

Abstract
Myotonic dystrophy (DM) is a multisystemic disease caused by CTG or CCTG expansion mutations. There is strong evidence that DM1 CUG and DM2 CCUG expansion transcripts sequester muscleblind-like (MBNL) proteins and that loss of MBNL function causes alternative splicing abnormalities that contribute to disease. Because MBNL1 loss is thought to play an important role in disease and localized AAV delivery of MBNL1 partially rescues skeletal muscle pathology in DM mice, there is strong interest in MBNL1 overexpression as a therapeutic strategy. We developed the first transgenic MBNL1 overexpression mouse model (MBNL1-OE) to test the safety and efficacy of multisystemic MBNL1 overexpression. First, we demonstrate that MBNL1 overexpression is generally well-tolerated in skeletal muscle. Second, we show the surprising result that premature shifts in alternative splicing of MBNL1-regulated genes in multiple organ systems are compatible with life and do not cause embryonic lethality. Third, we show for the first time that early and long-term MBNL1 overexpression prevents CUG-induced myotonia, myopathy and alternative splicing abnormalities in DM1 mice. In summary, MBNL1 overexpression may be a valuable strategy for treating the skeletal muscle features of DM.
INTRODUCTION
Myotonic dystrophy (DM) is an adult-onset autosomal dominant multisystemic disorder that affects approximately 1 in 8000 individuals (1). Two clinically similar disorders, DM types 1 and 2, result from microsatellite repeat expansions in functionally distinct genes. DM1 is caused by a CTG expansion in the 3′ UTR of dystrophia myotonica protein kinase (DMPK) gene and DM2 results from a CCTG intronic expansion in the CCHC-type zinc finger, nucleic acid binding protein (CNBP) gene (2–5). DM1 and DM2 patients have a diverse set of clinical features including myotonia, muscle wasting, insulin resistance, cardiac arrythmias, gastrointestinal dysfunction, posterior iridescent cataracts and cognitive dysfunction (1).
Strong evidence supports a pathogenic model in which CUG/CCUG expansion RNAs accumulate in DM patient cells and contribute to disease by dysregulating alternative splicing factors. These factors include CUGBP, Elav-like Family Member 1 (CELF1) and Muscleblind-like 1 (MBNL1) (6–9). CELF1 upregulation occurs through a protein kinase C-mediated hyperphosphorylation event (10), and reduced MBNL1 activity results from sequestration of MBNL1 by CUG and CCUG expansion transcripts (11–14). Although both CELF1 upregulation and MBNL1 sequestration likely play a role in DM, recent evidence suggests that >80% of splicing abnormalities in a DM mouse model are caused by Mbnl1 loss (15). Additionally, AAV-mediated delivery of recombinant MBNL1 to the tibialis anterior muscle of the HSALR-CTG250 (HSA, human skeletal actin) mouse model reverses myotonia and alternative splicing abnormalities (16). Together, these data suggest that MBNL1 upregulation may be an attractive therapeutic strategy for DM, although further work is needed to understand its safety and efficacy.
We have developed the first mouse model to investigate the safety and therapeutic potential of constitutive and long-term multisystemic MBNL1 overexpression. Our results show for the first time that high levels of MBNL1 overexpression are well-tolerated in skeletal muscle and that MBNL1 overexpression prevents CUGEXP-induced skeletal muscle myopathy.
RESULTS
Development of transgenic MBNL1-overexpression mice
We developed a transgenic mouse model that overexpresses the 40 kDa isoform of human MBNL1, which is the predominant isoform found in adult skeletal muscle and lacks exons 7 and 9 (16,17) (Fig. 1A). A humanized version of the highly conserved (99.5%) 40 kDa mouse cDNA (gift from M. Swanson) was tagged with a 3×Flag-epitope on the N-terminus to distinguish recombinant from endogenous Mbnl1 protein. To test the effects of early, long-term and high levels of MBNL1 expression, we used the ubiquitous β-actin promoter with the early CMV enhancer (18). Using a Tnnt3 minigene splicing assay (16), we show that the tagged-recombinant MBNL1 protein retains its function as an alternative splicing regulator and shifts Tnnt3 splicing to the adult pattern by exluding the fetal (F) exon (Fig. 1B).
Figure 1.
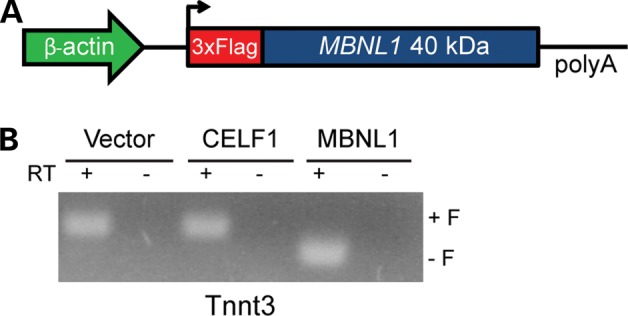
Transgene design and function. (A) Schematic diagram of the MBNL1-OE construct used to express the 40 kDa isoform, (−) exons 7 and 9, of human MBNL1 protein with a triple flag epitope on the N-terminus. Chicken β-actin promoter with early CMV enhancer (CAGG) drives transgene expression. (B) HEK293T cells transfected with 2 µg of Tnnt3 minigene plus 2 µg of either pcDNA3.1 empty vector, CUGBP1 (16) or MBNL1-OE vectors. Splicing of the Tnnt3 minigene was assayed using primers MSS1956 and MSS1938 (16).
Recombinant MBNL1 expression in MBNL1-OE mice
Pronuclear injection of the MBNL1-overexpression (MBNL1-OE) construct was performed on the FVB background. Because MBNL1 is an important regulator of alternative splicing, we expected that constitutive MBNL1 overexpression would cause embryonic lethality and that alternative conditional expression strategies might be required to generate a successful MBNL1-OE model. However, we were able to establish two transgenic lines (14685 and 14686) that constitutively overexpress MBNL1. The relative level of MBNL1 expressed in these mice was determined by immunoblot analyses (Fig. 2 and Supplementary Material, Fig. S1A). Adult animals from line 14685 overexpress total MBNL1 in skeletal muscle [gastrocnemius 9.2× (P = 0.0285); quadriceps 8.8× (P = 0.0081)] compared with controls. Although Flag-tagged recombinant protein is detectable in the 14685 brain, heart and lung, total MBNL1 levels were not significantly increased and no recombinant protein was detected in the kidney or liver (Fig. 2A). In contrast, 14686 mice showed broad multisystemic overexpression of MBNL1 [gastrocnemius 16.5×; (P < 0.001); quadriceps 8.95× (P = 0.00015); heart 7.3× (P = 0.00515); brain 2.9× (P = 0.0041); lung 6.7× (P = 0.0004); and liver 12.3× (P = 0.0114)], with variable levels of recombinant protein detected in the kidney (Fig. 2). Also, we observed a lower molecular weight N-terminal fragment. Because this band was positive for the N-terminal FLAG epitope but not detected by the A2764 antibody directed to the C-terminus, the fragment is likely caused by proteolytic cleavage of recombinant MBNL1. Consistent with the previous description of the β-actin promoter (19), both lines express recombinant MBNL1 proteins during embryonic development (Fig. 2B). Immunofluorescence analysis in transverse skeletal muscle sections shows that recombinant MBNL1 localizes to the nucleus, sometimes in a focal distribution (Fig. 2D and Supplementary Material, Fig. S2A). In summary, these results show significant overexpression of nuclear recombinant MBNL1 protein. The presence of aggregates and proteolytic cleavage fragments raises the possibility that not all recombinant protein expressed in these mice is functional.
Figure 2.

Recombinant MBNL1 expression and localization in MBNL1-OE mice. (A) Western blots of tissues from FVB, 14685 and 14686 mice at 6 months of age. Recombinant MBNL1 and endogenous Mbnl1 are detected using A2764 (αMbnl1) primary and HRP-conjugated α-rabbit secondary antibodies. αFlag-HRP (M2) and α-mouse-HRP detect recombinant MBNL1 only. GAPDH was used as a loading control. (B) Western blots of whole embryo protein lysates show expression of recombinant and endogenous MBNL1/Mbnl1 for 14685 at embryonic day 11 (E11) and 14686 at E13. (C) Relative levels of recombinant (MBNL1) and endogenous (Mbnl1) protein in 14685 (n = 7) versus wild-type littermates (n = 3) and 14686 (n = 3) versus wild-type littermates (n = 3) were quantified and normalized to the alpha-tubulin loading control. Endogenous Mbnl1 levels in wild-type controls were set to 1 to calculate a fold-change in total MBNL1/Mbnl1 levels. *P < 0.05; **P < 0.005; ***P < 0.001. (D) Immunofluorescence of recombinant MBNL1 in 14685 gastrocnemius muscle using α-Flag (F7425 Sigma) and α-rabbit Alexa Fluor 488 (Jackson Immunoresearch) antibodies. No recombinant protein is detected in wild-type controls. DAPI stain was used to detect nuclei.
Pheontypic effects of MBNL1 overexpression
To determine whether chronic, long-term MBNL1 overexpression is tolerated, we measured the lifespan of MBNL1-OE and WT mice. When MBNL1 overexpression is limited to skeletal muscle (line 14685), there was no increased mortality in animals aged 18 months (n = 35 WT, n = 28 OE; Fig. 3A). Although animals with multisystemic MBNL1 overexpression (line 14686) had no increased mortality at 20 (survival wt = 100%, OE = 100%), 40 (survival wt = 92%, OE = 96%) and 60 weeks (survival wt = 81%, OE = 87%), they showed increased mortality at 76 weeks of age (survival wt = 73%, OE = 52%; P = 0.02382; n = 26 WT, n = 23 OE; Fig. 3B). The deaths were often sudden, with the mice showing no signs of distress before being found dead in their cage. Whole-body mass comparisons of the 14685 line show that skeletal muscle overexpression does not affect mass or rates of growth (Fig. 3C). In contrast, multisystemic 14686 MBNL1-OE mice show significantly reduced body mass compared with wild-type littermates, although they gain mass at the same rate (Fig. 3D). The cause(s) of the late-onset mortality and reduced body mass phenotypes found in the 14686 line are not clear. These phenotypes may result from the high levels of multisystemic MBNL1 overexpression found only in the 14686 line, or an insertion effect. No overt changes in cage behavior were observed in either line, but some mice from line 14686 were susceptible to eye inflammation, which was resolved when treated with topical antibiotics.
Figure 3.

Lifespan and mass of MBNL1-OE mice. (A) Kaplan–Meier survival plot for 14685 (n = 35 WT; n = 28 OE) and (B) 14686 (n = 26 WT; n = 23 OE) mice up to 78 weeks of age. No statistically significant difference in survival at any time point was observed in line 14685. Mice from line 14686 have a significant increase in percent mortality beginning at week 76 (P = 0.023). (C) Mass of 14685 and (D) 14686 mice (n >3 for all time points). No significant difference in mass was observed for line 14685 at any time point. In contrast, 14686 mice were smaller at 3–8 weeks (P < 0.005) and for week 9 (P = 0.011). Asterisks (*) indicate statistically significant differences.
MBNL1 overexpression is generally well-tolerated in skeletal muscle
To determine whether MBNL1 overexpression is toxic to skeletal muscle, we analyzed transverse sections of gastrocnemius muscle by hematoxylin and eosin staining. Muscle from both lines appeared healthy and did not display any myopathic changes such as necrosis, fibrosis, split fibers, atrophy, hypertrophy or centrally located nuclei (Fig. 4A). Quantitative analyses showed no significant increase in centrally nucleated fibers (Fig. 4B) and no change in fiber cross-sectional area (Fig. 4C) in either line. Muscle function was assessed by forelimb grip strength analysis. 14685 mice displayed no difference in total grip strength compared with WT controls (Fig. 4D). 14686 mice showed a decrease in total grip strength (P = 0.006), but this difference was not significant when strength was normalized to muscle mass (Fig. 4D and E). Finally, overexpression of MBNL1 had no effect on rotarod performance in either line, suggesting that balance and motor coordination are unaffected by MBNL1 overexpression at 6 months of age (Fig. 4F).
Figure 4.

Muscle histology and function in MBNL1-OE mice. (A) Hematoxylin and eosin stains of skeletal muscle from 14685, 14686 and wild-type littermate controls. (B) Percentage of fibers with at least one central nuclei in skeletal muscle from wild-type (n = 6), 14685 (n = 4) and 14686 (n = 3) animals at 11–14 months. (C) Average fiber cross-sectional area in 11–14-month wild-type (n = 6), 14685 (n = 4) and 14686 (n = 3) mice. (D) Total forelimb grip strength in 4–7-month wild-type (n = 21), 14685 (n = 15) and 14686 (n = 11) mice. 14686 have a significant loss in forelimb grip strength compared with wild-type controls (P < 0.001). (E) Grip strength normalized to skeletal muscle mass in 4–7-month wild-type (n = 5) and 14686 (n = 5) mice. (F) Rotarod analysis at 6 months in 14685 (n = 10) versus wild-type littermates (n = 7) and 14686 (n = 8) versus wild-type littermates (n = 8).
MBNL1 overexpression causes a premature fetal-to-adult splicing transition
Endogenous Mbnl1 regulates a fetal-to-adult splicing transition that occurs between post-natal day 2 and 21 (20). In the absence of Mbnl1 or the presence of CUGEXP RNAs, this transition fails and fetal splicing patterns persist in adult tissues, resulting in the spliceopathy characteristic of DM (21–24). Because the 14685 and 14686 mice overexpress MBNL1 at both embryonic and neonatal time points (Fig. 2B and Supplementary Material, Fig. S3A), we hypothesized that MBNL1-regulated target mRNAs may prematurely shift to adult isoforms during early development even though the animals paradoxically appear relatively healthy. As predicted, 14686 neonates have a premature alternative splicing transition of Mbnl1 targets in skeletal muscle (Serca1, mZasp, mTitin and, variably, ClCn1), heart (Tnnt2, Sorbs1, RyR2 and Cacna1D) and brain (Mbnl2 and Sorbs1) (Fig. 5A–C). Most targets are shifted toward the adult isoform as early as embryonic day 13 (Supplementary Material, Fig. S4). The levels of recombinant MBNL1 are lower in line 14685 during development (Fig. 2B and Supplementary Material, Fig. S3A), and premature splicing changes were not observed.
Figure 5.

Neonatal splicing in MBNL1-OE mice. RT-PCR analysis of alternative splicing in 14686 and wild-type littermates at post-natal day 1. mRNA targets and specific alternatively spliced exons are listed on each gel. Mbnl1 targets were analyzed in skeletal muscle (A), brain (B) and heart (C).
Myotonia and alternative splicing in a mouse model of DM are corrected by MBNL1 overexpression
To determine whether long-term overexpression of MBNL1 which begins early in development (≤E11) will prevent DM skeletal muscle phenotypes, we crossed 14685 mice with HSALR-CTG250 mice (22) to generate mice that were hemizygous for both MBNL1-OE and HSALR-CTG250 transgenes (Fig. 6A). Because HSALR-CTG250 mice express a CUGEXP transcript specifically in skeletal muscle tissue, line 14685 was chosen for this experiment as these mice overexpress MBNL1 specifically in skeletal muscle and display no overt phenotype. As previously reported, CUGEXP RNA foci were present in the singly transgenic HSALR mice. We show RNA foci are also present in HSALR;MBNL1-OE doubly transgenic animals (Fig. 6B). Importantly, the MBNL1-OE transgene provided sufficient additional MBNL1 to prevent the CUGEXP-induced spliceopathy in doubly transgenic animals (Fig. 6C). Needle insertion electromyography (EMG) was performed on singly and doubly transgenic mice. Although robust myotonic discharges were prevalent (30–90% of needle insertions) in the HSALR skeletal muscle (tibialis anterior, gastrocnemius and paraspinal muscles; n = 8), the doubly transgenic HSALR;MBNL1-OE mice displayed less than one minor myotonic discharge per 30 needle insertions (n = 10). FVB mice did not display any myotonia (n = 3).
Figure 6.

MBNL1 overexpression corrects a DM skeletal muscle phenotype. (A) Schematic diagram of MBNL1-OE and HSALR cross. (B) RNA foci in skeletal muscle from HSALR and HSALR;MBNL1-OE mice detected with a Cy3 conjugated CAG8 oligonucleotide probe. No foci were detected in wild-type FVB mice. (C) RT-PCR splicing assays of FVB wild-type (WT), singly transgenic HSALR (HSA) and doubly transgenic HSALR;MBNL1-OE mice (D). (D) EMG trace from an HSALR gastrocnemius muscle showing a single needle insertion resulting in a ∼2s myotonic discharge and trace from an HSALR;MBNL1-OE doubly transgenic mouse gastrocnemius showing two normal needle insertion responses without electrical myotonia. (E) Hematoxylin and eosin staining of quadriceps muscle from 12-month FVB wild-type, singly transgenic HSALR and doubly transgenic HSALR;MBNL1-OE mice. Ringed fibers (asterisk) and multiple central nuclei (arrow) shown in the HSALR mouse. (F) Quantitation of centrally nucleated fibers in FVB wild-type, HSALR and HSALR;MBNL1-OE mice. Averages are shown ±SEM.
Overexpression of MBNL1 corrects the myopathy of HSALR muscle
Previous results show that delivery of MBNL1 to HSALR muscle at 4 weeks of age does not correct skeletal muscle myopathy (16). To test whether early and sustained overexpression of the 40 kDa isoform of MBNL1 prevents the HSALR-CUGEXP-induced myopathy, we performed hematoxylin and eosin staining of transverse sections of quadriceps muscles from ∼1-year-old HSALR singly transgenic, HSALR;MBNL1-OE doubly transgenic and FVB wild-type mice. Although the HSALR mice displayed classic symptoms of DM myopathy, including ringed fibers (asterisk), and fibers with multiple central nuclei (arrow), the HSALR;MBNL1-OE mice showed no signs of myopathy (Fig. 6E). Additionally, quantitation of central nuclei showed a significant decrease in the percentage of centrally nucleated fibers in the HSALR;MBNL1-OE doubly transgenic mice compared with age-matched HSALR mice. Although this correction was nearly complete (15.7 versus 6.9%, P = 0.04), the percentage of centrally nucleated fibers in the doubly transgenic animals was still higher than wild-type levels (6.9 versus 4.0%, P = 0.02). These results demonstrate that Mbn1 sequestration is a critical component of the myopathy in the HSALR mice, and that this myopathy can be substantially corrected by early and long-term upregulation of MBNL1.
DISCUSSION
Muscleblind sequestration is thought to play an important role in DM pathogenesis. We developed an MBNL1-OE mouse model to investigate the safety and efficacy of MBNL1 overexpression as a potential therapeutic strategy. Our data show: (i) MBNL1 overexpression is generally well-tolerated in skeletal muscle; (ii) the surprising result that premature shifts in alternative splicing of MBNL1-regulated genes in multiple organ systems are compatible with life and do not cause embryonic lethality; (iii) MBNL1 overexpression prevents CUGEXP-induced myotonia, myopathy and alternative splicing abnormalities in HSALR;MBNL1-OE doubly transgenic mice.
Previous studies have shown that localized AAV delivery of MBNL1 to tibialis anterior muscle in HSALR-CTG250 mice reverses myotonia and alternative splicing changes. These data suggested that MBNL1 overexpression may be an effective therapeutic strategy for DM. However, an unanswered question we now address is whether chronic multisystemic overexpression of this important regulator of alternative splicing has deleterious consequences. We show that, when MBNL1 overexpression (10-fold) is limited to skeletal muscle, the mice are healthy and develop no detectable phenotype up to 18 months of age. Similarly, higher levels of MBNL1 overexpression (17-fold) in a second transgenic line are also well-tolerated in skeletal muscle and cause no histological or functional abnormalities.
Because MBNL1 regulates a developmental splicing transition thought to be important in development (20,25), we analyzed splicing patterns of MBNL1 targets in embryos and neonates that overexpress MBNL1. We show that MBNL1 overexpression (17-fold in skeletal muscle, 9-fold in heart and 3-fold in brain) causes premature splicing transitions of MBNL1 target mRNAs in these tissues. Surprisingly, the skeletal muscle develops and functions normally in spite of these premature MBNL1-medated splicing shifts. This suggests either compensatory mechanisms allow muscle development despite the expression of adult isoforms of MBNL1-targets, or that MBNL1-regulated transcripts are not important for skeletal muscle development.
Because MBNL1 overexpression causes multisystemic changes in alternative splicing and overexpression of a similar alternative splicing factor, CELF1, causes embryonic lethality (20,25–31), we were surprised that the 14686 MBNL1-OE mice were viable. Line 14686 did, however, display a number of phenotypic abnormalities including reduced mass and late-onset sudden death. Because these phenotypic abnormalities only occurred in a single line, it is possible that transgene insertion effects cause the reduced mass and late-onset mortality. Future studies are needed to characterize the consequences of multisystemic overexpression to better understand the potential of MBNL1 as a multisystemic therapeutic strategy.
A number of strategies have been previously used to correct RNA gain of function effects in DM mouse models, including morpholinos, antisense oligonucleotides, small molecules and AAV-delivered MBNL1 (16,32–35). Although these methods have variably corrected alternative splicing abnormalities and myotonia, none has reversed the skeletal muscle myopathy. In addition to demonstrating safety of high levels of MBNL1-OE in skeletal muscle, we show overexpression of a single MBNL1 isoform (40 kDa) is sufficient to prevent CUGEXP-induced skeletal muscle phenotypes, including myotonia, alternative splicing changes and myopathy.
Skeletal muscle weakness is an important and intractable problem for DM patients, which often results in fatal respiratory complications. Finding strategies to prevent this incidious muscle wasting is of critical importance. Our results demonstrate the first succcessful prevention of DM1 myopathy in mice and highlights the promise of MBNL1 overexpression as therapeutic strategy for DM.
Finally, because MBNL1 sequestration has recently been implicated in SCA8, HD and HDL2, these mice will be a valuable tool to better understand the role of RNA gain-of-function effects in other disorders.
MATERIALS AND METHODS
Transgene construction
The pTRUF12Δ_MBNL1-40 kDa was obtained as a generous gift by Dr Maurice Swanson. An epitope tag containing three repeated FLAG epitope sequences with a strong Kozak and ATG codon was cloned into HindIII and XhoI sites upstream of the MBNL1 transgene coding sequence. For pronuclear injection, the transgene was linearized with Kpn1 and Sph1, gel-purified (Promega Wizard SV Gel and PCR Cleanup Kit) and dialyzed overnight. MBNL1-OE mice were genotyped using PCR with NEB Quick-Load Taq Mastermix with transgene-specific primers [OE R4 (5′-GATGAGCAAACCGACAGTCA-3′) and OE F6 (5′-ATCGCCTGTTTGATTCATT-3′)], with an initial denaturation at 95°C, then 30 cycles (95° for 30 s, 56° for 30 s, 68° for 30 s), followed by a final elongation at 68° for 6 min.
Fluorescence in situ hybridization/immunofluorescence
Mouse skeletal muscle was frozen in 2-methylbutane cooled in liquid nitrogen. Using a Leica cryostat, 8 µm transverse sections were cut. For RNA fluorescence in situ hybridization, tissue was fixed in 2% paraformaldahyde/1× phosphate buffered saline (PBS) for 30 min at 4°C and then washed in 1× PBS. Sections were then incubated in 2% acetone/1× PBS 5 min at 4°C, briefly washed in 1× PBS and incubated in pre-hybridization solution (30% formamide/2× SSC) for 10min at RT. Sections were then incubated 2h at 42°C with a CAG8 RNA probe (500 ng/ml) in a hybridization solution containing 30% formamide, 2× SSC, 0.2× BSA, 2 mm vanadyl ribonucleoside and 1 mg/ml yeast tRNA. Finally, sections were incubated in post-hybridization buffer (30% formamide/2× SSC) for 30 min at 45°C, washed 5 × 5 min in 1× SSC, briefly washed in 1× PBS and mounted with Prolong Gold antifade reagent with DAPI (Invitrogen). For immunofluorescence, tissue was fixed in 3% paraformaldehyde/1× PBS for 30 min at 4°C and washed 3 × 5min in 1× PBS. Tissue was blocked for 1 h at RT in 3% normal goat serum/1× PBS. Primary antibodies were diluted (α-MBNL1 A2764 1:1000; α-Flag F7425 1:200) in 3% NGS/1× PBS and sections were incubated in primary antibodies overnight at 4°C. Tissue was washed in 1× PBS and incubated in secondary antibody (DyLight 488-conjugated goat anti-rabbit IgG 1:5000) in 3% NGS/1× PBS 1 h at RT. Sections were washed in 1× PBS and mounted with Prolong Gold antifade reagent with DAPI (Invitrogen). Images were taken on a Leica TCS SP5 confocal microscope and are presented as a z-stack compressed image.
Western analysis
Mouse tissue was lysed in radioimmunoprecipitation buffer with protease inhibitors (Roche Complete Mini Tablets). Protein concentrations were determined using the Bio-Rad protein reagent. Ten micrograms (skeletal muscle) or 20μg (non-skeletal muscle) of protein was separated by electrophoresis on NuPAGE 4–12% Bis–Tris gels (Invitrogen) and transferred to nitrocellulose (GE Healthcare Amersham Hybond-ECL). Non-specific binding was blocked with 5% non-fat milk in PBS-T (PBS with 0.05% Tween 20) for 1h at room temperature and incubated with the primary antibody in 5% non-fat milk in PBS-T overnight at 4°C. Membranes were then washed three times in PBS-T and incubated for 1h at room temperature with secondary antibodies in 5% non-fat milk in PBS-T and visualized with ECL western blotting detection reagents (Amersham Biosciences).
The primary antibodies and concentrations used were as follows: MBNL1 A2764 (20) (gift from Dr Charles Thornton) 1:2000; FLAG (Sigma A8592) 1:5000; tubulin (Sigma T9026) 1:5000; and GAPDH (Chemicon MAB374) 1:40 000. The secondary antibodies used in conjunction with various primary antibodies were as follows: MBNL1 A2764 (1:5000 anti-rabbit IgG; GE Healthcare NA934V); FLAG and tubulin (1:5000 anti-mouse IgG; GE Healthcare NA931V); and GAPDH (1:10 000 anti-mouse IgG; GE Healthcare NA931V).
Lifespan/mass
To assess lifespan, entire litters of 14685 and 14686 were aged to 18 months without experimental intervention. Research and veterinary staff monitored animal health daily. Treatment of various conditions (fight wounds, skin lesions) was performed per veterinary recommendations regardless of mouse genotype. The 14686 OE mice appeared to have an increased frequency of eye infections, which were resolved with triple-antibiotic ointment. To monitor mass, individual cohorts of mice were weighed at various time points and data were compiled.
Muscle histology
Skeletal muscle was frozen in 2-methylbutane cooled with liquid nitrogen. Transverse sections of 8 µm were cut on a Leica cryostat. Samples to be compared were placed on the same microscope slide. Samples were incubated for 5min in harris hematoxylin; rinsed with cold tap water; immersed twice in eosin-y; washed consecutively with 80, 90 and 100% ethanol; and incubated three times for 2min in Xylenes. Samples were mounted with Cytoseal 60 (Richard Allan Scientific) and visualized on an Olympus BX51 upright light microscope.
To analyze fiber cross-sectional area and the percentage of centrally nucleated fibers of each muscle section, four representative, non-overlapping images were taken at 20× magnification. Using the Olympus CellSens Standard analysis software, 100 fibers from each image were outlined to determine the total fiber area. Centrally localized nuclei, as well as the total fiber number in each image, were manually counted to calculate the percentage of centrally nucleated fibers. All persons analyzing muscle histology were blinded to the genotypes of the mice.
Grip strength
Forelimb strength was measured using a grip strength monitor (Columbus Instruments). Each mouse was held by the base of its tail near the wire mesh pull-bar assembly and allowed to grasp the assembly. The mouse was then gradually drawn away from the assembly until the pull-bar was released. The force at release was recorded. This was repeated five times to obtain an average grip strength per mouse. For normalized grip strength, 14686 and wild-type mice were analyzed, and the combined mass of their tibialis anterior, gastrocnemious, quadriceps and triceps muscles was recorded. Total grip strength for each mouse was normalized to skeletal muscle mass. Data are presented as mean ± SEM.
Rotarod
For rotarod analysis, 6-month-old mice were allowed to habituate in a climate-controlled behavioral suite for 1h. Mouse was placed on an accelerating rotarod apparatus (Ugo Basile; 47600) initially rotating at 4 r.p.m. The rate of rotation was gradually increased to 40 r.p.m. over 5min and continued at the maximum rate of 40 r.p.m. for 5 additional minutes. The time that a mouse fell (latency to fall) from the rotating bar was recorded. Mice were rested for 10min following each trial. The trial was then repeated three additional times per day for 4 consecutive days. A mouse's latency to fall for each day was recorded as the mean latency of the four consecutive trials. Data are reported ± SEM.
Alternative splicing analysis
For splicing analysis in cell culture, HEK293T cells were plated in a six-well plate with DMEM (Gibco) and 10% FBS. Cells were transfected with 1 µg of Tnnt3 minigene construct and 1 µg of CUGBP1 or MBNL1 expression plasmids (16) using the Lipofectamine 2000 reagent (Invitrogen). RNA was purified with RNeasy Mini Kit (Qiagen) and cDNA was synthesized from 1 µg of template RNA using Superscript II First Strand Synthesis (Invitrogen) with random hexamer primers. Tnnt3 splicing analysis was carried out as described previously (16), using primers MSS1956 and MSS1938. For in vivo splicing assays, mouse tissue was snap-frozen in liquid nitrogen. RNA was isolated from frozen tissue using Trizol Reagent (Invitrogen). cDNA was synthesized from 2 µg of template RNA, using Superscript II First Strand Synthesis (Invitrogen) with random hexamer primers. Target-specific primers were as described previously for Mbnl2 E6, Serca1 E22, mZasp E11, mTitin E5, ClCn1 E7a (20), Tnnt2 E5 (21), nFix (15). Additional primers were as follows: RyR2 F[5′-CGGACCTGTCTATCTGCACCTTTGT-3′], R[5′-CATACCACTGTAGGAATGGCGTAGCA-3′]; Cacna1d F[5′-CATGCCCACCAGCGAGACTGAA-3′], R[5′-CACCAGGACAATCACCAGCCAGTAAA-3′]; and Sorbs1 E23 F[5′-CCAGCTGATTACTTGGAGTCCACAGAAG-3′], R[GTTCACCTTCATACCAGTTCTGGTCAATC-3′]. Each product was amplified for 32 cycles (95° for 30 s, 55° for 45 s, 72° for 30 s). PCR products were resolved on 2% agarose gels, stained with ethidium bromide and imaged on a GE ImageQuant 400.
Electromyography
EMG analysis was carried out on a Teca Synergy system (Viasys Healthcare). Mice were anesthetized with 100 mg/kg body mass ketamine and 10 mg/kg body mass xylazine. A 30 guage concentric needle electrode (CareFusion Teca Elite) was inserted at least 30 times for each muscle analyzed (gastrocnemius, tibialis anterior, quadriceps, triceps and paraspinals). The frequency of insertions yielding a myotonic discharge was recorded for each muscle. The individual performing EMG analysis was blinded to mouse genotypes.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
ACKNOWLEDGEMENTS
We would like to thank M. Swanson and T. Ebner for helpful discussions, reagents and comments on the manuscript. We also thank C. Thornton for the gift of the HSALR mice.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by funding from the NIH (PO1NS058901) and the Muscular Dystrophy Association.
REFERENCES
- 1.Harper P.S. Myotonic Dystrophy. 3rd edn. London: W.B. Saunders; 2001. [Google Scholar]
- 2.Liquori C.L., Ricker K., Moseley M.L., Jacobsen J.F., Kress W., Naylor S.L., Day J.W., Ranum L.P. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 3.Brook J.D., McCurrach M.E., Harley H.G., Buckler A.J., Church D., Aburatani H., Hunter K., Stanton V.P., Thirion J.P., Hudson T. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;69:385. doi: 10.1016/0092-8674(92)90418-c. [DOI] [PubMed] [Google Scholar]
- 4.Mahadevan M., Tsilfidis C., Sabourin L., Shutler G., Amemiya C., Jansen G., Neville C., Narang M., Barceló J., O'Hoy K. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992;255:1253–1255. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- 5.Fu Y.H., Pizzuti A., Fenwick R.G., King J., Rajnarayan S., Dunne P.W., Dubel J., Nasser G.A., Ashizawa T., de Jong P. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science. 1992;255:1256–1258. doi: 10.1126/science.1546326. [DOI] [PubMed] [Google Scholar]
- 6.Cooper T.A., Wan L., Dreyfuss G. RNA and disease. Cell. 2009;136:777–793. doi: 10.1016/j.cell.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee J.E., Cooper T.A. Pathogenic mechanisms of myotonic dystrophy. Biochem. Soc. Trans. 2009;37:1281–1286. doi: 10.1042/BST0371281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poulos M.G., Batra R., Charizanis K., Swanson M.S. Developments in RNA splicing and disease. Cold Spring Harb. Perspect. Biol. 2011;3:a000778. doi: 10.1101/cshperspect.a000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cardani R., Mancinelli E., Rotondo G., Sansone V., Meola G. Muscleblind-like protein 1 nuclear sequestration is a molecular pathology marker of DM1 and DM2. Eur. J. Histochem. 2006;50:177–182. [PubMed] [Google Scholar]
- 10.Kuyumcu-Martinez N.M., Wang G.S., Cooper T.A. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell. 2007;28:68–78. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller J.W., Urbinati C.R., Teng-Umnuay P., Stenberg M.G., Byrne B.J., Thornton C.A., Swanson M.S. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mankodi A., Urbinati C.R., Yuan Q.P., Moxley R.T., Sansone V., Krym M., Henderson D., Schalling M., Swanson M.S., Thornton C.A. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum. Mol. Genet. 2001;10:2165–2170. doi: 10.1093/hmg/10.19.2165. [DOI] [PubMed] [Google Scholar]
- 13.Yuan Y., Compton S.A., Sobczak K., Stenberg M.G., Thornton C.A., Griffith J.D., Swanson M.S. Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucleic Acids Res. 2007;35:5474–5486. doi: 10.1093/nar/gkm601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warf M.B., Berglund J.A. MBNL binds similar RNA structures in the CUG repeats of myotonic dystrophy and its pre-mRNA substrate cardiac troponin T. RNA. 2007;13:2238–2251. doi: 10.1261/rna.610607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Du H., Cline M.S., Osborne R.J., Tuttle D.L., Clark T.A., Donohue J.P., Hall M.P., Shiue L., Swanson M.S., Thornton C.A., et al. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat. Struct. Mol. Biol. 2010;17: 187–193. doi: 10.1038/nsmb.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanadia R.N., Shin J., Yuan Y., Beattie S.G., Wheeler T.M., Thornton C.A., Swanson M.S. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc. Natl Acad. Sci. USA. 2006;103:11748–11753. doi: 10.1073/pnas.0604970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tran H., Gourrier N., Lemercier-Neuillet C., Dhaenens C.M., Vautrin A., Fernandez-Gomez F.J., Arandel L., Carpentier C., Obriot H., Eddarkaoui S., et al. Analysis of exonic regions involved in nuclear localization, splicing activity, and dimerization of Muscleblind-like-1 isoforms. J. Biol. Chem. 2011;286:16435–16446. doi: 10.1074/jbc.M110.194928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niwa H., Yamamura K., Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 19.Okabe M., Ikawa M., Kominami K., Nakanishi T., Nishimune Y. ‘Green mice’ as a source of ubiquitous green cells. FEBS Lett. 1997;407:313–319. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- 20.Lin X., Miller J.W., Mankodi A., Kanadia R.N., Yuan Y., Moxley R.T., Swanson M.S., Thornton C.A. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 2006;15:2087–2097. doi: 10.1093/hmg/ddl132. [DOI] [PubMed] [Google Scholar]
- 21.Kanadia R.N., Johnstone K.A., Mankodi A., Lungu C., Thornton C.A., Esson D., Timmers A.M., Hauswirth W.W., Swanson M.S. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- 22.Mankodi A., Logigian E., Callahan L., McClain C., White R., Henderson D., Krym M., Thornton C.A. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 2000;289:1769–1773. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- 23.Gomes-Pereira M., Foiry L., Nicole A., Huguet A., Junien C., Munnich A., Gourdon G. CTG trinucleotide repeat ‘big jumps’: large expansions, small mice. PLoS Genet. 2007;3:e52. doi: 10.1371/journal.pgen.0030052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Orengo J.P., Chambon P., Metzger D., Mosier D.R., Snipes G.J., Cooper T.A. Expanded CTG repeats within the DMPK 3′ UTR causes severe skeletal muscle wasting in an inducible mouse model for myotonic dystrophy. Proc. Natl Acad. Sci. USA. 2008;105:2646–2651. doi: 10.1073/pnas.0708519105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalsotra A., Xiao X., Ward A.J., Castle J.C., Johnson J.M., Burge C.B., Cooper T.A. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc. Natl Acad. Sci. USA. 2008;105:20333–20338. doi: 10.1073/pnas.0809045105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boutz P.L., Stoilov P., Li Q., Lin C.H., Chawla G., Ostrow K., Shiue L., Ares M., Black D.L. A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev. 2007;21:1636–1652. doi: 10.1101/gad.1558107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee J.A., Tang Z.Z., Black D.L. An inducible change in Fox-1/A2BP1 splicing modulates the alternative splicing of downstream neuronal target exons. Genes Dev. 2009;23:2284–2293. doi: 10.1101/gad.1837009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Makeyev E.V., Zhang J., Carrasco M.A., Maniatis T. The microRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol. Cell. 2007;27:435–448. doi: 10.1016/j.molcel.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chawla G., Lin C.H., Han A., Shiue L., Ares M., Black D.L. Sam68 regulates a set of alternatively spliced exons during neurogenesis. Mol. Cell. Biol. 2009;29:201–213. doi: 10.1128/MCB.01349-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Q., Lee J.A., Black D.L. Neuronal regulation of alternative pre-mRNA splicing. Nat. Rev. Neurosci. 2007;8:819–831. doi: 10.1038/nrn2237. [DOI] [PubMed] [Google Scholar]
- 31.Ho T.H., Bundman D., Armstrong D.L., Cooper T.A. Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy. Hum. Mol. Genet. 2005;14:1539–1547. doi: 10.1093/hmg/ddi162. [DOI] [PubMed] [Google Scholar]
- 32.Mulders S.A., van den Broek W.J., Wheeler T.M., Croes H.J., van Kuik-Romeijn P., de Kimpe S.J., Furling D., Platenburg G.J., Gourdon G., Thornton C.A., et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc. Natl Acad. Sci. USA. 2009;106:13915–13920. doi: 10.1073/pnas.0905780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wheeler T.M., Sobczak K., Lueck J.D., Osborne R.J., Lin X., Dirksen R.T., Thornton C.A. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science. 2009;325:336–339. doi: 10.1126/science.1173110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Warf M.B., Nakamori M., Matthys C.M., Thornton C.A., Berglund J.A. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc. Natl Acad. Sci. USA. 2009;106:18551–18556. doi: 10.1073/pnas.0903234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee J.E., Bennett C.F., Cooper T.A. RNase H-mediated degradation of toxic RNA in myotonic dystrophy type 1. Proc. Natl Acad. Sci. USA. 2012;109:4221–4226. doi: 10.1073/pnas.1117019109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.