cell growth in the absence of the initiating virus. Other groups have suggested that in fact a small proportion of virally infected cells persist and secrete paracrine growth factors that maintain abnormal growth of a larger majority of uninfected cancer cells.47 Duration of exposure to an oncogene may play a role in the development of secondary oncogenic adaptations. For example, longer durations of infection are associated with a higher risk of neoplastic transformation in papillomavirus-induced cervical cancer;48 liver cancer incidence increases as the duration of Hepatitis C infection extends past 25 years;49 and in a mouse model of TAg-induced salivary epithelial cell dysplasia, abnormal activation of the cyclin-Cdk-Rb pathway by prolonged exposure to the viral oncoprotein maintains the dysplastic phenotype when TAg is downregulated.37 These in vivo phenomenon can be modeled in some tissue culture cells in vitro. For example, long-term exposure to estrogen can induce persistent changes in gene expression in the estrogen receptor alpha (ERα)-positive MCF7 cell line.50
Determine if adaptive change predicts response to therapy targeted to primary oncogenic stimulus
Experimentally, a conditional system, such as the tetracycline responsive gene expression system51 in which oncogene expression can be directly turned on and off in experimental animals, is helpful in establishing experiments that can test whether or not an oncogene induces oncogene addiction52–58 or a hit-and-run type of oncogenesis that leaves mutations or other genetic changes behind that then maintain carcinogenesis.33,36,37,54,55,59,60 In Myc-induced oncogenesis, investigations have found that p53 or thrombspondin-160 and CD4+ T cells61 can be required for regression following loss of Myc, with CD4+ T cells playing a role in regression following loss of Breakpoint cluster region-Abelson (BCR-ABL) as well.61 Activating mutations in K-ras2 can contribute to maintenance of Myc-induced carcinogenesis independent from expression of the myc oncogene.54,55,59 In lung tumors and lymphomas induced by K-ras(G12D) or Myc, levels of phosphorylated extracellular signal-regulated kinase (Erk) 1 and 2, Akt1, signal transducer and activator of transcription (STAT) 3/5, and p38 may predict whether or not a cancer will regress when the initiating oncogene is downregulated.56 In TAg-induced salivary dysplasia, upregulated expression of phosphorylated retinoblastoma (pRb) and transcription factor Dp-1 predict that dysplasia will reverse when TAg is downregulated, whereas low expression levels portend nonreversal.37
Removal of an oncogenic stimulus can result in the appearance of previously unnoticed pathophysiology
Conditional experimental systems also enable examination of cellular and biochemical changes that occur after the initiating oncogenic stimulus has been downregulated. In the case of TAg expression in striated duct cells in the submandibular salivary gland, dysplasia reverses when TAg is downregulated at four months of age but persists when TAg is downregulated at seven months of age.33,36,37 Before TAg downregulation at the two different ages, there are relatively few differences in gene expression as detected by cDNA array analysis,36 while elevated levels of pRB and Dp-1 in four-month-old mice have been the only differences on the protein level identified to date. In contrast, within days of TAg downregulation multiple differences in protein expression levels and extent of phosphorylation appear.37 Phosphorylated Rb levels increase in the seven-month-old mice but decrease in the four-month-old mice. p21 and p27 rise in four-month-old mice, paralleling the cell cycle arrest observed at that age when TAg is downregulated, but remain low in the seven-month-old mice that exhibit persistent cell cycle activation. Cdk4, Cdk6, and cyclin D1 levels rise initially at both ages after TAg downregulation, but this rise is reversed in the four-month-old mice, whereas it persists in the seven-month-old mice. Dp-1 levels fall in the four-month-old mice but increase in the seven-month-old mice. These experiments illustrate that underlying cellular and biochemical pathophysiology may only be revealed when an initiating oncogenic stimulus is removed. There are analogies in the response of breast cancer cells to tamoxifen, a mixed ERα antagonist/agonist used for primary and secondary prevention of ERα-positive breast cancer.62 Mucin4, which can reactivate the HER2 pathway, is upregulated in breast cancer cells following combined treatment with tamoxifen and targeted HER2 therapy.63 Moreover, tamoxifen-resistant cells are reported to demonstrate upregulation of a variety of proteins including methylated-DNA-protein-cysteine S-methyltransferase (MGMT),64 increased expression or phosphorylation of NF-κB pathway proteins p50, RelB, and p65,65 and downregulation of miRNA (miR)-375.66 It is possible that changes like these found in breast cancer cells can also occur in preneoplastic cells, compromising the impact of tamoxifen as a preventive. Measuring the cellular and biochemical changes that occur after targeting an oncogenic stimulus can provide a fuller understanding of the underlying pathophysiology in preneoplasia, as well as cancer, and may be useful in predicting which resistance pathways will develop.
Elucidating the pathological changes that are disease-driving
In the case of TAg-induced age-dependent reversible and irreversible hyperplasia and dysplasia, there are multiple molecular dissimilarities.37 Identification of the disease-driving molecular aberrations requires developing experimental approaches that can isolate and identify the key driver. In this case, abnormal activation of the Cdk4/6 pathway, and Cdk4 in particular, was found to be the culprit, but it was identified only through a series of experiments.37 First, related pathway molecules Cdk6 and cyclin D1 were eliminated as essential drivers by themselves because both molecules were significantly downregulated by administration of rexinoid X receptor (RxR) and peroxisome proliferator-activated receptor gamma (PPARγ) agonists, but dysplasia was not reversed. Successful reversal occurred only with the orally available Cdk4/6 inhibitor (PD-0332991), which downregulated Cdk4/Cdk6 and cyclin D1. This illustrated that Cdk4 is an essential driver of the dysplastic phenotype. Other experiments showed that the differences in p21, p27, and phosphatase 2A expression levels were immaterial in dysplasia reversal.36,37
Similar to the salivary gland experimental system, loss of cell cycle checkpoints also occurs during breast cancer progression. Some endocrine-resistant breast cancers in women are reported to exhibit higher levels of pRb, consistent with loss of cell cycle control.67 The same orally available Cdk4/6 inhibitor (PD-0332991), used successfully to reverse previously irreversible salivary dysplasia,37 has been suggested as an aide to antihormone therapy for ER-positive breast cancer.67–70 Activation of Cdk4/6 can occur secondary to estrogen pathway activation. But different primary drivers, even in the same pathway, may stimulate different secondary adaptations. For example, in genetically engineered mouse models, aromatase overexpression, but not ERα overexpression, stimulates increased levels of Cdk2.71 In preclinical studies, Cdk1/2 inhibitors have been shown to have activity in anti-estrogen–resistant breast cancer cells.72 As discussed earlier, levels of Erk1/2, Akt1, and STAT3/5 are possible predictors of reversal (versus nonreversal) when K-ras (G12D) or Myc are downregulated in lung tumors and lymphomas of experimental mice.56 Levels of Erk1/2 and STAT3/5 phosphorylation are significantly increased in mammary epithelial cell preneoplasia triggered by either ERα or aromatase overexpression, whereas phosphorylated Akt is increased in the aromatase over-expressing mice.71 Both of these mouse models demonstrate resistance to the ERα downregulator ICI 182, 780 (Faslodex®, AstraZeneca Pharmaceuticals LP, Wilmington, DE)73 when it is used as an agent to promote regression of preneoplasia, but the percentage of mice demonstrating resistance is higher in the aromatase overexpressing mice.71 It is possible that the higher degree of resistance found in the aromatase overexpressing mice is related to the fact that these mice demonstrate increased expression levels of Cdk2 and phosphorylated Akt, whereas the ERα overexpressing mice do not. Experiments analogous to those that identified Cdk4 as a primary driver in the TAg-induced salivary dysplasia could identify which, if any, of the identified molecular aberrations might be secondary adaptations contributing to antihormone resistance.
Cancer prevention approaches that target disease-initiating stimuli and secondary adaptations
The message from experimental studies of cancer progression and reversal is that when oncogene addiction is not the case, secondary pathophysiological adaptations induced by oncogene exposure may need to be identified and targeted for successful therapy (Table 1). Studies in salivary dysplasia indicate that these changes can occur not only in cancer cells but in preneoplastic cells as well. In some cases, preventive therapy may be more appropriately configured as “biomodulation” therapy that not only aims to disarm the initial disease-causing stimulus but also targets the secondary adaptations that can occur as a cell and tissue adapt to an abnormal growth stimulus.
Table 1.
Summary of published examples of secondary adaptations that are associated with disease maintenance following downregulation of an initiating cancer stimulus
| After downregulation/inactivation/antagonism of initiating stimulus | |||||||
|---|---|---|---|---|---|---|---|
| Initiating stimulus | disease maintenance after down-regulation of initiating stimulus | Accompany disease maintenance | Experimentally defined as disease-sustaining secondary adaptations | Disease regression factors | Experimental system | Tissue | References |
| SV40TAg | Low levels of pRb and Dp-1 | Increased levels of Dp-1, Cdk4, Cdk6, cyclin D1, and phosphatase 2A; low p21 and p27 | Increased levels of Cdk4 | Genetically engineered mice | Submandibular salivary gland | 37 | |
| Myc | Increased Erk1/2, Akt1, Stat3/5, p38 phosphorylation | Kras2 activating mutation | Kras2-activating mutation | p53, thrombospondin 1, CD4+ cells | Genetically engineered mice | Kras2 activating mutation: mammary tumors | 54–56, 59–61 |
| Disease regression factors: hematopoietic tumors, T cell acute lymphoblastic leukemia | |||||||
| Wnt-1 | Kras2-activating mutation | Genetically engineered mice | Mammary tumors | 54 | |||
| BCR-ABL | CD4+ cells | Genetically engineered mice | Pro-B cell leukemia | 61 | |||
| K-ras (G12D) | Increased Erk1/2, Akt1, Stat3/5, p38 phosphorylation | Genetically engineered mice | Lung and lymphatic tissue | 56 | |||
| ERα | Tamoxifen: increased Mucin4; increased MGMT; increased expression/ phosphorylation NF-κB pathway p50, RelB, and p65; decreased miR-375 | Increased NF-κB pathway activity or decreased miR-375 following tamoxifen | Mucin4: MCF7/HER2–18 xenografts | Breast cancer | 63–66, 76, 82 | ||
| Raloxifene: increased EGFR and Her2/Neu | Increased Her2/Neu following raloxifene | MGMT: breast cancer patients | |||||
| Estrogen deprivation: up-regulated PDGF/Abl pathway | NF-κB pathway, miR-375, EGFR/HER2 | ||||||
| PDGF/Abl: | |||||||
| MCF7 cell variants | |||||||
Development of such strategies might advance breast cancer prevention approaches. For example, tamoxifen is the only FDA-approved drug for chemoprevention of invasive breast cancer in premenopausal high-risk women; however, for every ten women who take tamoxifen, one to two will still develop invasive breast cancer. Both ERα+ and ERα− disease is found in these cases.62 Similarly, while tamoxifen is reported to reduce development of noninvasive breast cancer (ductal carcinoma in situ (DCIS)), it does not absolutely prevent it.74 In fact, some estimate as many as 40% of tamoxifen-treated breast cancer patients relapse.75 These observations are not inconsistent with the prevalence of preexisting and acquired tamoxifen resistance in breast cancers. Because tamoxifen is already approved, it is logical to look for approaches that might improve tamoxifen efficacy, as any clinical trial would need evaluation against tamoxifen as the standard for premenopausal women. Postmenopausal women have the option of selecting raloxifene but, like tamoxifen, resistance to raloxifene also has been reported in experimental models.76 Raloxifene-resistant tumor cells demonstrated increased expression of both epidermal growth factor receptor (Egrf) (ERBB) and v-erb-b2 erythroblastic leukemia viral oncogene homolog 2, neuro/glioblastoma derived oncogene homolog (avian) (ERBB2). Experiments using pharmaceuticals targeted to the proteins encoded by these two RNAs, gefitinib for ERBB (epidermal growth factor receptor) and trastuzumab for ERBB2 (HER2/Neu) revealed that the upregulated ERBB2 was a critical secondary adaptation driving tumor growth but not ERBB. More recently, aromatase inhibitors (AIs) have been shown to have a role in breast cancer prevention for postmenopausal women.77–81 Here, experimental models have shown that the platelet-derived growth factor (PDGF)/Abl pathway is upregulated in response to estrogen deprivation.82 Women with AI-resistant breast cancer have been shown to respond better to the combination of tamoxifen with everolimus, an oral inhibitor of the mammalian target of rapamycin, compared with tamoxifen alone,83 illustrating the theme that preemptively targeting both primary disease-initiating stimuli as well as predicted secondary adaptations can improve therapeutic outcome.
Thinking of cancer prevention as biomodulation with attention not only to the initiating stimulus but also encompassing consequent alterations in cellular and biochemical pathways may provide a more robust approach to developing effective chemoprevention. State-of-the-art approaches to accomplish this may include the application of RNAseq, proteomics, and metabolomics to identify the multiplexed changes that occur as part of the process of biomodulation in epithelial tissue that has become preneoplastic. Validation of specific targets within what may be a multitude of changes will be challenging and difficult to accomplish utilizing only previous clinical trial designs. More personalized and intensive comprehensive studies that incorporate serial testing of the response to candidate agents and include the use of novel approaches for primary cell culture, both epithelial and stromal, as well as evaluation of systemic factors such as immune response, may be required. The use of genetically engineered mouse models are likely to facilitate development of this comprehensive approach as they provide a renewable source of material for testing the positive and negative predictive value and sensitivity of specific algorithms used for the development of new chemopreventive approaches targeting both the original lesion and the secondary changes that occur in the course of cancer development.
Acknowledgments
NIH NCI RO1 CA112176 (P.A.F.), NIH NCI 2RO1 CA88041–1OS1 (P.A.F.), WCU (World Class University) program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (R31–10069) (P.A.F.), The Susan B. Komen Breast Cancer Foundation KG080359 (P.A.F.), NIH IG20 RR025828–01 (Rodent Barrier Facility Equipment), and NIH NCI 5P30CA051008 (Histology and Tissue; Genomic and Epigenomics and Animal Shared Resources).
Conflicts of interest
The author declares no conflicts of interest.
References
- 1.Simenhoff ML, et al. Biomodulation of the toxic and nutritional effects of small bowel bacterial overgrowth in end-stage kidney disease using freeze-dried Lactobacillus acidophilus. Miner. Electrolyte Metab. 1996;22:92–96. [PubMed] [Google Scholar]
- 2.Cerea G, et al. Biomodulation of cancer chemotherapy for metastatic colorectal cancer: a randomized study of weekly low-dose irinotecan alone versus irinotecan plus the oncostatic pineal hormone melatonin in metastatic colorectal cancer patients progressing on 5-fluorouracil-containing combinations. Anticancer Res. 2003;23:1951–1954. [PubMed] [Google Scholar]
- 3.Hill ME, et al. A phase I study of the biomodulation of capecitabine by docetaxel and gemcitabine (mGTX) in previously untreated patients with metastatic adenocarcinoma of the pancreas. Cancer Chemother. Pharmacol. 2011;67:511–517. doi: 10.1007/s00280-010-1348-3. [DOI] [PubMed] [Google Scholar]
- 4.Ardalan B, Luis R, Jaime M, Franceschi D. Biomodulation of fluorouracil in colorectal cancer. Cancer Invest. 1998;16:237–251. doi: 10.3109/07357909809039773. [DOI] [PubMed] [Google Scholar]
- 5.Sotos GA, Grogan L, Allegra CJ. Preclinical and clinical aspects of biomodulation of 5-fluorouracil. Cancer Treat. Rev. 1994;20:11–49. doi: 10.1016/0305-7372(94)90009-4. [DOI] [PubMed] [Google Scholar]
- 6.Bertino JR. Biomodulation of 5-fluorouracil with antifolates. Semin. Oncol. 1997;24:S18–52–S18–56. [PubMed] [Google Scholar]
- 7.Köhne CH, et al. Effective biomodulation by leucovorin of high-dose infusion fluorouracil given as a weekly 24-hour infusion: results of a randomized trial in patients with advanced colorectal cancer. J. Clin. Oncol. 1998;16:418–426. doi: 10.1200/JCO.1998.16.2.418. [DOI] [PubMed] [Google Scholar]
- 8.Harte RJ, et al. Tumor, normal tissue, and plasma pharmacokinetic studies of fluorouracil biomodulation with N-phosphonacetyl-L-aspartate, folinic acid, and interferon alfa. J. Clin. Oncol. 1999;17:1580–1588. doi: 10.1200/JCO.1999.17.5.1580. [DOI] [PubMed] [Google Scholar]
- 9.Morita T, Matsuzaki A, Suzuki K, Tokue A. Role of thymidine phosphorylase in biomodulation of fluoropyrimidines. Curr. Pharm. Biotechnol. 2001;2:257–267. doi: 10.2174/1389201013378662. [DOI] [PubMed] [Google Scholar]
- 10.Greenberg HS, et al. Radiosensitization with carotid intra-arterial bromodeoxyuridine +/– 5-fluorouracil biomodulation for malignant gliomas. Neurology. 1994;44:1715–1720. doi: 10.1212/wnl.44.9.1715. [DOI] [PubMed] [Google Scholar]
- 11.Deutsch E, Kaliski A, Maggiorella L, Bourhis J. New strategies to interfere with radiation response: ‘biomodulation’ of radiation therapy. Cancer Radiother. 2005;9:69–76. doi: 10.1016/j.canrad.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 12.Al-Watban FAH, Andres BL. Laser biomodulation of normal and neoplastic cells. Lasers Med. Sci. 2012;27:1039–1043. doi: 10.1007/s10103-011-1040-9. [DOI] [PubMed] [Google Scholar]
- 13.Pugliese LS, Medrado AP, Reis SR, Andrade Zde A. The influence of low-level laser therapy on biomodulation of collagen and elastic fibers. Pesqui. Odontol. Bras. 2003;17:307–313. doi: 10.1590/s1517-74912003000400003. [DOI] [PubMed] [Google Scholar]
- 14.van Breugel HH, Bär PR. Power density and exposure time of He-Ne laser irradiation are more important than total energy dose in photo-biomodulation of human fibroblasts in vitro. Lasers Surg. Med. 1992;12:528–537. doi: 10.1002/lsm.1900120512. [DOI] [PubMed] [Google Scholar]
- 15.Pires-Oliveira DAA, Oliveira RF, Machado AHA, et al. Laser biomodulation on L 929 cell culture. Photomed. Laser Surg. 2010;28:167–171. doi: 10.1089/pho.2008.2269. [DOI] [PubMed] [Google Scholar]
- 16.Ricci R, Pazos MC, Borges RE, Pacheco-Soares C. Biomodulation with low-level laser radiation induces changes in endothelial cell actin filaments and cytoskeletal organization. J. Photochem. Photobiol. B. 2009;95:6–8. doi: 10.1016/j.jphotobiol.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 17.Sagar SM, Wong RK. Chinese medicine and biomodulation in cancer patients—part one. Curr. Oncol. 2008;15:42–48. doi: 10.3747/co.2008.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McAllister SS, Weinberg RA. Tumor-host interactions: a far-reaching relationship. J. Clin. Oncol. 2010;28:4022–4028. doi: 10.1200/JCO.2010.28.4257. [DOI] [PubMed] [Google Scholar]
- 19.Polyak K, Kalluri R. The role of the microenvironment in mammary gland development and cancer. Cold Spring Harb. Perspect. Biol. 2010;2:a003244. doi: 10.1101/cshperspect.a003244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scheel C, Weinberg RA. Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin. Cancer Biol. 2012 doi: 10.1016/j.semcancer.2012.04.001. Apr 23. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arendt LM, Rudnick JA, Keller PJ, Kuperwasser C. Stroma in breast development and disease. Semin. Cell Dev. Biol. 2010;21:11–18. doi: 10.1016/j.semcdb.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Janakiram NB, Mohammed A, Bronze MS, Rao CV. Prophylactic vaccine approach for colon and pancreatic cancers: present and future. Curr Med Chem. 2012;19:3664–3678. doi: 10.2174/092986712801661022. [DOI] [PubMed] [Google Scholar]
- 23.Torti D, Trusolino L. Oncogene addiction as a foundational rationale for targeted anti-cancer therapy: promises and perils. EMBO Mol. Med. 2011;3:623–636. doi: 10.1002/emmm.201100176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kharas MG, Daley GQ. From hen house to bedside: tracing Hanafusa's legacy from avian leukemia viruses to SRC to ABL and beyond. Genes Cancer. 2010;1:1164–1169. doi: 10.1177/1947601911407327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antonescu CR. The GIST paradigm: lessons for other kinase-driven cancers. J. Pathol. 2011;223:251–261. doi: 10.1002/path.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng CJ, Slack FJ. The duality of OncomiR addiction in the maintenance and treatment of cancer. Cancer J. 2012;18:232–237. doi: 10.1097/PPO.0b013e318258b75b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bachireddy P, Rakhra K, Felsher DW. Immunology in the clinic review series; focus on cancer: multiple roles for the immune system in oncogene addiction. Clin. Exp. Immunol. 2012;167:188–194. doi: 10.1111/j.1365-2249.2011.04514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niller HH, Wolf H, Minarovits J. Viral hit and run-oncogenesis: genetic and epigenetic scenarios. Cancer Lett. 2011;305:200–217. doi: 10.1016/j.canlet.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 29.Pfeffer A, Schubbert R, Orend G, et al. Integrated viral genomes can be lost from adenovirus type 12-induced hamster tumor cells in a clone-specific, multistep process with retention of the oncogenic phenotype. Virus Res. 1999;59:113–127. doi: 10.1016/s0168-1702(98)00131-2. [DOI] [PubMed] [Google Scholar]
- 30.Nevels M, Täuber B, Spruss T, et al. ‘Hit-and-run’ transformation by adenovirus oncogenes. J. Virol. 2001;75:3089–3094. doi: 10.1128/JVI.75.7.3089-3094.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stevenson PG, May JS, Connor V, Efstathiou S. Vaccination against a hit-and-run viral cancer. J. Gen. Virol. 2010;91:2176–2185. doi: 10.1099/vir.0.023507-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen Y, Zhu H, Shenk T. Human cytomagalovirus IE1 and IE2 proteins are mutagenic and mediate ‘hit-and-run’ oncogenic transformation in cooperation with the adenovirus E1A proteins. Proc. Natl. Acad. Sci. U.S.A. 1997;94:3341–3345. doi: 10.1073/pnas.94.7.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ewald D, et al. Time-sensitive reversal of hyperplasia in transgenic mice expressing SV40 T antigen. Science. 1996;273:1384–1386. doi: 10.1126/science.273.5280.1384. [DOI] [PubMed] [Google Scholar]
- 34.Furth PA. SV40 rodent tumour models as paradigms of human disease: transgenic mouse models. Dev. Biol. Stand. 1998;94:281–287. [PubMed] [Google Scholar]
- 35.Furth PA, Li M, Hennighausen L. Studying development of disease through temporally controlled gene expression in the salivary gland. Ann. N. Y. Acad. Sci. 1998;842:181–187. doi: 10.1111/j.1749-6632.1998.tb09646.x. [DOI] [PubMed] [Google Scholar]
- 36.Tilli MT, et al. Loss of protein phosphatase 2A expression correlates with phosphorylation of DP-1 and reversal of dysplasia through differentiation in a conditional mouse model of cancer progression. Cancer Res. 2003;63:7668–7673. [PubMed] [Google Scholar]
- 37.Cabrera MC, et al. The CDK4/6 inhibitor PD0332991 reverses epithelial dysplasia associated with abnormal activation of the Cyclin-CDK-Rb pathway. Cancer Prev. Res. 2012;5:810–821. doi: 10.1158/1940-6207.CAPR-11-0532-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weggen S, et al. Low frequency of SV40, JC and BK polyomavirus sequences in human medulloblastomas, meningiomas and ependymomas. Brain Pathol. 2000;10:85–92. doi: 10.1111/j.1750-3639.2000.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khalili K, Del Valle L, Otte J, et al. Human neurotropic polyomavirus, JCV, and its role in carcinogenesis. Oncogene. 2003;22:5181–5191. doi: 10.1038/sj.onc.1206559. [DOI] [PubMed] [Google Scholar]
- 40.Barbanti-Brodano G, et al. Simian virus 40 infection in humans and association with human diseases: results and hypotheses. Virology. 2004;318:1–9. doi: 10.1016/j.virol.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 41.Hori R, et al. Detection of JC virus DNA sequences in colorectal cancers in Japan. Virchows Arch. 2005;447:723–730. doi: 10.1007/s00428-005-0014-3. [DOI] [PubMed] [Google Scholar]
- 42.Lawson W, Schlecht NF, Brandwein-Gensler M. The role of the human papillomavirus in the pathogenesis of Schneiderian inverted papillomas: an analytic overview of the evidence. Head Neck Pathol. 2008;2:49–59. doi: 10.1007/s12105-008-0048-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hessein M, et al. Hit-and-run mechanism of HBV-mediated progression to hepatocellular carcinoma. Tumori. 2005;91:241–247. doi: 10.1177/030089160509100306. [DOI] [PubMed] [Google Scholar]
- 44.Walker L, Levine H, Jucker M. Koch's postulates and infectious proteins. Acta Neuropathol. 2006;112:1–4. doi: 10.1007/s00401-006-0072-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ricciardiello L, et al. Induction of chromosomal instability in colonic cells by the human polyomavirus JC virus. Cancer Res. 2003;63:7256–7262. [PubMed] [Google Scholar]
- 46.Paschos K, Allday MJ. Epigenetic reprogramming of host genes in viral and microbial pathogenesis. Trends Microbiol. 2010;18:439–447. doi: 10.1016/j.tim.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tognon M, Corallini A, Martini F, et al. Oncogenic transformation by BK virus and association with human tumors. Oncogene. 2003;22:5192–5200. doi: 10.1038/sj.onc.1206550. [DOI] [PubMed] [Google Scholar]
- 48.Rodríguez AC, et al. Longitudinal study of human papillomavirus persistence and cervical intraepithelial neoplasia grade 2/3: critical role of duration of infection. J. Natl. Cancer Inst. 2010;102:315–324. doi: 10.1093/jnci/djq001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Strauss R, Törner A, Duberg A-S, et al. Hepatocellular carcinoma and other primary liver cancers in hepatitis C patients in Sweden—a low endemic country. J. Viral Hepat. 2008;15:531–537. doi: 10.1111/j.1365-2893.2008.00979.x. [DOI] [PubMed] [Google Scholar]
- 50.Englert NA, Spink BC, Spink DC. Persistent and non-persistent changes in gene expression result from long-term estrogen exposure of MCF-7 breast cancer cells. J. Steroid Biochem. Mol. Biol. 2011;123:140–150. doi: 10.1016/j.jsbmb.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 51.Furth PA, et al. Temporal control of gene expression in transgenic mice by a tetracycline-responsive promoter. Proc. Natl. Acad. Sci. U. S. A. 1994;91:9302–9306. doi: 10.1073/pnas.91.20.9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Z, et al. Inducible and repressable oncogene-addicted hepatocellular carcinoma in Tet-on xmrk transgenic zebrafish. J. Hepatol. 2012;56:419–425. doi: 10.1016/j.jhep.2011.07.025. [DOI] [PubMed] [Google Scholar]
- 53.Medina PP, Nolde M, Slack FJ. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature. 2010;467:86–90. doi: 10.1038/nature09284. [DOI] [PubMed] [Google Scholar]
- 54.Jang JW, Boxer RB, Chodosh LA. Isoform-specific ras activation and oncogene dependence during MYC- and Wnt-induced mammary tumorigenesis. Mol. Cell. Biol. 2006;26:8109–8121. doi: 10.1128/MCB.00404-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.D’Cruz CM, et al. c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nat. Med. 2001;7:235–239. doi: 10.1038/84691. [DOI] [PubMed] [Google Scholar]
- 56.Tran PT, et al. Survival and death signals can predict tumor response to therapy after oncogene inactivation. Sci. Transl. Med. 2011;3:103ra99. doi: 10.1126/scitranslmed.3002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Choi PS, et al. Lymphomas that recur after MYC suppression continue to exhibit oncogene addiction. Proc. Natl. Acad. Sci. U. S. A. 2011;108:17432–17437. doi: 10.1073/pnas.1107303108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Giuriato S, et al. Conditional TPM3-ALK and NPM-ALK transgenic mice develop reversible ALK-positive early B-cell lymphoma/leukemia. Blood. 2010;115:4061–4070. doi: 10.1182/blood-2008-06-163386. [DOI] [PubMed] [Google Scholar]
- 59.Tran PT, et al. Combined Inactivation of MYC and K-Ras oncogenes reverses tumorigenesis in lung adenocarcinomas and lymphomas. PLoS ONE. 2008;3:e2125. doi: 10.1371/journal.pone.0002125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Giuriato S, et al. Sustained regression of tumors upon MYC inactivation requires p53 or thrombospondin-1 to reverse the angiogenic switch. Proc. Natl. Acad. Sci. U. S. A. 2006;103:16266–16271. doi: 10.1073/pnas.0608017103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rakhra K, et al. CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell. 2010;18:485–498. doi: 10.1016/j.ccr.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fisher B, et al. Tamoxifen for the prevention of breast cancer: current status of the National Surgical Adjuvant Breast and Bowel Project P-1 study. J. Natl. Cancer Inst. 2005;97:1652–1662. doi: 10.1093/jnci/dji372. [DOI] [PubMed] [Google Scholar]
- 63.Chen AC, et al. Upregulation of mucin4 in ER-positive/HER2-overexpressing breast cancer xenografts with acquired resistance to endocrine and HER2-targeted therapies. Breast Cancer Res. Treat. 2012;134:583–593. doi: 10.1007/s10549-012-2082-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bobustuc GC, et al. MGMT inhibition restores ERα functional sensitivity to anti-estrogen therapy. Mol. Med. 2012 doi: 10.2119/molmed.2012.00010. April 27. [Epub ahead of print]. doi: 10.2119/molmed.2012.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yde CW, Emdal KB, Guerra B, Lykkesfeldt AE. NFκB signaling is important for growth of antiestrogen resistant breast cancer cells. Breast Cancer Res. Treat. 2012;135:67–78. doi: 10.1007/s10549-012-2053-1. [DOI] [PubMed] [Google Scholar]
- 66.Ward A, et al. Re-expression of microRNA-375 reverses both tamoxifen resistance and accompanying EMT-like properties in breast cancer. Oncogene. 2012 doi: 10.1038/onc.2012.128. April 16. [Epub ahead of print]. doi: 10.1038/onc.2012.128. [DOI] [PubMed] [Google Scholar]
- 67.Lange CA, Yee D. Killing the second messenger: targeting loss of cell cycle control in endocrine-resistant breast cancer. Endocr. Relat. Cancer. 2011;18:C19–C24. doi: 10.1530/ERC-11-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thangavel C, et al. Therapeutically activating RB: reestablishing cell cycle control in endocrine therapy-resistant breast cancer. Endocr. Relat. Cancer. 2011;18:333–345. doi: 10.1530/ERC-10-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sutherland RL, Musgrove EA. CDK inhibitors as potential breast cancer therapeutics: new evidence for enhanced efficacy in ER+ disease. Breast Cancer Res. 2009;11:112. doi: 10.1186/bcr2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Finn RS, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11:R77. doi: 10.1186/bcr2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Díaz-Cruz ES, Sugimoto Y, Gallicano GI, et al. Comparison of increased aromatase versus ERα in the generation of mammary hyperplasia and cancer. Cancer Res. 2011;71:5477–5487. doi: 10.1158/0008-5472.CAN-10-4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Johnson N, et al. Pre-clinical evaluation of cyclin-dependent kinase 2 and 1 inhibition in anti-estrogen-sensitive and resistant breast cancer cells. Br. J. Cancer. 2010;102:342–350. doi: 10.1038/sj.bjc.6605479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Robertson JF. Faslodex (ICI 182, 780), a novel estrogen receptor downregulator–future possibilities in breast cancer. J. Steroid Biochem. Mol. Biol. 2001;79:209–212. doi: 10.1016/s0960-0760(01)00138-8. [DOI] [PubMed] [Google Scholar]
- 74.Wickerham DL, et al. The use of tamoxifen and raloxifene for the prevention of breast cancer. Recent Results Cancer Res. 2009;181:113–119. doi: 10.1007/978-3-540-69297-3_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guler G, et al. Wwox and Ap2gamma expression levels predict tamoxifen response. Clin. Cancer Res. 2007;13:6115–6121. doi: 10.1158/1078-0432.CCR-07-1282. [DOI] [PubMed] [Google Scholar]
- 76.O’Regan RM, et al. Development and therapeutic options for the treatment of raloxifene-stimulated breast cancer in athymic mice. Clin. Cancer Res. 2006;12:2255–2263. doi: 10.1158/1078-0432.CCR-05-2584. [DOI] [PubMed] [Google Scholar]
- 77.Smith J, et al. A pilot study of letrozole for one year in women at enhanced risk of developing breast cancer: effects on mammographic density. Anticancer Res. 2012;32:1327–1331. [PubMed] [Google Scholar]
- 78.Pujol P, et al. Uptake of a randomized breast cancer prevention trial comparing letrozole to placebo in BRCA1/2 mutations carriers: the LIBER trial. Fam. Cancer. 2012;11:77–84. doi: 10.1007/s10689-011-9484-4. [DOI] [PubMed] [Google Scholar]
- 79.Litton JK, Bevers TB, Arun BK. Exemestane in the prevention setting. Ther. Adv. Med. Oncol. 2012;4:107–112. doi: 10.1177/1758834012438214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Decensi A, Dunn BK, Puntoni M, et al. Exemestane for breast cancer prevention: a critical shift? Cancer Discov. 2012;2:25–40. doi: 10.1158/2159-8290.CD-11-0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Goss PE, et al. Exemestane for breast-cancer prevention in postmenopausal women. N. Engl. J. Med. 2011;364:2381–2391. doi: 10.1056/NEJMoa1103507. [DOI] [PubMed] [Google Scholar]
- 82.Weigel MT, et al. Preclinical and clinical studies of estrogen deprivation support the PDGF/Abl pathway as a novel therapeutic target for overcoming endocrine resistance in breast cancer. Breast Cancer Res. 2012;14:R78. doi: 10.1186/bcr3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bachelot T, et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: a GINECO Study. J. Clin. Oncol. 2012;30:2718–2724. doi: 10.1200/JCO.2011.39.0708. [DOI] [PubMed] [Google Scholar]