

Abstract
Treatment-related toxicity can be life-threatening and is the primary cause of interruption or discontinuation of chemotherapy for acute lymphoblastic leukemia (ALL), leading to an increased risk of relapse. Mercaptopurine is an essential component of continuation therapy in all ALL treatment protocols worldwide. Genetic polymorphisms in thiopurine S-methyltransferase (TPMT) are known to have a marked effect on mercaptopurine metabolism and toxicity; however, some patients with wild-type TPMT develop toxicity during mercaptopurine treatment for reasons that are not well understood. To identify additional genetic determinants of mercaptopurine toxicity, a genome-wide analysis was performed in a panel of human HapMap cell lines to identify trans-acting genes whose expression and/or single-nucleotide polymorphisms (SNPs) are related to TPMT activity, then validated in patients with ALL. The highest ranking gene with both mRNA expression and SNPs associated with TPMT activity in HapMap cell lines was protein kinase C and casein kinase substrate in neurons 2 (PACSIN2). The association of a PACSIN2 SNP (rs2413739) with TPMT activity was confirmed in patients and knock-down of PACSIN2 mRNA in human leukemia cells (NALM6) resulted in significantly lower TPMT activity. Moreover, this PACSIN2 SNP was significantly associated with the incidence of severe gastrointestinal (GI) toxicity during consolidation therapy containing mercaptopurine, and remained significant in a multivariate analysis including TPMT and SLCO1B1 as covariates, consistent with its influence on TPMT activity. The association with GI toxicity was also validated in a separate cohort of pediatric patients with ALL. These data indicate that polymorphism in PACSIN2 significantly modulates TPMT activity and influences the risk of GI toxicity associated with mercaptopurine therapy.
INTRODUCTION
Approximately 80% of children with acute lymphoblastic leukemia (ALL) can be cured with combination chemotherapy that almost always includes the purine anti-metabolite mercaptopurine (1,2). However, treatment-related toxicity with this medication can be life-threatening and is the primary cause of interruption or discontinuation of chemotherapy, which can increase the risk of disease recurrence. Germline polymorphisms in genes encoding drug-metabolizing enzymes can significantly influence the efficacy and toxicity of antileukemic therapy (3). For mercaptopurine, methylation of the thiol moiety, which is catalyzed by the enzyme thiopurine S-methyltransferase (TPMT), yields inactive 6-methyl-mercaptopurine (4,5). Although genetic polymorphisms in the gene encoding human TPMT are known to markedly influence mercaptopurine metabolism and toxicity, there are patients with wild-type TPMT who also develop toxicity for reasons that are not fully understood (6,7).
Mercaptopurine is used together with high-dose methotrexate in post-remission consolidation therapy for pediatric ALL; this treatment regimen is associated with gastrointestinal (GI) toxicity that can be severe and constitutes an important adverse effect of ALL consolidation therapy (2).
In a previous study investigating the genetic basis of inter-individual differences in methotrexate pharmacokinetics in children with ALL, we showed that single-nucleotide polymorphisms (SNPs) in solute carrier organic anion transporter family, member 1B1 (SLCO1B1) are associated with methotrexate clearance and GI toxicity during consolidation therapy (8).
Hapmap cell lines (from Utah residents with ancestry from northern and western Europe; CEU) are a model system suitable for genome-wide studies of genetic polymorphisms influencing pharmacological phenotypes that can be measured in cell lines, despite limited statistical power due to the relatively small sample size (9,10). We previously used a genome-wide approach to rank 17 542 genes for their association with TPMT activity in HapMap cells: a TPMT haplotype significantly predicted TPMT phenotype; however, haplotypes of 96 genes ranked higher than TPMT, indicating the possibility that genetic polymorphism in other genes could influence TPMT activity (11). This prior genome-wide SNP analysis provided important insights into trans-acting genes influencing TPMT activity, but it is known that the incorporation of gene expression with genome-wide SNP analyses can reduce false positives and increase the robustness of discoveries (12), consistent with trait-associated SNPs being more likely to be expression quantitative trait loci (13).
Therefore, to identify additional genetic variability influencing TPMT activity, a genome-wide analysis was performed to identify trans-genes whose expression or SNPs are related to TPMT activity in a panel of human HapMap cell lines, with subsequent validation of their effects on TPMT activity and mercaptopurine toxicity in patients receiving mercaptopurine therapy.
RESULTS
TPMT activity in HapMap (CEU) cells
TPMT activity was measured in the 30 HapMap CEU trios (Fig. 1). The median value was 28.5 U/109 cells, with a range of 9.1–90.4. Genotype for SNP rs1142345, the most common SNP defining a non-functional TPMT allele in humans (TPMT*3A and TPMT*3C both contain rs1142345), was assessed in all HapMap CEU cell lines considered. As depicted in Figure 1 and as we have previously reported (11), the four cell lines from subjects heterozygous for the rs1142345 SNP (i.e. AG genotype) had significantly lower TPMT activity than the 83 cell lines with the AA wild-type genotype for this SNP (P-value Welch test = 0.00026).
Figure 1.
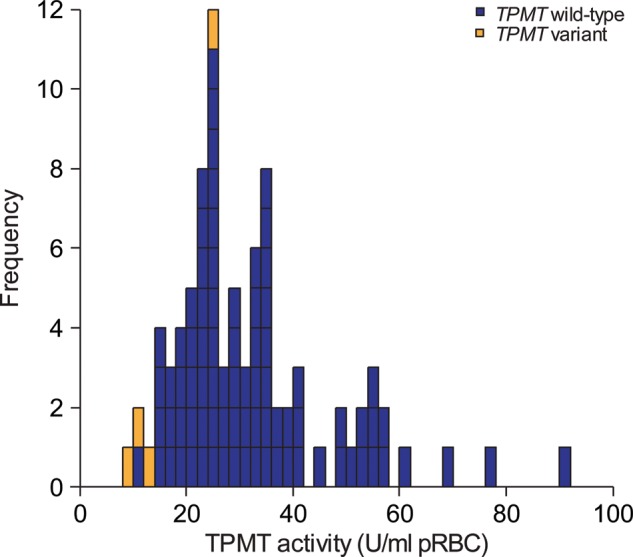
Distribution of TPMT activity measured in 87 HapMap CEU cell lines. Each rectangle represents the TPMT activity value measured in one cell line. Blue rectangles represent cell lines from individuals with wild-type TPMT, whereas orange rectangles represent cell lines from individuals with variant TPMT (rs1142342 AG genotype); the four cell lines from subjects heterozygous for the rs1142345 SNP (i.e. AG genotype) had significantly lower TPMT activity than the 83 cell lines with the AA wild-type genotype for this SNP (P-value Welch test = 0.00026) (11).
Genes whose expression or SNPs were significantly associated with TPMT activity in HapMap cells
A summary of genes whose mRNA expression or SNPs were associated with TPMT activity in the HapMap cells is depicted in Figure 2. Association between gene expression and TPMT activity was analyzed for 15 661 genes: the expression of 38 genes was significantly correlated with TPMT activity with a P-value <5 × 10−5 (Table 1). Among these 38 genes, only one gene also had a SNP annotated to its sequence that was significantly associated with TPMT activity at a P-value <5 × 10−4: protein kinase C and casein kinase substrate in neurons 2 (PACSIN2) mRNA expression (P-value linear model = 1.20 × 10−5, correlation estimate = 0.36) and SNP rs2413739 (P-value linear model = 4.6 × 10−4) were both significantly associated with TPMT activity.
Figure 2.

Diagram describing the analysis strategy to identify the most important trans-genes whose expression and SNPs are associated with TPMT activity in the HapMap CEU population. Among the genes whose expression is highly associated with TPMT activity (false discovery rate 1.6%), PACSIN2 is the only one that has significant SNPs (P-value <0.0005) associated with TPMT activity.
Table 1.
Genes whose expression was statistically associated with TPMT activity (P-value lower than 5 × 10−5) in 87 Human HapMap CEU cell lines
| Transcript ID | Chromosome | Gene ID | Gene symbol | Reference sequence | Correlation estimate | P-value |
|---|---|---|---|---|---|---|
| 3764471 | chr17 | 9110 | MTMR4 | NM_004687 | 0.55 | 1.48E−07 |
| 3454006 | chr12 | 91010 | FMNL3 | NM_198900, NM_175736 | 0.52 | 5.27E−07 |
| 3496409 | chr13 | 2262 | GPC5 | NM_004466 | 0.48 | 6.91E−07 |
| 3290785 | chr10 | 8030 | CCDC6 | NM_005436 | 0.49 | 1.07E−06 |
| 3456212 | chr12 | 7786 | MAP3K12 | NM_006301 | 0.47 | 1.35E−06 |
| 3403595 | chr12 | 50856 | CLEC4A | NM_016184,NM_194450,NM_194447, NM_194448 | 0.39 | 1.85E−06 |
| 3275729 | chr10 | 3559 | IL2RA | NM_000417 | 0.44 | 2.46E−06 |
| 3662417 | chr16 | 1071 | CETP | NM_000078 | 0.39 | 2.91E−06 |
| 3832256 | chr19 | 10653 | SPINT2 | NM_021102 | 0.44 | 3.20E−06 |
| 3063795 | chr7 | 55262 | C7orf43 | NM_018275 | 0.46 | 5.96E−06 |
| 3156307 | chr8 | 5747 | PTK2 | NM_005607,NM_153831 | 0.43 | 6.57E−06 |
| 3883819 | chr20 | 22839 | DLGAP4 | NM_183006,NM_001042486,NM_014902 | 0.40 | 7.88E−06 |
| 3338968 | chr11 | 55191 | NADSYN1 | NM_018161 | 0.46 | 1.08E−05 |
| 3426257 | chr12 | 8835 | SOCS2 | NM_003877 | 0.44 | 1.11E−05 |
| 3962678 | chr22 | 11252 | PACSIN2 | NM_007229 | 0.38 | 1.20E−05 |
| 3965784 | chr22 | 5600 | MAPK11 | NM_002751 | 0.37 | 1.42E−05 |
| 3621080 | chr15 | 9333 | TGM5 | NM_201631, NM_004245 | 0.34 | 1.44E−05 |
| 3608638 | chr15 | 9899 | SV2B | NM_014848 | 0.39 | 1.47E−05 |
| 3802980 | chr18 | 1824 | DSC2 | NM_004949, NM_024422 | 0.41 | 1.50E−05 |
| 3470193 | chr12 | 1240 | CMKLR1 | NM_004072 | 0.40 | 1.53E−05 |
| 3479438 | chr12 | 55743 | CHFR | NM_018223 | 0.49 | 1.69E−05 |
| 2803329 | chr5 | 10409 | BASP1 | NM_006317 | 0.47 | 1.78E−05 |
| 3362263 | chr11 | 23258 | RAB6IP1 | NM_015213 | 0.45 | 2.27E−05 |
| 3830484 | chr19 | 2867 | FFAR2 | NM_005306 | 0.35 | 2.32E−05 |
| 2766492 | chr4 | 201895 | C4orf34 | NM_174921 | 0.39 | 2.72E−05 |
| 3476097 | chr12 | 8099 | CDK2AP1 | NM_004642 | 0.40 | 2.85E−05 |
| 3833793 | chr19 | 53916 | RAB4B | NM_016154 | 0.45 | 3.14E−05 |
| 3405032 | chr12 | 2120 | ETV6 | NM_001987 | 0.50 | 3.23E−05 |
| 3240987 | chr10 | 1326 | MAP3K8 | NM_005204 | 0.48 | 3.25E−05 |
| 2412312 | chr1 | 22996 | C1orf34 | NM_001080494 | 0.41 | 3.52E−05 |
| 3802924 | chr18 | 1825 | DSC3 | NM_001941, NM_024423 | 0.38 | 3.65E−05 |
| 2485688 | chr2 | 23177 | CEP68 | NM_015147 | 0.39 | 3.77E−05 |
| 3662612 | chr16 | 89970 | RSPRY1 | NM_133368 | 0.48 | 3.82E−05 |
| 2556752 | chr2 | 200734 | SPRED2 | NM_181784 | 0.47 | 3.84E−05 |
| 3753860 | chr17 | 256957 | C17orf66 | NM_152781 | 0.31 | 4.31E−05 |
| 2500275 | chr2 | 10018 | BCL2L11 | NM_138621, NM_006538 | -0.40 | 4.47E−05 |
| 3470597 | chr12 | 54434 | SSH1 | NM_018984 | 0.38 | 4.66E−05 |
| 3338192 | chr11 | 595 | CCND1 | NM_053056 | 0.36 | 4.72E−05 |
P-values are from linear mixed effects model with formula: log2(TPMT activity) ∼ gene expression, adjusted for relatedness.
Relation of PACSIN2 SNP and TPMT activity in patients
The effect on erythrocyte TPMT activity of the PACSIN2 SNP identified in the HapMap population (rs2413739) was assessed in ALL patients who either had a wild-type TPMT genotype or were heterozygous for the most common variant TPMT alleles. TPMT activity and genotype was evaluated in 299 patients; the PACSIN2 genotype was successfully determined for 286 of the 299 patients for whom TPMT activity and the genotype were measured (for 13 patients, PACSIN2 genotyping could not be determined because of technical reasons). This analysis showed that both the TPMT and PACSIN2 variants were significantly associated with TPMT activity (Fig. 3). Indeed SNPs in both genes had a significant association with TPMT enzyme activity in a univariate analysis (P-values from linear model: TPMT = 6.0 × 10−16; PACSIN2 = 0.027). Moreover, these SNPs (TPMT, PACSIN2) were also significantly related to TPMT activity in a multivariate analysis that combined the two SNPs in a multilocus genotype (P-values from linear model, TPMT = 3.0 × 10−16 and PACSIN2 = 0.011); this multivariate model explained 22.4% of variability in TPMT activity. Effects of TPMT and PACSIN2 genotypes were significant in patients after adjusting for ethnicity, evaluated as self-reported race, addressing the possibility of a potential confounding effect of ancestry (P-values from linear model adjusted for ethnicity, TPMT = 6.4 × 10−16; PACSIN2 = 0.034). The PACSIN2 SNP was significantly related to TPMT activity both in patients with the wild-type TPMT genotype (explaining 1.8% of variability, P-value from linear model = 0.031) and in those with the variant TPMT genotype (explaining 30.8% of variability, P-value from linear model = 0.0091); the difference in the median TPMT activity in patients with wild-type versus variant PACSIN2 genotypes (SNP rs2413739) was 1.2 U/ml packed erythrocytes for patients with wild-type TPMT and 2.6 U/ml packed erythrocytes for those with variant TPMT (Fig. 3).
Figure 3.

The box plot shows TPMT activity in 286 patients with ALL as a function of the TPMT rs1142345 [A719G]/PACSIN2 rs2413739 multilocus genotype. There were no GG genotypes for TPMT rs1142345 and therefore all variants genotypes for TPMT are heterozygous. SNPs in both genes had a significant association with TPMT enzyme activity in a univariate analysis (P-values from linear model with formula TPMT activity–genotype, considering an additive effect for the genotype: TPMT = 6.0 × 10−16; PACSIN2 P = 0.027). Moreover, these SNPs (TPMT, PACSIN2) were also significantly related to TPMT activity in a multivariate analysis that combined the two SNPs in a multilocus genotype (TPMT P-value from linear model = 3.0 × 10−16 and PACSIN2 P-value from linear model = 0.011). The PACSIN2 genotype had a significant effect even in the subgroup with the wild-type TPMT genotype (P-value from linear model = 0.031) and in the subgroup with the variant TPMT genotype (P-value from linear model = 0.0091).
PACSIN2 SNP had a significant association with GI toxicity in patients treated with mercaptopurine
The entire cohort of Total 13B patients was considered for the genetic association discovery study for GI toxicity in patients: of 247 children with newly diagnosed ALL enrolled on the Total 13B study, 208 were evaluable herein. We excluded one patient who died shortly after diagnosis and did not have a remission DNA sample, five patients with Down syndrome, and one patient with cystic fibrosis because their underlying diseases can influence toxicity; we also excluded 32 patients who came off study for various reasons: induction failure (n = 1), ALL relapse (n = 12), second cancer (n = 3), stem cell transplantation (n = 3), non-compliance (n = 3), non-GI toxicity (n = 3), refused therapy (n = 2), death unrelated to disease or therapy (n = 2) or other reasons (n = 3) (14). Among the 208 children with ALL receiving therapy according to St Jude Total 13B protocol and evaluable for the toxicity during consolidation therapy, 16 (7.7%) developed severe (Grade 3–4) GI toxicity; the TPMT genotype was available for all 208 patients enrolled, whereas PACSIN2 and SLCO1B1 genotypes were determined in 189 (for 19 patients, PACSIN2 and/or SLCO1B1 could not be determined because of technical reasons). For these 189 children with complete genotyping data for TPMT, SLCO1B1 and PACSIN2, the frequency of GI toxicity was 8.5%; among these patients, deficiency in TPMT activity predisposed to an increased incidence of severe GI toxicity during consolidation therapy which comprised methotrexate (2 g/m2/week) and mercaptopurine (75 mg/m2/day, in all patients during consolidation therapy, regardless of TPMT genotype). Indeed, among nine patients with a variant TPMT allele, the frequency of GI toxicity was 33%, compared with 7.2% in patients with wild-type TPMT (P-value from logistic regression model = 0.0058, Fig. 4). PACSIN2 SNP rs2413739 also had a significant association with GI toxicity during consolidation therapy: the frequency of toxicity was 2.1, 9.1 and 13.2%, respectively, for the CC, CT and TT allele (P-value from logistic regression model = 0.046, Fig. 4). An SLCO1B1 SNP (rs11045879) that we have previously reported to be associated with GI toxicity in ALL patients (8) was also associated with the incidence of GI toxicity: none of the patients with the SLCO1B1 CC or CT genotype had GI toxicity, whereas 11.8% of the patients with the wild-type SLCO1B1 TT genotype had GI toxicity (P-value from logistic regression model = 0.012, Fig. 4).
Figure 4.
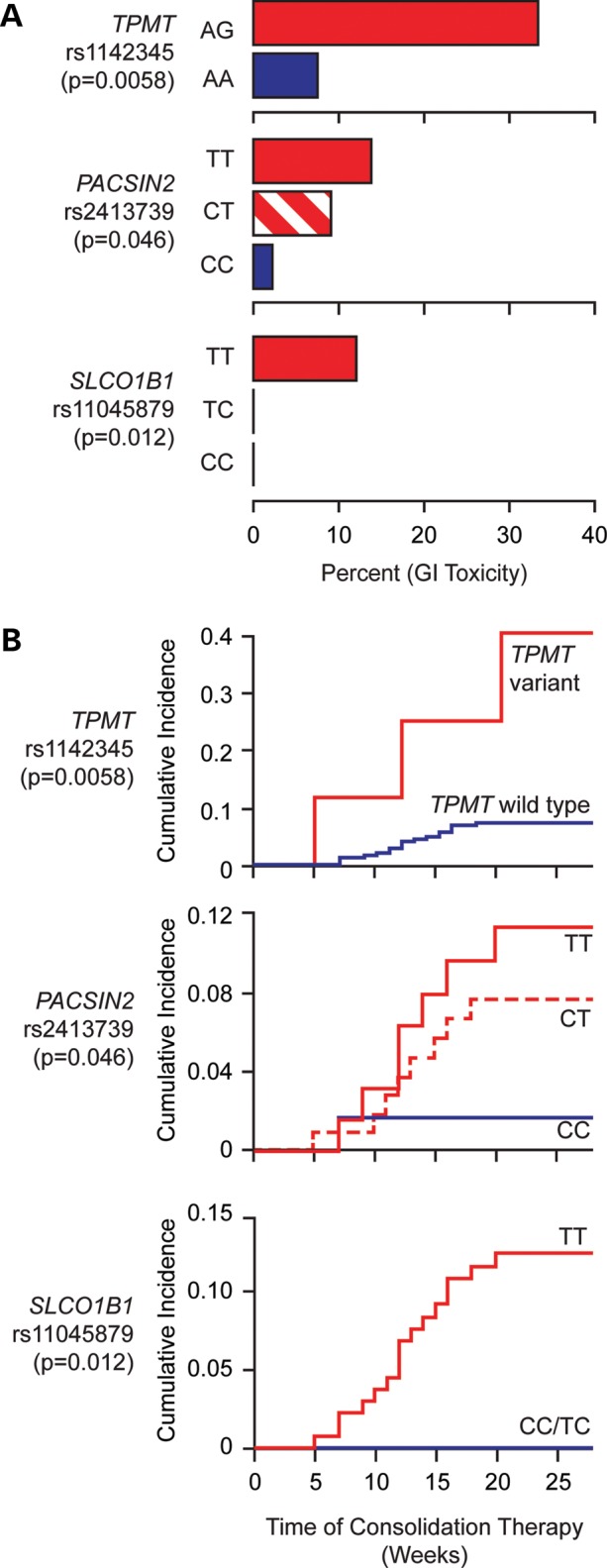
The frequency of severe (Grade 3–4) GI toxicity during consolidation therapy in patients with ALL treated according to the St Jude Total 13B protocol as a function of TPMT rs1142345, PACSIN2 rs2413739, SLCO1B1 rs11045879 genotypes. Panel A shows barplots reporting the percentage of patients developing GI toxicity and the corresponding genotypes. Panel B represents cumulative incidence plots of GI toxicity and the corresponding genotypes for the same patients. There were no GG genotypes for TPMT rs1142345 and therefore all variants genotypes for TPMT are heterozygous. Among nine patients with a variant TPMT allele, the frequency of gastrointestinal toxicity was 33%, compared with 7.2% in patients with the wild-type TPMT (P-value from logistic regression model = 0.0058); PACSIN2 SNP rs2413739 also had a significant association with GI toxicity during consolidation therapy: the frequency of toxicity was 2.1, 9.1 and 13.2%, respectively, for the CC, CT and TT allele (P-value from logistic regression model = 0.046). An SLCO1B1 SNP (rs11045879) that we have previously reported to be associated with GI toxicity in ALL patients (9) was also associated with the incidence of GI toxicity: none of the patients with the SLCO1B1 CC or CT genotype had GI toxicity, whereas 11.8% of the patients with the wild-type SLCO1B1 TT genotype had GI toxicity (P-value from logistic regression model = 0.012).
TPMT, SLCO1B1 and PACSIN2 multi-locus genotype and GI toxicity
Multivariate logistic regression models along with classification and regression trees analyses (CART) were used to assess the effects of TPMT, SLCO1B1 and PACSIN2 SNPs and their interaction in predisposing to GI toxicity. As summarized in Table 2, all three SNPs had a significant relation with the GI toxicity in the multivariate analysis; the effect of the considered SNPs on the incidence of GI toxicity was confirmed after adjusting for ethnicity as defined by self-reported race, addressing the possibility of a potential confounding effect determined by ancestry (Table 2). CART analysis to evaluate interactions between the genotypes in reference to their relation to the GI toxicity (Fig. 5) revealed that the TPMT genotype best distinguished GI toxicity probability (split 1 on CART, Fig. 5), with the presence of the variant TPMT allele conferring an increased probability of GI toxicity (33.3%) compared with patients with wild-type TPMT (7.2%, P-value from logistic regression model on subgroup = 0.0058). Among the nine patients with variant TPMT, no additional effect of PACSIN2 and SLCO1B1 was evident, although the number of these patients was relatively low. Among the 180 patients with wild-type TPMT, the most important genotype to distinguish the probability of GI toxicity was the SLCO1B1 SNP (split 2 on CART, Fig. 5): none of the 52 patients with a C (variant) allele for the SLCO1B1 SNP developed toxicity, whereas the probability among the 128 patients with the TT genotype was 10.2% (P-value from logistic regression model on subgroup = 0.017). Among the 128 patients with a wild-type allele for TPMT and TT SCLO1B1 genotypes (split 3 on CART) the probability of GI toxicity in patients with the PACSIN2 TT allele was two times higher than in those with a C allele (15.8 versus 7.8%), albeit not statistically significant given the small number of patients (P-value from a logistic regression model on subgroup = 0.17).
Table 2.
Multivariate logistic regression model for the effects of PACSIN2, SLCO1B1 and TPMT genotypes on the incidence of GI toxicity
| Multivariate logistic regression model for the incidence of GI toxicity |
Multivariate logistic regression model for the incidence of GI toxicity, adjusted for ethnicitya |
|||||
|---|---|---|---|---|---|---|
| Effectb | 95% Confidence interval | P-value | Effectb | 95% Confidence interval | P-value | |
| PACSIN2 rs2413739 genotype (T versus C) | 2.06 | 1.01–4.56 | 0.040 | 2.09 | 1.03–4.58 | 0.037 |
| SLCO1B1 rs11045879 genotype (T versus C) | 12.61 | 1.69–1613.81 | 0.017 | 11.49 | 1.56–1466.31 | 0.0016 |
| TPMT rs1142345 genotype (A versus G) | 6.03 | 1.23–27.26 | 0.0085 | 6.58 | 1.11–33.33 | 0.045 |
aEthnicity was defined by self-reported race (white for 143 patients, black for 37 patients, Asian for 1 patient and other for 8 patients).
bThe coefficient or effect size represents the odds ratio for toxicity associated with each variable listed. For example, a patient with a G allele for TPMT rs1142345 would have an odd ratio toxicity of 6.03 in comparison with a patient with the A allele.
Figure 5.
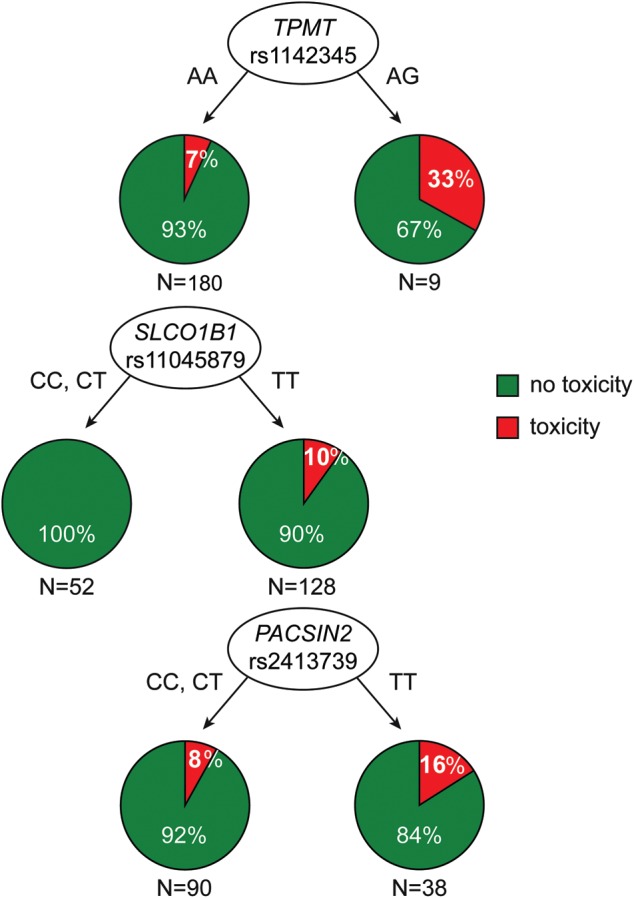
Classification and regression tree analysis for genetic predictors influencing those patients at an increased risk for GI toxicity. The three SNPs have independent effects, with TPMT being ranked before SLCO1B1 and PACSIN2.
Validation of TPMT, SLCO1B1 and PACSIN2 SNPs on mercaptopurine-related GI toxicity in an independent cohort of patients
To validate the effect of TPMT, SLCO1B1 and PACSIN2 SNPs on the incidence of severe GI toxicity during consolidation therapy of pediatric ALL, we assessed an independent set of patients who were treated on the Associazione Italiana di Ematologia ed Oncologia Pediatrica (Italy); Berlin-Frankfurt-Münster (Germany, Austria, Switzerland) ALL Study Group (AIEOP-BFM-ALL) 2000 protocol. PACSIN2 SNP rs2413739 also showed a correlation with the incidence of GI toxicity in this cohort; the frequency of cases with GI toxicity was 28, 32 and 51% in the CC, CT and TT genotypes, respectively (P-value from logistic regression model = 0.032, Fig. 6), as did SNP rs737782, in LD with rs2413739, with frequencies of GI toxicity of 26, 31 and 51% in the CC, CG and GG genotypes, respectively (P-value from a logistic regression model = 0.015, Fig. 6). However, for SNP rs5996259, not in LD with rs2413739, there was not an association with toxicity (P-value from a logistic regression model = 0.32). The TPMT and SLCO1B1 SNPs did not show a significant association with the incidence of GI toxicity in the Italian cohort, perhaps due to the lower dosage of mercaptopurine used in this protocol (25 versus 75 mg/m2/day).
Figure 6.

The frequency of severe (Grade 3–4) GI toxicity during consolidation therapy in the validation cohort, the Italian AIEOP-BFM-ALL 2000 and genotypes in PACSIN2; PACSIN2 SNP rs2413739 and rs737782 resulted associated with the incidence of GI toxicity in this cohort (P-values from logistic regression model, 0.032 and 0.015, respectively).
Knock-down of PACSIN2 was associated with reduction in TPMT activity and mRNA in a human B-lineage leukemia cell line
Knock-down of PACSIN2 was performed in the NALM6 B-lineage ALL human cell line, heterologously expressing either TPMT*1 or TPMT*3A. Through the heterologous expression of TPMT*1, enzymatic activity increased approximately six times in comparison with TPMT activity in native NALM6 (mean activity 176.8 versus 28.9 U/109 cells, P-value Welch test = 1.1 × 10−6), whereas TPMT*3A expression lead to only a modest (<2-fold) increase in TPMT activity (41.5 versus 28.9 U/109 cells, P-value Welch t-test = 0.028), due to the known rapid degradation of TPMT protein encoded by TPMT*3A in comparison with TPMT*1 (15).
After knock-down of PACSIN2, a significant (30%) decrease in TPMT activity was observed in cells heterologously expressing TPMT*1 (123.8 U/109 cells in PACSIN2 knock-down versus 176.8 U/109 in a knock-down scrambled vector control, P-value Welch t-test = 0.0036), but less so in cells expressing TPMT*3A (47.3 versus 41.5 U/109 cells, P-value Welch t-test = 0.090) (Fig. 7).
Figure 7.

TPMT activity in NALM6 cells heterologously expressing TPMT*1 or TPMT*3A and with knock-down of PACSIN2. After knock-down of PACSIN2, a significant (30%) decrease in TPMT activity was observed in cells heterologously expressing TPMT*1 (P-value Welch t-test = 0.0036).
Measurement of TPMT mRNA in these cells confirmed the presence of the knock-down of PACSIN2 and showed an effect (reduction in 30%) on the expression of TPMT mRNA comparable with the effect on TPMT activity (Supplementary Material, Fig. S1).
Pathway analysis of genes modified after PACSIN2 knock-down identifies autophagy as significant process
For global gene expressions in NALM6 with PACSIN2 knock-down in comparison with a scrambled vector control, 13 990 Affymetrix Human Genome U133 Plus 2.0 Array mRNA probe sets passed the filtering criteria based on signal intensity and probe sets present/absent call. Of these, 20 probe sets changed at least 50% [P-value LPE test <1 × 10−3, false discovery rate (FDR) = ∼65%] (Tables 3). To test the reliability of microarray data, we validated four genes [PACSIN2, CSMD1, KIAA0226L/protein phosphatase 1, regulatory (inhibitor) subunit 2 pseudogene 4 (PPP1R2P4), SH3-domain GRB2-like endophilin B1 (SH3GLB1)], by real-time PCR, confirming changes as assessed by microarray analysis (Supplementary Material, Fig. S2).
Table 3.
Genes whose mRNA was differentially expressed after knock-down of PACSIN2 in human NALM6 leukemia cells
| Gene name | Gene symbol | Probe set ID | Ratio KD versus Ctrl | P-value | False discovery rate |
|---|---|---|---|---|---|
| CUB and Sushi multiple domains 1 | CSMD1 | 231223_at | 3.3 | 1.87E−10 | 0 |
| Not annotated to a gene | NA | 1562093_at | 19.2 | 2.28E−10 | 0 |
| Protein kinase C and casein kinase substrate in neurons 2 | PACSIN2 | 201651_s_at | 0.4 | 1.54E−07 | 7.00E−04 |
| Microtubule-associated protein 2 | MAP2 | 225540_at | 9.6 | 5.72E−07 | 0.002 |
| Not annotated to a gene | NA | 216532_x_at | 2.9 | 1.05E−05 | 0.0293 |
| Transcription elongation factor A (SII)-like 4 | TCEAL4 | 202371_at | 3.2 | 2.90E−05 | 0.0674 |
| KIAA0226L/protein phosphatase 1, regulatory (inhibitor) subunit 2 pseudogene 4 | KIAA0266L, PPP1R2P4 | 44790_s_at | 0.5 | 4.13E−05 | 0.0822 |
| Inhibitor of DNA binding 2, dominant negative helix-loop-helix protein | ID2 | 201565_s_at | 2.6 | 7.61E−05 | 0.1327 |
| Granzyme K (granzyme 3; tryptase II) | GZMK | 206666_at | 0.5 | 8.91E−05 | 0.1379 |
| Not annotated to a gene | NA | 241883_x_at | 4.9 | 0.000117 | 0.1632 |
| Secreted phosphoprotein 1 | SPP1 | 209875_s_at | 2.1 | 0.000166 | 0.2107 |
| Coiled-coil domain containing 88A | CCDC88A | 219387_at | 2.0 | 0.000205 | 0.2233 |
| Protein phosphatase 1, regulatory (inhibitor) subunit 2 pseudogene 4 | KIAA0266L, PPP1R2P4 | 219471_at | 0.5 | 0.000208 | 0.2233 |
| Not annotated to a gene | NA | 1560525_at | 3.5 | 0.000331 | 0.3293 |
| Intersectin 1 (SH3 domain protein) | ITSN1 | 209298_s_at | 3.6 | 0.000588 | 0.4962 |
| SH3-domain GRB2-like endophilin B1 | SH3GLB1 | 210101_x_at | 0.5 | 0.000596 | 0.4962 |
| Granzyme A (granzyme 1, cytotoxic T-lymphocyte-associated serine esterase 3) | GZMA | 205488_at | 0.4 | 0.00064 | 0.4962 |
| Zinc finger protein 75a | ZNF75A | 227670_at | 3.8 | 0.000641 | 0.4962 |
| tRNA methyltransferase 11 homolog (S. cerevisiae) | TRMT11 | 218877_s_at | 2.4 | 0.00074 | 0.5426 |
| odz, odd Oz/ten-m homolog 2 (Drosophila) | ODZ2 | 231867_at | 2.8 | 0.000927 | 0.646 |
Enrichment analysis using MetaCore GeneGO revealed that genes within certain functional pathways were over-represented among those differentially expressed after knock-down of PACSIN2 (Table 4): the most highly ranked GeneGo pathways included vesicle formation (rank #1, P-value = 0.014), osteopontin and granzyme signaling (rank #2, P-value = 0.022) and autophagy (rank #3, P-value = 0.023). Highly ranked GeneOntology processes included cellular membrane organization (rank #1, P-value = 0.00010), membrane organization (rank #2, P-value = 0.00010) and cellular component organization (rank #3, P-value = 0.00016). GeneGo biological process networks included proteolysis in cell-cycle and apoptosis (rank #1, P-value = 0.010) and actin filaments in cytoskeleton (rank #2, P-value = 0.020); complete results for enrichment analysis are provided in Supplementary Material, Table S1a–c.
Table 4.
Enrichment analysis of mRNA differentially expressed in human NALM6 leukemia cells, between PACSIN2 controls and knock-down
| MetaCore GeneGo | Ranking | Pathway name | P-value | Ratio probe sets significant/in pathway | Significant mRNAs in pathway |
|---|---|---|---|---|---|
| GeneGo pathway maps | #1 | wtCFTR and delta508 traffic/clathrin-coated vesicles formation (norm and CF) | 1.36E−02 | 1/19 | SH3GLB1 |
| #2 | Development_Osteopontin signaling in osteoclasts and | 2.15E−02 | 1/30 | SPP1 | |
| #2 | Apoptosis and survival_Granzyme A signaling | 2.15E−02 | 1/30 | GZMA | |
| #3 | Autophagy | 2.29E−02 | 1/32 | SH3GLB1 | |
| GeneGoProcess networks | #1 | Proteolysis: proteolysis in cell cycle and apoptosis | 1.01E−02 | 2/125 | GZMA, GZMK |
| #2 | Cytoskeleton: actin filaments | 1.95E−02 | 2/176 | PACSIN, PACSIN2 | |
| GeneOntology Processes | #1 | Cellular membrane organization | 1.01E−04 | 5/653 | SH3GLB1, CCDC88A, ITSN1, PACSIN, PACSIN2 |
| #2 | Membrane organization | 1.03E−04 | 5/656 | SH3GLB1, CCDC88A, ITSN1, PACSIN, PACSIN2 | |
| #3 | Negative regulation of cellular component organization | 1.61E−04 | 4/375 | MAP2, SPP1, PACSIN, PACSIN2 |
DISCUSSION
This study has documented that PACSIN2 modulates TPMT activity and that a common SNP in PACSIN2 is significantly associated with the incidence of mercaptopurine-related GI toxicity in children with ALL. Although the importance of TPMT genetic polymorphisms in determining the mercaptopurine response and hematological toxicity in children with ALL is very well established (mechanistically and clinically) (6,16), this is the first in vivo evidence that genetic variation in a second gene (PACSIN2) also influences TPMT activity. PACSIN2 was identified by genome-wide SNP and gene expression analyses of the human HapMap cells (9,10,12,13) for which TPMT activity was also determined (11), and the association was validated in patients. Indeed, the highest ranking gene for association with TPMT activity in the HapMap cell lines based on gene expression and SNPs (after adjusting for TPMT genotype) was PACSIN2, which carries a SNP (rs2413739) that is significantly associated with both TPMT activity and severe GI toxicity in children receiving mercaptopurine for the treatment of ALL. Moreover, knock-down of PACSIN2 mRNA in human leukemia cells (NALM6) heterologously expressing TPMT*1 resulted in significantly lower TPMT activity and mRNA.
PACSIN2, also called syndapin II, is a member of the ‘protein kinase C and casein kinase substrate in neurons’ family of proteins; they are involved in linking the actin cytoskeleton with vesicle formation by regulating tubulin polymerization and exert their function mainly through protein–protein interactions with different substrates, such as dynamin or N-WASP (17). There are data indicating that PACSIN2 has a role in various biological processes, including endocytosis (18), cell-cycle control (19) and autophagy (20).
To identify additional genes and biological processes that are potentially altered by PACSIN2, we identified genes whose expression changed significantly in human leukemia cells in which the PACSIN2 had been knocked-down, revealing actin and cytoskeleton (17,21), membrane organization (22) and autophagy (20) as processes/pathways significantly altered by knock-down of PACSIN2. Identification of autophagy was driven largely by the reduction in expression of SH3GLB1 by PACSIN2 knock-down. SH3GLB1, also known as endophilin B1/BIF-1, interacts with the UVRAG-Beclin 1, and contributes to the activation of autophagy (23); it is plausible that SH3GLB1 has a role in the fission of membranes from the Golgi during autophagosome formation (24). The effects of PACSIN2 on autophagy is further supported by significant changes we observed in a second gene C13orf18 (also known as KIAA0226-like/PPP1R2P4), which has recently been shown to be involved in autophagy (25). The PACSIN2 role in autophagy may also be involved in the mechanism of its effects on TPMT activity, as autophagy has been linked to degradation of the TPMT protein codified by variant TPMT*3A and to a lesser extent wild-type TPMT*1 (26). Taken together, our results indicate that PACSIN2 may influence TPMT activity through an effect on TPMT mRNA levels and/or TPMT protein degradation. Future studies are warranted to fully elucidate the molecular mechanisms by which PACSIN2 alters TPMT activity.
The PACSIN2 SNP rs2413739 is located in an intronic region of the gene and, therefore, it is not clear how this SNP influences PACSIN2 mRNA and function. Interestingly, the UCSC Genome Browser (27) indicates that this SNP is located in an area enriched for a marker of acetylation of lysine 27 of the histone 3 protein (H3K27Ac histone mark) as determined by ChIP-Seq analyses (27,28) (Supplementary Material, Fig. S3). Therefore, the SNP rs2413739 may modulate transcription of the PACSIN2 gene by influencing the level of acetylation or the effects of the acetylation of this marker, that is thought to enhance transcription possibly by blocking the spread of the repressive histone mark H3K27Me3 (29).
In addition to showing a significant association of this PACSIN2 SNP on TPMT activity in cell lines and patients, we also show that patients with the PACSIN2 rs2413739 C genotype had higher TPMT activity and lower GI toxicity than patients with the T genotype. Furthermore, the association of this PACSIN2 SNP with GI toxicity during consolidation therapy of ALL with mercaptopurine was identified in patients treated according to the St Jude Total 13B protocol and validated in an independent cohort of Italian patients. In the St Jude Total 13B protocol, patients were treated for a short period of time (2 weeks) with relatively high doses of mercaptopurine (75 mg/m2/day, regardless of TPMT genotype) and methotrexate (2 g/m2 with folic acid rescue), which resulted in ∼8% of patients exhibiting Grade 3–4 GI toxicity; in this context, PACSIN2 effects on GI toxicity were significant and independent from those of TPMT and SLCO1B1. In the validation cohort, the post-remission consolidation phase (protocol M) was quite different and involved lower mercaptopurine doses (25 mg/m2/day) and a longer treatment time (8 versus 2 weeks) and the overall incidence of severe GI toxicity was lower (∼4% of patients versus ∼8%); in this context the PACSIN2 SNP was significantly related to the incidence of GI toxicity, whereas no effects of TPMT and SLCO1B1 could be detected. Patients with high TPMT activity may be protected from mercaptopurine toxicity (7,30) and in this study, the observation that PACSIN2 was associated with GI toxicity in both cohorts may indicate that its effects on patients with high TPMT activity are clinically relevant and evident across different protocols of consolidation therapy for pediatric ALL: indeed the most common TPMT coding SNPs do not identify patients with high TPMT activity (only low TPMT activity), whereas the PACSIN2 rs2413739 CC genotype does. Polymorphisms in the promoter region of TPMT have been previously associated with increased TPMT activity, although the clinical relevance of these observations is not clear (31). It is also plausible that PACSIN2 may influence mercaptopurine sensitivity independent of its effects on TPMT; indeed it has been shown recently to interact with Rac1 (32), which is implicated in the pharmacodynamics of thiopurines (33).
We have shown previously that during maintenance therapy of pediatric ALL according to Total 13B protocol, adjustment of mercaptopurine dose on the basis of TPMT phenotype or genotype mitigates the increased incidence of Grade 3–4 febrile neutropenia in patients with variant TPMT genotypes, allowing the effect of other genetic polymorphisms, such as those in inosine-triphosphate-pyrophosphatase to emerge (34). To explore whether the association of PACSIN2 SNP with dose-dependent mercaptopurine toxicity is via its effects on TPMT activity, we evaluated whether the PACSIN2 genotype was associated with the incidence of severe febrile neutropenia during Total 13B maintenance therapy and we could not find a significant association (P-value of a weighted logistic regression model considering time at risk = 0.41, Supplementary Material, Fig. S4). This suggests that the association of the PACSIN2 genotype with mercaptopurine-related dose-dependent toxicity occurs through modulation of TPMT activity, and not through a direct effect of PACSIN2 on cell sensitivity to mercaptopurine. However, this does not rule out tissue-specific effects of PACSIN2 on mercaptopurine sensitivity occurring in GI tissue and not in white blood cells.
In summary, the current study has identified a new genetic polymorphism influencing TPMT activity and mercaptopurine toxicity in children being treated for ALL, providing new insights into the pharmacogenomics of ALL chemotherapy.
MATERIALS AND METHODS
Cell culture
Eighty-nine Epstein–Barr virus-transformed lymphoblastoid cell lines, forming the 30 CEPH family trios genotyped in the International HapMap project, were obtained from the Coriell Institute of Medicine (http://www.ccr.coriell.org). Cell lines were cultured in RPMI-1640 media with 15% fetal bovine serum. Two cell lines with poor growth were excluded from the study.
TPMT activity in cell lines
Aliquots were obtained for each cell line while in the log phase and cytosolic cell lysates were prepared, as previously reported in detail (11). TPMT activity was measured adapting a previously described radiochemical assay (35). Briefly, the level of TPMT activity for each cell line was determined by measuring the incorporation of radioactively labeled S-adenosyl-l-methionine into mercaptopurine's inactive metabolite, methylmercaptopurine (11,35). One unit of enzyme activity represents the formation of 1 nmol of 6-methylmercaptopurine per hour of incubation. TPMT activity in cell lines was normalized per billion of cells evaluated.
Genotypes and gene expression in cell lines
SNP genotypes for CEU cell lines were downloaded from release 22 on the International HapMap project Website (http://www.HapMap.org). Gene expression measured by the Affymetrix GeneChip Human Exon 1.0 ST Array was downloaded from the website:
Ethical
We studied children enrolled as patients in two single-institution (St Jude Total 13B and Total 15) and one multi-institution (AIEOP-BFM-ALL 2000) clinical protocol for the treatment of newly diagnosed ALL. All patients and/or their parents provided informed consent for the institutional review board-approved protocols.
TPMT activity in patients
TPMT activity was measured in erythrocytes from pediatric patients with ALL enrolled at St Jude Children's Research Hospital in Total 13B or Total 15 protocols (36,37) using a previously described method (35); one unit of enzyme activity represents the formation of 1 nmol of 6-methylmercaptopurine per hour of incubation. TPMT activity in patients was normalized per milliliter of packed erythrocytes. TPMT activity was measured after patients were in complete remission from ALL (i.e. at least 60 days from diagnosis) and at least 90 days from a red blood cell transfusion. There were a total of 323 measurements for 299 patients; for patients with multiple measurements, the median value was used.
Toxicity in patients
Toxicity was evaluated during consolidation therapy for ALL according to St Jude Total 13B protocol and classified according to NCI criteria (8,36): patients developing relevant GI toxicity during consolidation therapy (Grade 3–4 mucositis and diarrhea) were defined as cases and they were compared with all other patients in the cohort who did not develop mucositis and or diarrhea and served as controls in the genetic analyses; other GI toxicity (hepatotoxicity, vomiting and pancreatic toxicity) were uncommon (1 Grade 3 hepatotoxicity, 1 Grade 3 vomiting and no pancreatic toxicity) and therefore were not considered in the present analysis. Consolidation therapy for Total 13B consisted of 2 weekly doses of high-dose methotrexate (2 g/m2 over 2 h, followed by leucovorin rescue) and daily mercaptopurine (75 mg/m2/day for 2 weeks) (38). During consolidation therapy, the dosage of mercaptopurine therapy was the same for all patients, regardless of the TPMT genotype; mercaptopurine dosages were subsequently adjusted during continuation therapy based on the TPMT genotype (6,34). The entire cohort of Total 13B patients was considered for the genetic association discovery study: of 247 children with newly diagnosed ALL enrolled on the Total 13B study, 208 were evaluable herein. We excluded one patient who died shortly after diagnosis and did not have a remission DNA sample, five patients with Down syndrome, and one patient with cystic fibrosis because their underlying diseases can influence toxicity; we also excluded 32 patients who came off study for various reasons: induction failure (n = 1), ALL relapse (n = 12), second cancer (n = 3), stem cell transplantation (n = 3), non-compliance (n = 3), non-GI toxicity (n = 3), refused therapy (n = 2), death unrelated to disease or therapy (n = 2) or other reasons (n = 3) (14). Validation cohort consisted of patients enrolled on the multi-institution AIEOP-BFM-ALL 2000 protocol. Post-remission consolidation therapy (protocol M) for the AIEOP-BFM-ALL 2000 protocol consisted of 8 weeks of treatment with four administrations of high-dose methotrexate (2 g/m2 over 24 h followed by leucovorin rescue) and daily mercaptopurine (25 mg/m2/day for 8 weeks) (39). Sixty-seven patients without Down syndrome who developed Grade 3–4 mucositis and/or diarrhea during consolidation therapy served as cases and each of them was matched with two controls from the same protocol based on sex, age, ALL lineage and ALL risk classification. The toxicity was classified by the NCI criteria (39).
Genotypes in patients
Genotyping of St Jude patients was performed for the major non-functional variant alleles of TPMT (TPMT*2, TPMT*3A and TPMT*3C, defined by SNPs rs1142345, rs1800460 and rs1800462) using methods that we have previously described in detail (6). Genotyping of SNP rs11045879 in SLCO1B1 in St Jude patients was obtained from Affymetrix 500K Array Set and Affymetrix Genome-Wide Human SNP Array 6.0. SNPs rs2413739 of PACSIN2 for St Jude patients was genotyped independently with two different methods, by DNAPrint Genomics (Sarasota, FL) and by iPlex Sequenom assay done at the University of Chicago. Genotyping of the Italian validation cohort was performed at the University of Washington in St Louis, by Sequenom technology. Quality control of genotyping was verified by repeated analysis of the same samples, using different genotyping approaches; moreover, in the validation cohort, in addition to the SNPs in TPMT, SLCO1B1 and PACSIN2 genotyped in the St Jude cohort, two additional PACSIN2 SNPs were genotyped: rs737782 and rs5996520, which were, respectively, in LD or not in LD with PACSIN2 rs2413739.
Knock-down of PACSIN2 in NALM6 heterologously expressing TPMT
The B lineage human leukemia cell line NALM6 was used to study effects of modulation of PACSIN2 expression on TPMT activity.
The heterologous expression of TPMT*1 and TPMT*3A proteins was obtained by transfection using Nucleofector (Lonza) of native NALM6 with the appropriate plasmid (Origene), according to the manufacturer instructions. For selection of stably transfected cells, after transfection, culture medium supplemented with G418 was used.
PACSIN2 knockdown was achieved in NALM6 heterologously expressing TPMT by RNA interference using a lentiviral vector-based shRNA approach from the MISSION™ TRC-Hs library (Sigma). Lentiviral particles corresponding to the MISSION shRNA SHVRS-NM_007229 target set were used as well as the MISSION Non-Target shRNA control. The specificity and efficacy of the shRNA PACSIN2 procedure were controlled by western blotting and real-time PCR, after transduction and puromycin selection.
TPMT activity was measured in these cell lines using the previously described radiochemical assay (11,35). PACSIN2 and TPMT mRNA were quantified by real-time PCR using the commercial TaqMan Gene Expression Assay (Hs00200589_m1 and Hs00909010_g1, respectively) from Applied Biosystems.
Microarray analysis
Global gene expression in NALM6 stably transfected with shRNA sequences for the knock-down of PACSIN2 or scrambled sequences (control) were evaluated using microarray analysis. Total RNA of cells harvested during logarithmic growth was isolated using Trizol reagent. For quality control, RNA purity and integrity were evaluated by measuring the OD 260/280 ratio and analyzed on Agilent Bioanalyzer. Gene expression data was measured for 30 708 transcripts by the use of the Affymetrix Human Genome U133 Plus 2.0 Array, according to the manufacturer's instructions. Validation of a panel of differentially expressed genes was performed using gene expression real-time PCR assays from Applied Biosystem (Hs00899130_m1 for CSMD1, Hs00228336_m1 for C13orf18/KIAA0266L/PPP1R2P4, Hs00211220_m1 for SH3GLB1).
Statistical analysis
Analysis was done using the software R. To identify trans-genetic factors associated with TPMT activity in the HapMap cell lines, a linear mixed effects model adjusted for relatedness was used, with log10 of TPMT activity as the dependent variable and gene expression and SNPs as the independent variables. Trans-genes were selected if they had SNPs associated with TPMT activity, using a cut-off of 5 × 10−4 for the P-value and then ranked according to the P-value for the association of gene expression with TPMT activity.
Effects of candidate SNPs on TPMT activity and GI toxicity in St Jude patients were evaluated by linear (for TPMT activity) or logistic regression (for toxicity) models, with formula phenotype–genotype; P-values are from the ANOVA test applied to these models, evaluating an additive effect of the genotype on the phenotype of interest. To check for the potential confounding effects of ancestry, analysis was performed even with models adjusted for ethnicity, determined as self-reported race [we have previously shown that in this population of patients, self-reported race was highly concordant with ethnicity determined using ancestry informative markers (14)]. CART analysis was used to evaluate interaction between genetic factors associated with toxicity in St Jude patients.
Validation of the effect of genotypes on GI toxicity in the Italian cohort was done using logistic regression models, with formula phenotype–genotype and P-values are from the ANOVA test applied to these models.
Comparison of measurements (e.g. TPMT activity, gene mRNA expression) in different genetically modified NALM6 cell lines was performed by Welch's t-test and P-values were adjusted for multiple testing by Holm's method (40).
Gene expression data from the Affymetrix chip was filtered by setting detection >500 and considering only probe sets called present by the MAS5 algorithm in at least 50% of samples. Selected gene signal values were log-2 transformed and normalized by the IQR method (so that inter-quartile ranges on all chips are set to their widest range), as implemented in the pre-process function of the LPE R package. The significance of the expression data was determined using the LPE test from the same R package and fold change, in which the null hypothesis was that no difference exists among the two groups (41). The FDR was controlled by adjusting P-values using the Benjamini–Hochberg algorithm (42). Pathway analysis of the genes differentially expressed was performed using MetaCore/GeneGo; enrichment analysis was performed evaluating GeneGo pathways, GeneOntology processes and GeneGo process networks.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
ACKNOWLEDGEMENTS
We thank the patients and their parents for their participation in this study and our clinical staff for providing outstanding protocol-based patient care. We thank Natalya Lenchik, Yan Wang, Siamac Salehy, Margaret Needham, May Chung and Yaqin Chu for their outstanding technical assistance. We thank Nancy Kornegay and Mark Wilkinson for their computer and database expertise, the staff from the St Jude Hartwell Center for Bioinformatics and Biotechnology and in particular Jay Morris, Geoff Neale and Jennifer Peters for assistance with microarray and in vitro experiments, and the staff of St Jude Biomedical Communications and in particular Julie Groff for the preparation of the figures.
Conflict of Interest statement. W.E.E. is an inventor on a patent awarded to SJCRH for the molecular diagnosis of TPMT deficiency based on the TPMT genotype and has received patent royalties. M.V.R. has also received such royalties. The other authors declared no conflict of interest.
FUNDING
Funding sources include grants from the National Institutes of Health (R37 CA36401 to W.E.E., M.V.R., C.-H.P.; R01 CA78224 to W.E.E., M.V.R., C.-H.P.); NIH Pharmacogenomics Research Grant (U01 GM 92666 to M.V.R., W.E.E., C.C., W.Y., C.-H.P.), NCI Cancer Center Support Grant to St Jude (CA21765), F32 CA141752 to S.W.P., the American Lebanese Syrian Associated Charities (ALSAC) and the Associazione Genitori Malati Emopatici Neoplastici Friuli Venezia Giulia (AGMEN).
REFERENCES
- 1.Pui C.H., Carroll W.L., Meshinchi S., Arceci R.J. Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J. Clin. Oncol. 2012;29:551–565. doi: 10.1200/JCO.2010.30.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pui C.H., Evans W.E. Treatment of acute lymphoblastic leukemia. N. Engl. J. Med. 2006;354:166–178. doi: 10.1056/NEJMra052603. [DOI] [PubMed] [Google Scholar]
- 3.Paugh S.W., Stocco G., McCorkle J.R., Diouf B., Crews K.R., Evans W.E. Cancer pharmacogenomics. Clin. Pharmacol. Ther. 2011;90:461–466. doi: 10.1038/clpt.2011.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang L., Weinshilboum R. Thiopurine S-methyltransferase pharmacogenetics: insights, challenges and future directions. Oncogene. 2006;25:1629–1638. doi: 10.1038/sj.onc.1209372. [DOI] [PubMed] [Google Scholar]
- 5.Krynetski E., Evans W.E. Drug methylation in cancer therapy: lessons from the TPMT polymorphism. Oncogene. 2003;22:7403–7413. doi: 10.1038/sj.onc.1206944. [DOI] [PubMed] [Google Scholar]
- 6.Relling M.V., Gardner E.E., Sandborn W.J., Schmiegelow K., Pui C.H., Yee S.W., Stein C.M., Carrillo M., Evans W.E., Klein T.E. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin. Pharmacol. Ther. 2011;89:387–391. doi: 10.1038/clpt.2010.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Relling M.V., Hancock M.L., Rivera G.K., Sandlund J.T., Ribeiro R.C., Krynetski E.Y., Pui C.H., Evans W.E. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J. Natl. Cancer. Inst. 1999;91:2001–2008. doi: 10.1093/jnci/91.23.2001. [DOI] [PubMed] [Google Scholar]
- 8.Trevino L.R., Shimasaki N., Yang W., Panetta J.C., Cheng C., Pei D., Chan D., Sparreboom A., Giacomini K.M., Pui C.H., et al. Germline genetic variation in an organic anion transporter polypeptide associated with methotrexate pharmacokinetics and clinical effects. J. Clin. Oncol. 2009;27:5972–5978. doi: 10.1200/JCO.2008.20.4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peters E.J., Kraja A.T., Lin S.J., Yen-Revollo J.L., Marsh S., Province M.A., McLeod H.L. Association of thymidylate synthase variants with 5-fluorouracil cytotoxicity. Pharmacogenet. Genomics. 2009;19:399–401. doi: 10.1097/FPC.0b013e328329fdec. [DOI] [PubMed] [Google Scholar]
- 10.Wheeler H.E., Dolan M.E. Lymphoblastoid cell lines in pharmacogenomic discovery and clinical translation. Pharmacogenomics. 2012;13:55–70. doi: 10.2217/pgs.11.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones T.S., Yang W., Evans W.E., Relling M.V. Using HapMap tools in pharmacogenomic discovery: the thiopurine methyltransferase polymorphism. Clin. Pharmacol. Ther. 2007;81:729–734. doi: 10.1038/sj.clpt.6100135. [DOI] [PubMed] [Google Scholar]
- 12.Gamazon E.R., Huang R.S., Cox N.J., Dolan M.E. Chemotherapeutic drug susceptibility associated SNPs are enriched in expression quantitative trait loci. Proc. Natl. Acad. Sci. USA. 2010;107:9287–9292. doi: 10.1073/pnas.1001827107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nicolae D.L., Gamazon E., Zhang W., Duan S., Dolan M.E., Cox N.J. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kishi S., Cheng C., French D., Pei D., Das S., Cook E.H., Hijiya N., Rizzari C., Rosner G.L., Frudakis T., et al. Ancestry and pharmacogenetics of antileukemic drug toxicity. Blood. 2007;109:4151–4157. doi: 10.1182/blood-2006-10-054528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tai H.L., Fessing M.Y., Bonten E.J., Yanishevsky Y., d'Azzo A., Krynetski E.Y., Evans W.E. Enhanced proteasomal degradation of mutant human thiopurine S-methyltransferase (TPMT) in mammalian cells: mechanism for TPMT protein deficiency inherited by TPMT*2, TPMT*3A, TPMT*3B or TPMT*3C. Pharmacogenetics. 1999;9:641–650. doi: 10.1097/01213011-199910000-00011. [DOI] [PubMed] [Google Scholar]
- 16.Stanulla M., Schaeffeler E., Flohr T., Cario G., Schrauder A., Zimmermann M., Welte K., Ludwig W.D., Bartram C.R., Zanger U.M., et al. Thiopurine methyltransferase (TPMT) genotype and early treatment response to mercaptopurine in childhood acute lymphoblastic leukemia. J.A.M.A. 2005;293:1485–1489. doi: 10.1001/jama.293.12.1485. [DOI] [PubMed] [Google Scholar]
- 17.Kessels M.M., Qualmann B. The syndapin protein family: linking membrane trafficking with the cytoskeleton. J. Cell Sci. 2004;117:3077–3086. doi: 10.1242/jcs.01290. [DOI] [PubMed] [Google Scholar]
- 18.Modregger J., Ritter B., Witter B., Paulsson M., Plomann M. All three PACSIN isoforms bind to endocytic proteins and inhibit endocytosis. J. Cell Sci. 2000;113(Pt 24):4511–4521. doi: 10.1242/jcs.113.24.4511. [DOI] [PubMed] [Google Scholar]
- 19.Meng H., Tian L., Zhou J., Li Z., Jiao X., Li W.W., Plomann M., Xu Z., Lisanti M.P., Wang C., et al. PACSIN 2 represses cellular migration through direct association with cyclin D1 but not its alternate splice form cyclin D1b. Cell Cycle. 2011;10:73–81. doi: 10.4161/cc.10.1.14243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szyniarowski P., Corcelle-Termeau E., Farkas T., Hoyer-Hansen M., Nylandsted J., Kallunki T., Jaattela M. A comprehensive siRNA screen for kinases that suppress macroautophagy in optimal growth conditions. Autophagy. 2011;7:892–903. doi: 10.4161/auto.7.8.15770. [DOI] [PubMed] [Google Scholar]
- 21.Qualmann B., Kelly R.B. Syndapin isoforms participate in receptor-mediated endocytosis and actin organization. J. Cell. Biol. 2000;148:1047–1062. doi: 10.1083/jcb.148.5.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Q., Navarro M.V., Peng G., Molinelli E., Goh S.L., Judson B.L., Rajashankar K.R., Sondermann H. Molecular mechanism of membrane constriction and tubulation mediated by the F-BAR protein Pacsin/Syndapin. Proc. Natl. Acad. Sci. U. S. A. 2009;106:12700–12705. doi: 10.1073/pnas.0902974106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takahashi Y., Coppola D., Matsushita N., Cualing H.D., Sun M., Sato Y., Liang C., Jung J.U., Cheng J.Q., Mule J.J., et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell. Biol. 2007;9:1142–1151. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takahashi Y., Meyerkord C.L., Hori T., Runkle K., Fox T.E., Kester M., Loughran T.P., Wang H.G. Bif-1 regulates Atg9 trafficking by mediating the fission of Golgi membranes during autophagy. Autophagy. 2011;7:61–73. doi: 10.4161/auto.7.1.14015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Behrends C., Sowa M.E., Gygi S.P., Harper J.W. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li F., Wang L., Burgess R.J., Weinshilboum R.M. Thiopurine S-methyltransferase pharmacogenetics: autophagy as a mechanism for variant allozyme degradation. Pharmacogenet. Genomics. 2008;18:1083–1094. doi: 10.1097/FPC.0b013e328313e03f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raney B.J., Cline M.S., Rosenbloom K.R., Dreszer T.R., Learned K., Barber G.P., Meyer L.R., Sloan C.A., Malladi V.S., Roskin K.M., et al. ENCODE whole-genome data in the UCSC genome browser (2011 update) Nucleic Acids Res. 2011;39:D871–D875. doi: 10.1093/nar/gkq1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosenbloom K.R., Dreszer T.R., Pheasant M., Barber G.P., Meyer L.R., Pohl A., Raney B.J., Wang T., Hinrichs A.S., Zweig A.S., et al. ENCODE whole-genome data in the UCSC Genome Browser. Nucleic Acids Res. 2010;38:D620–D625. doi: 10.1093/nar/gkp961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McLeod H.L., Coulthard S., Thomas A.E., Pritchard S.C., King D.J., Richards S.M., Eden O.B., Hall A.G., Gibson B.E. Analysis of thiopurine methyltransferase variant alleles in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 1999;105:696–700. doi: 10.1046/j.1365-2141.1999.01416.x. [DOI] [PubMed] [Google Scholar]
- 31.Roberts R.L., Gearry R.B., Bland M.V., Sies C.W., George P.M., Burt M., Marinaki A.M., Arenas M., Barclay M.L., Kennedy M.A. Trinucleotide repeat variants in the promoter of the thiopurine S-methyltransferase gene of patients exhibiting ultra-high enzyme activity. Pharmacogenet. Genomics. 2008;18:434–438. doi: 10.1097/FPC.0b013e3282f85e47. [DOI] [PubMed] [Google Scholar]
- 32.de Kreuk B.J., Nethe M., Fernandez-Borja M., Anthony E.C., Hensbergen P.J., Deelder A.M., Plomann M., Hordijk P.L. The F-BAR domain protein PACSIN2 associates with Rac1 and regulates cell spreading and migration. J. Cell Sci. 2011;124:2375–2388. doi: 10.1242/jcs.080630. [DOI] [PubMed] [Google Scholar]
- 33.Tiede I., Fritz G., Strand S., Poppe D., Dvorsky R., Strand D., Lehr H.A., Wirtz S., Becker C., Atreya R., et al. CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J. Clin. Invest. 2003;111:1133–1145. doi: 10.1172/JCI16432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stocco G., Cheok M.H., Crews K.R., Dervieux T., French D., Pei D., Yang W., Cheng C., Pui C.H., Relling M.V., et al. Genetic polymorphism of inosine triphosphate pyrophosphatase is a determinant of mercaptopurine metabolism and toxicity during treatment for acute lymphoblastic leukemia. Clin. Pharmacol. Ther. 2009;85:164–172. doi: 10.1038/clpt.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McLeod H.L., Lin J.S., Scott E.P., Pui C.H., Evans W.E. Thiopurine methyltransferase activity in American white subjects and black subjects. Clin. Pharmacol. Ther. 1994;55:15–20. doi: 10.1038/clpt.1994.4. [DOI] [PubMed] [Google Scholar]
- 36.Pui C.H., Pei D., Sandlund J.T., Ribeiro R.C., Rubnitz J.E., Raimondi S.C., Onciu M., Campana D., Kun L.E., Jeha S., et al. Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia. 2010;24:371–382. doi: 10.1038/leu.2009.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pui C.H., Campana D., Pei D., Bowman W.P., Sandlund J.T., Kaste S.C., Ribeiro R.C., Rubnitz J.E., Raimondi S.C., Onciu M., et al. Treating childhood acute lymphoblastic leukemia without cranial irradiation. N. Engl. J. Med. 2009;360:2730–2741. doi: 10.1056/NEJMoa0900386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pui C.H., Sandlund J.T., Pei D., Campana D., Rivera G.K., Ribeiro R.C., Rubnitz J.E., Razzouk B.I., Howard S.C., Hudson M.M., et al. Improved outcome for children with acute lymphoblastic leukemia: results of Total Therapy Study XIIIB at St Jude Children's Research Hospital. Blood. 2004;104:2690–2696. doi: 10.1182/blood-2004-04-1616. [DOI] [PubMed] [Google Scholar]
- 39.Schrappe M., Valsecchi M.G., Bartram C.R., Schrauder A., Panzer-Grumayer R., Moricke A., Parasole R., Zimmermann M., Dworzak M., Buldini B., et al. Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study. Blood. 2011;118:2077–2084. doi: 10.1182/blood-2011-03-338707. [DOI] [PubMed] [Google Scholar]
- 40.Holm S. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 1979;6:65–70. [Google Scholar]
- 41.Jain N., Thatte J., Braciale T., Ley K., O'Connell M., Lee J.K. Local-pooled-error test for identifying differentially expressed genes with a small number of replicated microarrays. Bioinformatics. 2003;19:1945–1951. doi: 10.1093/bioinformatics/btg264. [DOI] [PubMed] [Google Scholar]
- 42.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B. 1995;57:289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.