

Abstract
Abnormal presence of autophagic vacuoles is evident in brains of patients with Parkinson's disease (PD), in contrast to the rare detection of autophagosomes in a normal brain. However, the actual cause and pathological significance of these observations remain unknown. Here, we demonstrate a role for mitochondrial metabolism in the regulation of the autophagy-lysosomal pathway in ex vivo and in vitro models of PD. We show that transferring mitochondria from PD patients into cells previously depleted of mitochondrial DNA is sufficient to reproduce the alterations in the autophagic system observed in PD patient brains. Although the initial steps of this pathway are not compromised, there is an increased accumulation of autophagosomes associated with a defective autophagic activity. We prove that this functional decline was originated from a deficient mobilization of autophagosomes from their site of formation toward lysosomes due to disruption in microtubule-dependent trafficking. This contributed directly to a decreased proteolytic flux of α-synuclein and other autophagic substrates. Our results lend strong support for a direct impact of mitochondria in autophagy as defective autophagic clearance ability secondary to impaired microtubule trafficking is driven by dysfunctional mitochondria. We uncover mitochondria and mitochondria-dependent intracellular traffic as main players in the regulation of autophagy in PD.
INTRODUCTION
Parkinson's disease (PD) is a common neurodegenerative movement disorder, characterized by a dramatic loss of midbrain dopaminergic neurons in the substantia nigra pars compacta (SNpc), and the presence of ubiquitylated α-synuclein-containing intracytoplasmic inclusions called Lewy bodies (LBs) in surviving SNpc neurons (1).
Aging is considered the greatest risk factor for sporadic PD (sPD). Accumulation of mitochondrial DNA (mtDNA) mutations and mitochondria-driven oxidative stress is thought to represent a bridge between sPD and the natural aging process. Mitochondrial association with sPD was established when a mitochondrial NADH dehydrogenase (complex I) activity deficit was identified in the SNpc of post-mortem PD patient brains (2) and in PD patient platelets (3). In addition, mtDNA involvement in complex I defects observed in PD platelets was further recognized after transference of platelet mitochondria into mtDNA-deficient cell lines and validated in the resultant cell lines known as ‘cybrids’ (4). Data obtained using this ex vivo cellular model have shown that several pathogenic features observed in PD subject brains are actually recapitulated by sPD cybrids (5,6). Moreover, the generation of fibrillar and vesicular protein inclusions in sPD cybrids replicating most antigenic and structural features of LBs was reported (7,8).
The presence of LB-like structures in sPD cybrids suggests that mitochondrial dysfunction associated with defective protein handling may account for PD pathogenesis. Relevant to PD pathology, autophagy represents a major mechanism by which intracellular long-lived proteins, protein aggregates (such as α-synuclein oligomers) and entire cytoplasmic organelles (such as mitochondria) are directly degraded within lysosomes. It is now considered that constitutive basal autophagic activity is a main quality control (QC) process that selectively disposes aberrant protein aggregates and damaged organelles for degradation (9,10). Thus, the regulation of QC autophagy may be critical to restrain the neurodegenerative process (11,12).
In line with this, growing evidence has suggested a role for autophagy deregulation in PD. Increased number of autophagosomes has been observed in cultured cells treated with mitochondrial complex I inhibitors such as 1-methyl-4-phenylpyridinium (MPP+), rotenone and 6-OHDA (13,14), and in postmortem PD patient brains (15). Although these changes have been commonly interpreted as an abnormal induction of autophagy, the actual origin and significance of these observations for the disease pathogenesis remain elusive.
Here, we investigated cause-and-effect relationships between mitochondrial dysfunction, microtubule network disruption and accumulation of autophagosomes and autophagy substrates. Using sPD cybrid cells, mtDNA-depleted cells and MPP+-treated primary cortical neurons, we characterized basal and induced autophagic responses and the clearance of autophagy cargos. We found that changes in mitochondrial function per se have a severe impact on autophagy since autophagosomes are actually actively formed but inefficiently cleared in sPD cells. Molecular dissection of each of the steps revealed that microtubule disruption rather than abnormal induction of autophagy gives rise to the characteristic patterns of autophagic pathology observed in PD.
RESULTS
Hybrid cells harboring sPD patient mitochondria and mtDNA-depleted cells accumulate morphologically abnormal mitochondria and nonfused autophagic vacuoles
To directly explore the functional consequences of an altered mitochondrial function over the autophagic-lysosomal system in the context of sPD, we modeled PD by creating transmitochondrial cytoplasmic hybrid cell lines (cybrids) in which endogenous mtDNA from sPD or control (CT) subject platelets was transferred to human teratocarcinoma (NT2) cells after complete depletion of endogenous mtDNA (Rho0 cells). By this approach, it is possible to follow the effects of mtDNA heteroplasmy within a nuclear and environmentally controlled context, thus providing a rational basis for the propagation of PD-related mitochondrial dysfunction, including a decline in mitochondrial ATP synthesis capacity and other mitochondria-dependent processes.
In this study, we used the same cohort of cybrids that was previously characterized in terms of mitochondrial bioenergetics (16). In this cohort, sPD cybrids were found to have a reduced complex I function, though citrate synthase activity, a commonly used quantitative mitochondrial enzyme marker, was not altered. In addition, basal oxygen consumption between sPD and CT cybrids was comparable. However, when mitochondria were challenged via chemical-induced uncoupling into a state of maximum oxygen consumption, sPD cybrids revealed an increase in proton leak in conjunction with a reduced respiratory reserve capacity and ATP production, compared with CT cells. In addition, pathways influenced by aerobic metabolism were also altered in these sPD cybrids. sPD cybrids showed reduced sirtuin-1 (SIRT1) phosphorylation, reduced peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1α) levels and increased NF-κB activation (16).
In this study, to investigate potential sequence variations that may be functionally relevant and cause mitochondrial dysfunction in sPD, we sequenced cybrid mtDNA derived from three sPD patients and three CT subjects and performed an mtDNA screening analysis of seven mtDNA genes coding for complex I subunits. As shown in Supplementary Material, Table S1, we identified different patterns of mtDNA variations, in both CT and sPD patients. However, no consistent differences were found in terms of mutation patterns or localizations and no PD-related mtDNA variations were detected in sPD cybrids. Our results do not rule out the possible involvement of mtDNA on mitochondrial dysfunction seen in sPD cybrids since other genes outside the regions investigated may be involved in the disease or that mtDNA involvement is related to either deletion events or copy number alterations. We then decided to evaluate whether the phenotypic biochemical changes found earlier in these cybrids are correlated with the mitochondrial content. We performed a comparative analysis for different mitochondrial markers in order to assess the total mitochondrial pool of CT versus sPD cybrids. As shown in Figure 1A and B, we have found variability among the levels of the proteins studied, but no statistically significant differences in sPD cybrids in comparison with CT cells were detected in the most significant structural proteins of mitochondria.
Figure 1.
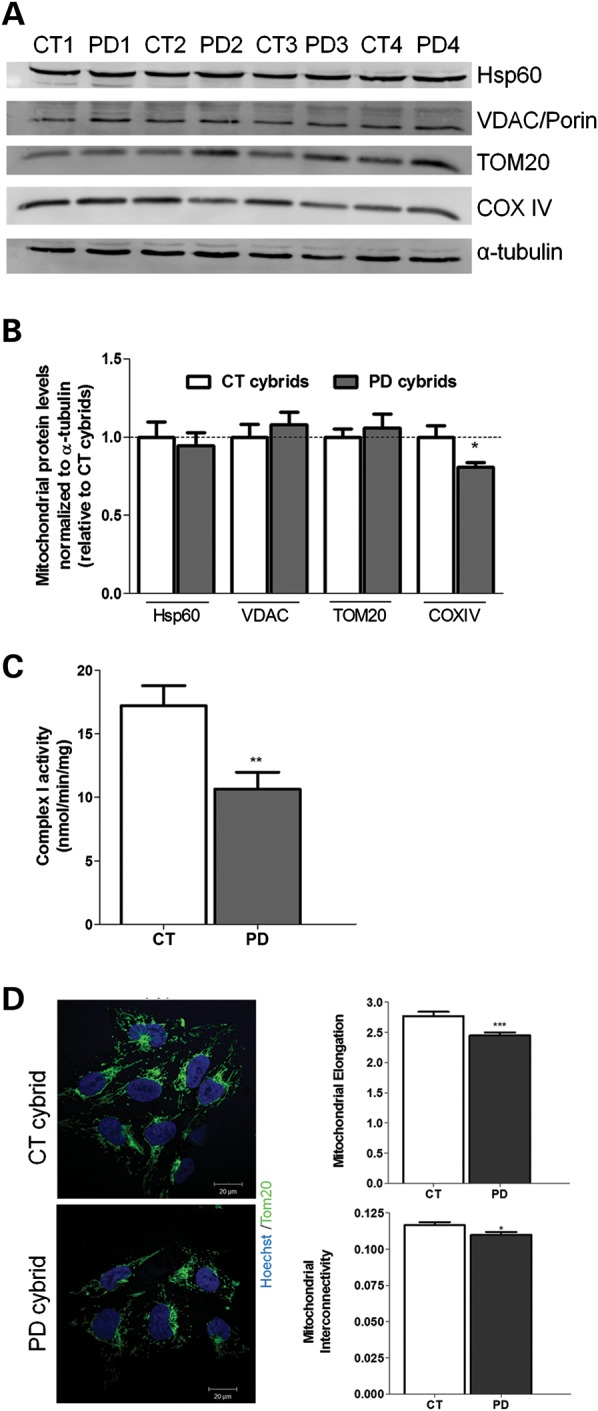
Mitochondrial deficits are related with an altered ETC complex I activity. (A) Immunoblot for mitochondrial markers (Hsp60, VDAC/Porin, TOM20, COXIV) from CT and sPD cybrids. (B) Densitometric analysis of mitochondrial marker levels (n= 8, *P< 0.05, versus CT cybrids). (C) Mitochondrial respiratory chain complex I activity. Data are reported in nmol/min/mg as the mean ± SEM (n= 12; **P< 0.01, versus CT cybrids). (D) TOM20 immunostaining (green) of CT and sPD cybrids showing alterations in mitochondria distribution, elongation and interconnectivity in sPD cybrids. Hoechst 33342- stained nuclei are in blue (n= 3, ***P< 0.001, mitochondrial elongation sPD versus CT cybrids; *P< 0.05, mitochondrial interconnectivity sPD versus CT cybrids). Scale bar: 20 µm.
We have also evaluated complex I activity, and, as previously characterized by our group (16,17), sPD cybrid cell lines show a stable decrease in this mitochondrial activity (Fig. 1C). To evaluate mitochondrial distribution and morphology, we used TOM20 staining and determined indices of mitochondrial interconnectivity and mitochondrial elongation. Mitochondrial interconnectivity is calculated as the mean area/perimeter ratio and is consistent with the degree of mitochondrial branching, namely the connectivity/dynamics between mitochondria (18,19). A higher mitochondrial interconnectivity means a higher access and communication between mitochondria. We have found that, in sPD cybrids, mitochondrial distribution was more perinuclear than in CT cybrids (Fig. 1D). Moreover, sPD cybrids showed a decrease in mitochondria elongation and interconnectivity, both features correlating with increased mitochondrial fragmentation. In fact, in sPD cybrids, mitochondria appear as small dots instead of a mitochondrial interconnected net.
The aforementioned alteration in sPD cybrid biochemical features is in agreement with an altered mitochondrial ultrastructure and may account for the clinical phenotype of the patients included in this study. Accordingly, in this study, we have examined 15 cell profiles of each cell line and categorized them as to whether they contained predominantly normal or abnormally shaped mitochondria and autophagy-related structures. Regarding mitochondrial structure, we have found that sPD cybrids had many morphologically abnormal mitochondria comparative to CT cybrids. In general, Figure 2A shows that mitochondria in CT cybrids were small and round or rod-like and elongated (as shown in Fig. 2Aa–d). The matrices were dark and uniform and the cristae were homogeneously distributed (as shown in Fig. 2Ab–d). sPD cybrid mitochondria were mostly found enlarged with oval rather than rod-like profiles (as shown in Fig. 2Ah). These abnormal mitochondria contained pale and patchy matrices, disrupted cristae (as shown in Fig. 2Ai–k). Some of sPD cybrid mitochondria contained inclusions of electron-dense amorphous substances within matrices that are thought to represent deposits of calcium and inorganic phosphates (denoted as filled triangles in Fig. 2Aj and k) (8,20,21).
Figure 2.

Mitochondrial deficits induced the accumulation of morphologically abnormal mitochondria and nonfused autophagic vacuoles. (A) Electron micrographs of CT and sPD cybrids. Lower inserts show higher magnification images to illustrate morphological features of mitochondria and individual examples of autophagic vacuoles in both CT and sPD cybrids. Black arrows, autophagolysosomes; white arrows, autophagosomes; arrows heads, electron-dense amorphous inclusions within mitochondria. Scale bars: 2 µm (top) and 0.5 µm (middle and bottom). (B) Electron micrographs of NT2 Rho0 cells. Higher magnification fields show morphological features of mitochondria and autophagic vacuoles in Rho0 cells. (a) Dark arrows: swollen pale mitochondria with discontinuous cristae; Dark arrow heads: enlarged autophagosome enclosing mitochondria and other materials; (b and c) dark arrow heads: abnormal membranous structures. Scale bars: 2 µm (top) and 0.5 µm (bottom). (C) Quantification of autophagosomes (APG) and autophagolysosomes (APGL) in cybrid cell lines and mtDNA-depleted (Rho0) cells. The total number of vesicles was quantified from 15 cell profiles for each cell line. (n= 3, *P< 0.05, ***P< 0.001, versus APG CT cybrids; *P< 0.05, **P< 0.01, versus APG+APGL CT cybrids; ##P< 0.01, versus APG+APGL sPD cybrids).
In order to verify whether these changes in sPD cybrid mitochondrial ultrastructure are exclusively due to the transferred sPD patient mitochondria, we have extended our analysis to the cybrid parental cell line (Rho0 cells). These cells, which lack functional mitochondria due to mtDNA depletion, were found to have high accumulation of swollen pale mitochondria with discontinuous cristae (denoted as arrows in Fig. 2B a). In addition, mitochondrial shape showed a rather circular or oval contour and mitochondria were found apart from each other (Fig. 2B). Cristae-like structures were perfectly discernible in Rho+ cell line mitochondria (Fig. 2A), whereas in Rho0 cells (Fig. 2B) we observed few cristae-like structures with an atypical structure going from one point to another of the mitochondrial peripheral contour (denoted as filled triangles in Fig. 2Bb and c). Indeed, these observations suggest that abnormal mitochondria features found in sPD cybrids are clearly due to the transferred mitochondria.
In addition, ultrastructural analysis revealed that sPD cybrid cells also contain cytosolic vesicles (denoted as white arrows in Fig. 2A h) that appear to be autophagic vacuoles enclosing dark bodies and other cytoplasmic inclusions identical to the cytoplasm surrounding the vacuoles, which are compatible with autophagosomes (Fig. 2Al–n). In contrast, vesicles containing amorphous materials looking total or partially degraded similar to autophagolysosomes and/or lysosomes were more prominent and easily detectable in CT cybrids comparative to sPD cybrids (denoted as arrows in Fig. 2Aa versus g). Quantitative analysis of the electron micrographs showed an increase in the number of autophagosomes (APG) in those cells compared with CT cybrids (Fig. 2C).
Similarly, Rho0 cells accumulated a large number of autophagic vacuoles (Fig. 2C), but apparently no lysosomes or cytoplasmic inclusions (Fig. 2Ba). Interestingly, for these cells, we have also detected a great number of mitochondria inside ‘giant’ autophagosomes, indicating a relatively high index of autophagic sequestration (denoted as filled triangles in Fig. 2Ba and d).
Since newly formed autophagosomes mature and eventually fuse with lysosomes, thereby delivering their contents to lysosomes for degradation, the exacerbated accumulation of autophagosomes observed in sPD cybrids and more evident in Rho0 cells prompted us to assess the effects of sPD-mitochondrial dysfunction on the autophagic activity.
Mitochondrial deficits in cells harboring PD patient mitochondria or in mtDNA-depleted cells compromise QC autophagic response
We next evaluated autophagosome formation by directly measuring the cellular distribution of endogenous LC3B and by monitoring the autophagic flux, as determined by comparing the accumulation of autophagosomes after the inhibition of lysosomal proteolysis with NH4Cl/leupeptin (NL) relative to the steady-state levels of autophagosomes (22).
Under basal conditions, sPD cybrids show an increase in both the number and size of endogenous LC3B puncta relative to CT cybrids. (Fig. 3Aa versus c, and B), but when lysosomal degradation was inhibited this number increased more markedly in CT cybrids (Fig. 3Ab versus d, and B), indicating a higher basal autophagic activity for CT cybrids. Removal of serum further activated autophagy in a higher extent in CT cybrids than in sPD cybrids, as depicted by the higher increase in the number of bright puncta without treatment (Fig. 3Aa versus e, compared with c versus g, and B). Similar results were obtained when lysosomal degradation ability was inhibited (Fig. 3Ab versus f, compared with d versus h, and B).
Figure 3.

QC autophagic response is impaired in cells harboring sPD patient mitochondria. (A) LC3B immunostaining (green) of CT and sPD cybrids maintained in the presence [Serum (+)] or absence [Serum (−)] of serum and inhibitors of lysosomal proteolysis [NL (+); NL (−)]. Hoechst 33342-stained nuclei are in blue. (B) Mean number of LC3B-positive vesicles per cell profile (n= 3, *P< 0.05, ***P< 0.001 versus S+ CT cybrids; ##P< 0.01 versus S− CT cybrids). Scale bar: 10 µm. (C) Immunoblot for endogenous LC3B from CT and sPD cybrids after culture in serum (+) or serum (−) conditions and treatment with NL. (D) Densitometric analysis of endogenous levels of LC3B (n= 18, ***P< 0.001 versus S+ CT cybrids; ###P< 0.001 versus S+ sPD cybrids; $$$P< 0.001 versus S− CT cybrids; &&&P< 0.001 versus S− sPD cybrids). (E) Determination of autophagic vacuole (AV) levels. Values of LC3-II in the absence of NL represent the steady-state AV content (n= 18, *P< 0.05, ***P< 0.001 versus S+ CT cybrids). (F) Assessment of autophagic flux, calculated as the ratio of LC3-II densitometric value of NL-treated samples over the corresponding untreated samples (n= 18, **P< 0.01 versus S+ CT cybrids).
These immunofluorescence results were confirmed by determining the autophagic flux by immunoblot. By this approach, we have also detected an increase in the basal levels of LC3B-II, a phosphatidylethanolamine-conjugated form that is localized to autophagosomes, in sPD cybrids when compared with CT cybrids (Fig. 3C and D). As in the LC3B puncta staining data, the increase in LC3B-II upon inhibition of lysosomal degradation was higher for CT cybrids under normal nutrient conditions as well as upon induction of autophagy by starvation [serum (-) conditions]. Accordingly, we observed that, although the basal levels of autophagic vacuoles are increased in sPD cybrids, their rate of autophagic degradation is significantly lower when compared with CT cybrids (Fig. 3E and F, respectively). Also, when compared with CT cybrids, sPD cybrids showed increased basal levels of the autophagic substrate p62 which were not potentiated after lysosomal inhibition, demonstrating an impaired lysosomal turnover of protein cargo via autophagy in these cells (Fig. 10A and B).
Figure 10.

α-Synuclein and p62 degradation by autophagy is impaired in cells harboring mitochondrial dysfunction. (A) Immunoblot for p62 from CT and sPD cybrid cells cultured in S+ or S− conditions and treated with or without lysosomal inhibitors (NL). (B) Densitometric analysis of p62 levels (n= 6, **P< 0.01 versus S+ CT cybrids; #P< 0.05, versus S− CT cybrids). (C and E) Immunoblots for α-synuclein oligomeric forms from CT and sPD cybrid cells cultured in S+ or S− conditions and treated with or without lysosomal inhibitors (NL). Representative blots of Triton X-100-soluble oligomeric species (C) and Triton X-100-insoluble, SDS-resistant oligomeric species (E). The arrow indicates band of tetrameric form of α-synuclein. (D and F) Densitometric analysis of α-synuclein-soluble oligomers content (D) (n= 12, *P< 0.05, **P< 0.01, versus S+ CT cybrids; ##P< 0.01, versus S+ sPD cybrids) and α-synuclein-insoluble oligomers content (F) (n= 12, ***P< 0.001, versus S+ CT cybrids).
We obtained similar results with the parental cell line NT2 when treated with MPP+ (1 mm, 24 h), a classic inhibitor of complex I (Supplementary Material, Fig. S1A–D). These results were also corroborated in differentiated sPD cybrid cell lines that exhibit processes similar to neuronal cells (Supplementary Material, Fig. S2A and B). Consistent with our results in cybrid cells and MPP+-treated NT2 cells, LC3B content increased significantly more in neuronal-like CT cybrid cells when exposed to lysosomal inhibition for 4 h, indicating that these cells were more prone to develop an autophagic response rather than nsPD cybrids (Supplementary Material, Fig. S2A and B). The increased accumulation of LC3-II obtained for all those cellular models was also evident in sPD patient peripheral blood lymphocytes (Supplementary Material, Fig. S3A and B). Moreover, under serum starvation conditions, CT cybrids activated nonselective ‘bulk’ autophagy in a greater extent than sPD cybrids, although no alterations were observed in the autophagic flux.
Similar results were obtained for cells lacking the mitochondrial genome (Rho0 cells) and thereby a dysfunctional mitochondrial respiratory system. In the absence of such system, these cells require alternative mechanisms for the maintenance of an appropriate redox state and energy supply. If sufficient pyruvate and uridine are provided, these cells can grow without a functional mitochondrial electron transport chain and oxidative phosphorylation. Theoretically, this would increase the glycolytic pathway flux for continual regeneration of NAD+, including that regenerated by the plasma membrane oxidoredutase (23), ensuring that glycolytic pathway will provide sufficient ATP to sustain essential cellular metabolic activities and cell viability (24).
In agreement with the EM data in Figure 2B, Rho0 cells showed a higher number of autophagic vacuoles in comparison with their parental counterpart cell line containing a full complement of mtDNA (Rho+ cells), distinguished as LC3B-positive puncta by immunofluorescence (data not shown) and by autophagic vacuole basal levels (Supplementary Material, Fig. S4A and B). However, this was not accompanied by an increase in the number of these organelles in response to lysosomal inhibition (Supplementary Material, Fig. S4A and B), supporting a lower autophagic flux in Rho0 cells (Supplementary Material, Fig. S4C).
To evaluate whether mitochondrial dysfunction could lead to the impairment in the autophagic response due to a bioenergetic failure, we have also compared the autophagic response of Rho0 cells grown in the presence [pyr/urd (+/+)] or absence [pyr/urd (−/−)] of pyruvate and uridine. Subsequently, autophagy was induced exposing cells to serum starvation conditions. Under these conditions, cells were also maintained in non-supplemented media to determine the extent to which the autophagic response depends on ATP production derived from glycolysis. No statistically significant differences were observed between cells grown in pyr/urd (+/+) or in pyr/urd (−/−) media for 24 h (Fig. 4A a versus c and e versus g, and B). Moreover, the shift to a punctuate pattern of LC3B upon inhibition of lysosomal-degradative ability was not statistically different between cells subject to both types of pyr/urd treatment, under both normal growing (Fig. 4A b versus d and B) and serum removal conditions (Fig. 4Af versus h, and B). We also checked the effect of growing cells in pyr/urd (−/−) medium for 48 h on the autophagic response, but no significant changes were observed (data not shown). These results were supported by our biochemical data showing significantly higher LC3B-II levels only upon serum withdrawal (Fig. 4C–E), which was correlated with an impaired autophagic response/activity (Fig. 4F).
Figure 4.

QC autophagic response is impaired in mtDNA-depleted cells. (A) LC3B immunostaining (green) of Rho0 cells maintained in serum (+) or serum (−) conditions and NL treatment, following growth in the presence [pyr/urd (+/+)] or absence [pyr/urd (−/−)] of pyruvate and uridine. Hoechst 33342-stained nuclei are in blue. (B) Mean number of LC3B-positive vesicles per cell profile [n= 3, **P< 0.01 versus S+ pyr/urd (+/+); ###P< 0.001 versus S− pyr/urd (+/+)]. Scale bar: 10 µm. (C) Immunoblot for endogenous LC3B from Rho0 cells maintained in serum (+) or serum (−) and NL treatment following growth in pyr/urd (+/+) or pyr/urd (−/−) conditions. (D) Densitometric analysis of LC3B endogenous levels [n= 5, *P< 0.05, **P< 0.01, versus S+ pyr/urd (+/+); ##P< 0.01, versus S+ pyr/urd (−/−); &&P< 0.01, versus S− pyr/urd (+/+); $$P< 0.01, versus S− pyr/urd (−/−)]. (E) Determination of autophagic vacuole (AV) levels [n= 5, *P< 0.05, versus S+ pyr/urd (+/+)]. (F) Assessment of autophagic flux (n= 5).
Overall, these data support that the defect on both QC and ‘bulk’ autophagy observed in PD cells was not due to changes on ATP availability through glycolysis.
Autophagic degradation is impaired in MPP+-treated primary cortical neurons
To extend these findings and further confirm our previous observation that mitochondrial deficits can compromise the degradative ability of the autophagic system, we used MPP+-treated primary cortical neurons, a well-established in vitro model of PD-associated neuronal dysfunction directly caused by mitochondrial impairment (25–27). In this model, we have also found a rapid accumulation of LC3B-positive vesicles within 4 h after blocking of lysosomal proteolysis, indicating a constitutive autophagic flux in primary cortical neurons (Fig. 5Aa versus b and e versus f). However, we could not observe an increase in the number of LC3B-positive vesicles in MPP+-treated cells (50 µm, 24 h) when compared with CT cells (Fig. 5Aa versus C, and B), both in CT conditions or in the presence of rapamycin, an mTOR inhibitor that induces autophagy (Fig. 5Ae versus G, and B). These data were also verified by immunoblotting, showing no significant effect on the levels of LC3B-II when cells were exposed to MPP+ (Fig. 5C–E). This was accompanied by a significant reduction on the autophagic flux (Fig. 5F), similar to the results obtained using cellular models of mitochondrial dysfunction. Rapamycin was able to develop an autophagic response, which indicates that the decreased flow observed by MPP+ treatment is specific. Even in the presence of rapamycin, MPP+-treated cortical neurons were unable to develop an efficient autophagic response, since autophagic flux was decreased when compared with rapamycin treatment and did not reach CT values.
Figure 5.

MPP+-induced mitochondrial dysfunction mediates autophagy-lysosome pathway impairments in primary cortical neurons. (A) LC3B immunostaining (green) of primary cortical neurons treated with MPP+ for 24 h. In the last 4 h, cells were co-treated with or without rapamycin and lysosomal inhibitors (NL). Beta-III-tubulin (red) and Hoechst 33342 (blue) co-staining were used as a neuronal and nuclei markers, respectively. Scale bar: 10 µm. (B) Mean number of LC3B-positive vesicles per cell profile (n= 3, **P< 0.01, ***P< 0.001, versus CT; ###P< 0.001, versus MPP+-treated cells; &P< 0.05, &&P< 0.01, versus rapamycin (Rap)-treated cells; $$$P< 0.001, versus Rap+MPP+-treated cells. (C) LC3B immunoblot of the same cells after MPP+ (24 h) or rapamycin (Rap, 4 h) treatment. (D) Densitometric analysis of LC3B endogenous levels (n= 13, *P< 0.05, ***P< 0.001, versus untreated cells; ###P< 0.001, versus MPP+-treated cells; &&P< 0.01, versus Rap-treated cells; $$$P< 0.001, versus Rap+MPP+-treated cells). (E) Determination of autophagic vacuole (AV) levels. Values of LC3-II in the absence of NL represent the steady-state AV content (n= 13, *P< 0.05, versus CT). (F) Assessment of autophagic flux, calculated as the ratio of LC3-II densitometric value of NL-treated samples over the corresponding untreated samples (n= 13, *P< 0.05, **P< 0.01, versus CT; &P< 0.05, versus Rap-treated cells).
Although with minor differences between chronic and acute models, together these data suggest that mitochondrial impairment leads to alterations in the autophagy system, as mainly translated by a reduction in the autophagic flux.
Induction of autophagy is not primarily affected in sPD transmitochondrial cybrids
We further tried to understand the nature of the changes in the autophagy-lysosome pathway found in the context of our sPD transmitochondrial cybrid model. Since this pathway encompasses multiple steps, changes not only in the clearance but also in the initiation could concur for the accumulation of autophagic vesicles observed. In order to clarify if an abnormal induction of autophagy could also be involved and contribute to the observed autophagic vesicles accumulation, we started by analyzing the rates of LC3B-II synthesis. We evaluated possible changes in synthesis in the presence of lysosomal proteolytic activity inhibitors at three different time points (Fig. 6A). The changes in LC3B-II levels when degradation is clamped would reflect the synthesis of autophagosomes in the experimental time frame if the autophagy system is under steady state (22). According to our previous data on LC3B-II flux (Fig. 3C–E), the basal levels of LC3B-II were significantly higher in sPD cybrids in comparison with CTs (after 4 h). However, no significant differences between sPD and CT cybrids were observed for the levels of LC3B-II along this time course when lysosomal activity was inhibited (Fig. 6B). Since LC3-II levels were maximal at the earliest time point considered (2 h), we also determined changes in LC3-II formation between CT and sPD cybrids at earlier time points (30 min and 1 h). As expected, no changes were found between these two cell lines (data not shown). In addition, Figure 6C shows that the rates of autophagosome synthesis were similar in CT and sPD cybrids when comparisons were defined for 4 versus 2 h as well as 6 versus 4 h, further supporting the notion that the system was under steady state. These results indicate that the initiation step is not affected in sPD cybrids.
Figure 6.

AV synthesis is not primarily affected in sPD cybrid cells. (A) LC3B immunoblot for autophagosome synthesis assessment in CT and sPD cybrids treated with or without lysosomal inhibitors (NL) at three different time points (2, 4 and 6 h) (n =4). (B) Time course for autophagic vacuole (AV) levels. AV levels correspond to the densitometry values of LC3-II for each condition at each time point (n =4, ***P< 0.001, versus CT cybrids). (C) Assessment of autophagic synthesis. Rates of AV synthesis were determined by comparing LC3-II levels at two different time points after the addition of NL (4 versus 2 and 6 versus 4 h) (n =4).
We next decided to explore some of the key proteins belonging to the PI3-kinase complex machinery that controls the nucleation of autophagic vesicles. One of these proteins is Beclin-1. Beclin-1 is a key autophagy protein that acts as an essential activator of autophagy and for this reason its cellular abundance has been correlated with autophagic activity (28). Surprisingly, we did not find significant differences in the total cellular levels of this protein in sPD cybrids in comparison with CT cybrids (Fig. 7A and B) in the presence of serum. However, when cells were challenged to induce autophagy by serum withdrawal, a large increase in the levels of Beclin-1 was observed for CT cybrids, which was not found for sPD cybrids, demonstrating the inefficient capability of these cells to efficiently prompt a ‘bulk’ autophagic response, as previously observed in Figure 3C and F.
Figure 7.

Autophagic induction is not compromised in sPD cybrid cells. (A) Immunoblot for Beclin-1 from CT and sPD cybrids after culture in serum (+) or serum (−) conditions and treatment with or without NL. (B) Densitometric analysis of endogenous levels of Beclin-1 (n= 8, ***P< 0.001, versus S+ CT cybrids). (C) Immunoblot for Bcl-2 from CT and sPD cybrids after culture in serum (+) or serum (−) conditions and treatment with NL. (D) Densitometric analysis of endogenous levels of Bcl-2 (n= 6, *P< 0.05, **P< 0.01, ***P< 0.001, versus S+ CT cybrids). (E) Representative immunoblots for Bcl-2 and Beclin-1 cellular subcompartmentalization in mitochondria (M)- and cytosol (C)-enriched fractions. (F) and (G) Densitometric analysis of the levels of Bcl-2 (F) (n= 3, **P< 0.01, versus S+ CT cybrids; ##P< 0.01, versus S− CT cybrids) and Beclin-1 (G) (n= 3). (H) Co-immunoprecipitation of Beclin-1 and Bcl-2 in CT and sPD cybrids maintained in serum (+) or serum (−) conditions. Levels of Beclin-1 (top) and Bcl-2 (bottom) in the input, immunoprecipitate (IP) and flow through (FT) are shown. (I) Determination Bcl-2/Beclin-1 physical interaction (n= 3).
Another hypothesis is that the cellular availability of Beclin-1, rather than just total cellular abundance, might be the key point to abnormal induction of autophagy (12). Beclin-1 interacts with the class III phosphatidylinositol 3-kinase (Vps34) to localize other autophagy proteins to the pre-autophagosomal membrane (29). In conditions in which autophagy is inhibited, Beclin-1 is inactivated by its interaction with Bcl-2, which functions as a brake to autophagy and autophagic cell death by affecting the interaction between Beclin-1 and Vps34. Thus, we sought to determine whether Bcl-2 has a role in modulating the autophagic response observed in our sPD models. Consistent with data from other study involving sPD cybrids (30), we have found that our sPD cybrids exhibited significantly increased basal levels of Bcl-2 relative to CT cybrids (Fig. 7C and D). Additionally, alterations in the total levels of Bcl-2 were also associated with lysosomal inhibition and serum removal, more remarkably in CT cybrids, which showed dramatic increases in the levels of this protein in such conditions (Fig. 7C and D).
We next assessed the cellular subcompartmentalization of Bcl-2 and Beclin-1 in cytosol and mitochondria-enriched fractions. We have observed that the content of Bcl-2 was increased in the mitochondria-enriched fractions of sPD cybrids relative to CT cybrids (Fig. 7E and F), which is in accordance with the results obtained for the whole-cell lysates (Fig. 7C and D). Conversely, no major differences were detectable in the levels of Beclin-1 between subcellular fractions of CT and sPD cybrids (Fig. 7E and G). However, the changes in the total cellular abundance obtained for both proteins approximately match the sum of their cytosolic and mitochondrial enrichment.
As well, the targeting of Beclin-1 by Bcl-2 represents the crucial point to understand the mechanism of autophagy regulation. Therefore, we have also investigated the physical interaction between Beclin-1 and Bcl-2. When normalized to Beclin-1, the amount of Bcl-2 associated with Beclin-1 did not differ in sPD and CT cybrids (Fig. 7H and I). In addition, the anti-apoptotic protein Bcl-2 inhibits starvation-induced autophagy via targeting the BH3-like domain in Beclin-1 (31,32).
Taken together, these data strongly suggest that the defects in autophagic flux and more remarkably in the autophagic response under serum starvation conditions are not due to defects in the nucleation complex machinery, and the initiation step of autophagy is not primarily affected in our models of sPD.
Deficient intracellular traffic results in incomplete autophagosome degradation and reduced autophagosome and mitochondrial movements in sPD transmitochondrial cybrids
Autophagic failure found in our sPD cybrids model can begin from a delayed or interrupted traffic of autophagosomes along the microtubule system, which provides the tracks necessary for an efficient mobilization of cargos through the autophagy pathway. In support of this hypothesis, we have shown earlier that mitochondrial deficits induce alterations in microtubule network, mainly characterized by an increase in free/polymerized tubulin ratio that was responsible for high levels of α-synuclein oligomeric forms (33).
In order to correlate our previous findings with defective autophagic clearance, we have modulated microtubule-dependent trafficking of autophagosomes by using taxol (a microtubule polymerizing agent) and nocodazole (a microtubule depolymerizing agent) in concentrations that did not interfere with cell viability (data not shown), in the presence or absence of lysosomal inhibitors. Taxol was effective in promoting autophagy turnover in sPD cybrids, as reflected by decreased autophagosome content (Fig. 8A and B) and improved autophagic flux (Fig. 8A and C). In contrast, nocodazole considerably enhanced autophagosome accumulation in CT cybrids (Fig. 8A and B) associated with a decreased autophagic flux similar to what was observed in sPD cybrids (Fig. 8A and C). These findings suggest that autophagosomes clearance may be dependent on the functional status of the microtubule-dependent intracellular traffic. If so, the dynamic interaction of autophagic vacuoles with the microtubule system tracks may be altered in our sPD models hampering autophagosome transport and so the fusion with lysosome. Accordingly, we observed a decreased degree of co-localization between LC3-positive vacuoles and Lamp-1-positive vacuoles in sPD cybrids, an effect that was reversed by taxol treatment. This result may indicate an impairment of autophagosome–lysosome fusion, but could also point to a disruption in the autophagosomal intracellular trafficking (Supplementary Material, Fig. S5A and B). In contrast, nocodazole treatment decreased LC3B/Lamp-1 co-localization in both CT and sPD cybrids (Supplementary Material, Fig. S5A and B).
Figure 8.

Disruption of microtubule network results in a deficient autophagic turnover and reduced autophagic vesicle movements in sPD cybrid cells. (A) Immunoblot for endogenous LC3B from CT and sPD cybrids after treatment with taxol (Tax) or nocodazole (Noc) for 24 h. In the last 4 h, cells were co-treated with or without lysosomal inhibitors (NL). (B) Determination of autophagic vacuole (AV) levels. Values of LC3-II in the absence of NL represent the steady-state AV content (n= 4, *P< 0.05, ***P< 0.001, versus CT cybrids; ##P< 0.01 versus sPD cybrids). (C) Assessment of autophagic flux, determined as the ratio of LC3-II densitometric value of NL-treated samples over the corresponding untreated samples (n= 4, *P< 0.05, **P< 0.01, versus CT cybrids; ###P< 0.001 versus sPD cybrids). (D) Accumulation of AVs in CT and sPD cybrid cells. LC3B immunostaining was used to determine AV size distribution from CT and sPD cybrids after treatment with or without lysosomal inhibitors (NL). AV size distribution was graphed as percent of total vacuoles within the indicated size ranges (n= 4, *P< 0.05, **P< 0.001, versus CT cybrids). (E) Representative kymograph images (out of three experiments) of AV movement in CT and sPD cybrid cells treated with nocodazole (24 h) or taxol (24 h). Scale bars: 5 µm. (F) Number of movable AVs when compared with those of total AVs (n= 3, *P< 0.05, versus CT cybrids). (G1 and 2). Cumulative data for AV transport velocity in CT and sPD cybrid cells treated with Noc (G1) or treated with taxol (G2) (n= 3). (H) Average transport velocity of AVs (μm/s) (n= 3, **P< 0.01, versus CT cybrids; #P< 0.05, versus sPD cybrids).
Moreover, changes in autophagy progression between CT and sPD cybrids were also addressed by measuring the vesicle size distribution profile in the absence and presence of NL (34). Comparing the vesicle size distribution in these conditions reveals a further increase in larger vesicles >1.5 μm, which under normal autophagy conditions are supposed to be quickly degraded (34). In addition, we observed that in sPD cells there is a significant increase of the largest size autophagic vacuoles (>2.25 μm) indicating an accumulation of these structures which positively correlates with a decreased vesicular trafficking (Fig. 8D). In this context, we have also monitored autophagic vacuoles movements. For each observed autophagic vacuole, we recorded whether it moved or remained static (Fig. 8E), and subsequently the relative percentages of stationary or movable events were calculated (Fig. 8F). As shown in Figure 8F, sPD cybrids exhibited a significant decreased number of movable autophagic vacuoles. In addition, microtubule disruption by nocodazole promoted a decrease in the number of movable autophagic vacuoles to similar values in both CT and sPD cybrids.
We have also determined autophagic vacuole transport velocity as described previously (35). Cumulative percentage data displayed a left shift in autophagic vacuole transport velocity in sPD cybrids compared with basal CT cybrids (Fig. 8G1 and 2). The shift in curve profiles was minimized when cells were treated with nocodazole or taxol (Fig. 8G1 and 2). Intriguingly, the effect of taxol was more pronounced for sPD cybrids, resulting in a deflection of the cumulative curve to a similar profile observed for CT cybrids (Fig. 8G2). Concerning average velocities, sPD cybrids demonstrated a significant reduction in autophagic vacuole movement velocity similar to what was observed for CT cybrids treated with nocodazole. On the other hand, taxol was able to restore autophagic vacuole velocity in sPD cybrids (Fig. 8H). We believe that this process relies on a dynamic state of mitochondria at several levels of organization and interaction, such as the distribution of mitochondria within a cell, and the dynamic interaction with the vesicular trafficking and turnover mechanisms, as both autophagic vacuoles and mitochondria are driven by dynein and kinesin motors and depend on microtubule system tracks for transport within the cell (36).
This premise led us to analyze alterations in mitochondrial movements in sPD cybrids. Similar to what was performed for autophagic vacuoles, we recorded whether mitochondria moved or remained static (Fig. 9A), and subsequently the relative number of stationary or movable events was calculated (Fig. 9B). As shown in Figure 9B, sPD cybrids exhibited a significant decreased number of movable mitochondria in the fields studied. In addition, microtubule disruption by nocodazole, disruption of vesicular traffic by neutralizing acidic vesicles with NH4Cl/leupetin (37) and serum removal further promoted a decrease in the number of movable mitochondria to similar values in both CT and sPD cybrids. In contrast, taxol was able to re-establish the number of movable mitochondria in sPD cybrid cells.
Figure 9.

Disruption of microtubule network results in reduced mitochondrial movements in sPD cybrid cells. (A) Representative kymograph images (out of three experiments) of mitochondrial movement in CT and sPD cybrid cells treated with nocodazole (24 h), taxol (24 h) and lysosomal inhibitors (NL, 4 h) or subjected to starvation (starved, 6 h). Scale bars: 5 µm. (B) Number of movable mitochondria when compared with those of total mitochondria (n= 3, *P< 0.05, **P< 0.01, ***P< 0.001, versus CT cybrids; ##P< 0.01, ###P< 0.001, versus sPD cybrids). (C1–4) Cumulative data for mitochondrial transport velocity in CT and sPD cybrid cells treated with Noc (C1), NL (C2), subjected to starvation (C3) or treated with taxol (C4) (n= 3). (D) Average transport velocity of mitochondria (μm/s) (n= 3, *P< 0.05, ***P< 0.001, versus CT cybrids; #P< 0.05,##P< 0.01,###P< 0.001, versus sPD cybrids).
We have also measured mitochondrial transport velocity as described previously (38). As observed previously for autophagic vacuoles, cumulative percentage data displayed a left shift in mitochondrial transport velocity in sPD cybrids when compared with basal CT cybrids (Fig. 9C1–4). In addition, this difference in curve profiles was abolished when cells were treated with nocodazole (Fig. 9C1) and under starvation (Fig. 9C3). Interestingly, the effect of lysosomal degradation inhibition was more pronounced for CT cybrids, resulting in a cumulative curve that was left-shifted relative to the sPD cybrid subjected to the same treatment (Fig. 9C2). Such effects were completely counteracted by taxol, reflected by a pull-down change in the cumulative curve profile (Fig. 9C4). Surprisingly, this change was more obvious for sPD cybrids rather than for CT cybrids. Regarding average velocities, sPD cybrids demonstrated a significant decline in mitochondrial movement velocity. As expected, taxol promoted an increase in moving mitochondria velocity in both CT and sPD cybrids (Fig. 9D). A dramatic negative effect was found with nocodazole and even more remarkably following autophagic modulation (serum removal and lysosomal degradation inhibition) (Fig. 9D).
Altogether, our data suggest that a correlation exists between mitochondrial motility and autophagic turnover, both processes being dependent on the dynamic stability and functional integrity of microtubule network.
Alterations in autophagic activity and defective microtubule-dependent transport result in a poor α-synuclein aggregate clearance
Autophagy deregulation has long been implicated in cellular aging and α-synuclein toxicity during PD. Indeed, in PD, α-synuclein accumulation has been linked to alterations in chaperone-mediated autophagy and lysosomal system functioning (39–42). Therefore, we next investigated the impact of alterations in the autophagic activity due to mitochondrial deficits and altered microtubule transport in autophagy substrates and PD-related α-synuclein clearance.
We started by evaluating the levels of p62/SQSTM1, a selective substrate for autophagy as this protein is normally localized to the autophagosome via LC3 interaction and is continuously degraded by the autophagy–lysosome system (43). According to our previous data on LC3B-II flux (Fig. 3C–E), we have verified that p62 levels were significantly increased in sPD cybrids relative to CT cybrids (Fig. 10A and B).
We then analyzed the α-synuclein solubility and its propensity for oligomerization in the presence or absence of lysosomal proteolytic activity inhibitors by performing a Triton X-100 detergent fractionation. Overall, Figure 10C and E shows that Triton-soluble and -insoluble α-synuclein band patterns, resulting from α-synuclein oligomerization, were comparable. However, it was clear that α-synuclein was preferentially accumulated in oligomeric forms, in the detergent-soluble fraction (Fig. 10C and D). Interestingly, among them, we detected a band corresponding to the tetrameric form (denoted by an arrow in Fig. 10C) that has been recently shown as a helically folded structure in cells under physiological conditions (44). We have observed that the generation of SDS-resistant and high molecular weight-soluble species of α-synuclein was enhanced in sPD cybrids relative to CT cybrids (Fig. 10D and F). Moreover, we have also detected in sPD cybrids that lysosomal inhibition did not potentiate α-synuclein oligomers accumulation within cells (Fig. 10C–F). Likewise, activation of nonselective autophagy upon serum removal was not able to efficiently shift back to a completely soluble non-oligomerized phenotype, further supporting the argument that the final steps of the autophagy-lysosome pathway are compromised in these cells.
Impairment of autophagic turnover prompts apoptosis in cells harboring mitochondrial dysfunction
Autophagy is well recognized as a survival mechanism especially during conditions of nutrient limitation. Based on the bulk degradation of cytoplasmic material, autophagy is utilized to generate both nutrients and energy in starving cells. Under these conditions, autophagy is critical for maintaining cell survival. We have previously reported that, in our sPD cybrid model and MPP+-treated cells, both mitochondria and endoplasmic reticulum-dependent apoptotic pathways are initiated during particular stressful circumstances of PD pathology (17,45,46).
Here, we examined whether autophagic degradation impairment observed in our cellular models is also related to the apoptotic events described formerly. In order to check whether autophagy could be activated in cells with mitochondrial deficits as an attempt to prevent cell suicide by apoptosis, we used pharmacological modulators of autophagy and determined their roles on caspase-3-mediated apoptosis.
We observed that treatment of our cells with 3-methyladenine (3-MA), a pharmacological compound that inhibits the early sequestration events in autophagy, further stimulated caspase-3 activation in all models studied (Fig. 11A–C). Interestingly, co-incubation with MPP+ significantly enhanced this activation in NT2 cells (Fig. 11C).
Figure 11.

Impaired autophagic turnover triggers caspase-3 over-activation in cells harboring mitochondrial deficits. Caspase-3 activation was addressed by Ac-DEVD-pNA cleavage in CT and sPD cybrids (A) (n= 12; ***P< 0.001, versus CT cybrids); NT2 Rho0 cells (B) (n= 5; **P< 0.01, ***P< 0.001, versus untreated Rho0 cells) treated with or without lysosomal inhibitors (NL), 3-MA or rapamycin (Rap) for 4 h; and in MPP+-treated NT2 Rho+ cells treated with or without 3-MA for 4 h (C) (n= 6–8; *P< 0.001, versus untreated cells; ###P< 0.001, versus MPP+-treated cells; &&P< 0.01, versus 3-MA-treated cells) or Rap for 6 h (D) as indicated (n= 6–8; ***P< 0.001, versus untreated cells; #P< 0.01, versus Rap-treated cells; &&&P< 0.001, versus MPP+-treated cells).
Rapamycin-induced autophagy did not prevent caspase-3 activation in sPD cybrids, which indicates that the autophagy-lysosomal pathway is impaired in this model as described previously (Fig. 11A). However, rapamycin per se was able to further activate caspase-3 in Rho0 cells (Fig. 11B). In MPP+-treated cells, rapamycin counteracted the deleterious effects of MPP+ on caspase-3 activation (Fig. 11D). Perhaps surprisingly, these results indicate different apoptotic outcomes upon modulation of authophagy in the different paradigms of mitochondrial dysfunction.
In an acute model, such as MPP+-treated cells, activation of autophagy by rapamycin can still protect cells. In chronic models with great compromise of mitochondrial function (Rho0 cells), both activation of autophagy by rapamycin or inhibition by 3-MA could be harmful to cells. Additionally, in an intermediate chronic stage, such as cybrids, cells may eventually succumb to biochemical processes typically associated with apoptosis when autophagy is inhibited by 3-MA, in accordance with NT2 cells, but has no additive effect on sPD cybrids. Moreover, when autophagy is stimulated by rapamycin, sPD cybrids are unable to efficiently proceed with the pathway probably due to vesicular traffic impairment, and still activate apoptosis. The mechanisms underlying these differences in cellular responses are unclear but might be related with different capacities of the cell models to compensate for autophagic dysfunction.
DISCUSSION
In this study, we have demonstrated that a prolonged metabolic failure due to mitochondrial dysfunction, either in cellular models harboring sPD subjects mtDNA (sPD cybrids) or knock-down of all mtDNA (Rho0 cells) or in MPP+-treated cortical neurons, causes a functional decline in the activity of the autophagic system. Consistently, in our models, autophagosomes are actively formed but some of their structural components and autophagic substrates are unable to be efficiently degraded within lysosomes. We propose that these alterations in autophagosome clearance can originate from a decreased efficiency in the mobilization of autophagosomes from their site of formation toward lysosomal compartments along the microtubule network, thereby impeding the assumed neuroprotective functions of autophagy.
Progress in the understanding of autophagy has emphasized its importance in cellular homeostasis, with significant consequences for the development of new therapeutic strategies for neurodegenerative disorders such as PD. In fact, ultrastructural examination has revealed an abnormal presence of autophagic vacuoles in myelinized neurons of the SNpc in PD patients, in contrast to the rare detection of autophagosomes in normal brains during aging (15).
Although the presence of accumulating autophagosomes could represent an aberrant activation of autophagy, we provide evidence that autophagy is not over-stimulated in our models and, instead, defective clearance of the autophagic vacuoles might account for those observations. In our study, the rapid accumulation of autophagosomes within a few hours after blocking of lysosomal proteolysis, even in primary cortical neurons, evidences a proper basal level of autophagic activity. However, and of significant relevance to PD pathology, the data on autophagic flux indicate that formed autophagosomes are not efficiently eliminated by lysosomal degradation. The situation in cortical neuron cultures treated with MPP+ was somewhat different than in other cellular models, but essential features of the effects of PD-related mitochondrial impairment in the lysosomal pathway were confirmed in this primary neuron setting.
The aforementioned results raise the possibility that alterations in mitochondrial energy metabolism play a role in the modulation of autophagy and, therefore, multiple defects in mitochondria-dependent metabolism might be the initial events in sPD-related autophagy pathology.
Mitochondria are the site of many biochemical reactions fundamental for normal cellular functioning, particularly cellular energy production. Our data further confirm and extend prior studies showing that ATP levels are reduced in sPD cybrids relative to those of CT cybrids (7,17). In addition, mitochondrial respiration and pathways influenced by aerobic metabolism are also altered in sPD cybrids. sPD cybrids show reduced SIRT1 phosphorylation, reduced PGC-1α levels and increased NF-κB activation (16). In this context, although our findings indicate that ATP may be required to the autophagic response, especially during serum starvation, the lack of effect of the pyruvate/uridine deprivation on the autophagic response in Rho0 cells leads us to hypothesize that an imbalance in glycolytic ATP production is unlikely to represent a primary signal for autophagy deregulation, reinforcing the concept that mitochondrial dysfunction has a main role on the control of the autophagic response. In fact, it is well established that autophagy is an energy-sensitive process (47–49). Although the various steps of the autophagic process may differ in their response to the energy status of the cell, early evidence indicated that all steps of this pathway are responsive to relatively small changes of intracellular ATP (50). Whether or not autophagosomes accumulate under ATP-depleted conditions would depend on the relative effects of energy depletion on sequestration and post-sequestrational steps of the autophagic pathway. In view of that, it was described that rapid mitochondrial depolarization with ATP loss did not induce mitochondrial degradation by autophagy, implying that additional mechanisms may regulate mitophagy in neurons (51).
Our results clearly support the idea that autophagy failure stemming from mitochondrial dysfunction is not translated into defects in autophagosome formation but mainly in the autophagosomes trafficking along the microtubule network toward the lysosomal compartment. Accordingly, in all of our paradigms, LC3B was observed in punctuated structures in both basal or serum deprivation conditions, supporting that the recruitment to the pre-autophagosomal structure is not affected. In addition, we did not find significant differences in the total cellular levels and subcompartimentalization of Beclin-1, a principal regulator in autophagosome formation. However, sPD cybrids exhibited increased basal levels of Bcl-2 mainly associated with increased targeting to mitochondria. Thus, the anti-autophagic activity of Bcl-2 was not verified as the mild changes observed in the binding and sequestration of Beclin-1 by Bcl-2 were not expected to disturb the formation of the class III PI3-kinase complex that is critical to the induction of autophagy. Even under serum starvation conditions, Bcl-2/Beclin-1 association in sPD cybrids was similar to that in CT cybrids allowing Beclin-1 to proceed with autophagy.
Autophagosome and lysosome movement relies on microtubules as tracks and dynein as a motor (52,53). Thus, tubulin cytoskeleton is fundamental to maintain the spatial organization of autophagy-lysosome pathway by conducting the trafficking of organelles and vesicles involved in different interactions during this process. We have previously described that sPD cybrids have mitochondria-dependent cytoskeletal changes, which manifest as microtubule depolymerization and increase in free tubulin levels (33). In this study, our results using modulators of microtubules assembly and intracellular traffic point to the fundamental requirement of an efficient microtubule network for the transport and so the clearance of autophagic cargos.
We confirm that autophagic flux is microtubule-dependent as depolymerization of microtubules with nocodazole inhibited the degradation of autophagosomes within lysosomes and microtubule stabilization with taxol potentiated autophagosome clearance and cargo motility. In addition, our results on live-cell imaging of autophagic vacuoles and mitochondrial transport clearly demonstrate that mitochondrial deficits as the ones described in our sPD cybrids (7,16,17) significantly impair autophagic vacuoles and mitochondrial motility, which alter autophagic and mitochondrial dynamics due to disruption of microtubule network trafficking. Moreover, these findings are in accordance with previous observations involving dyneins as essential motors (52,54), as starvation induced a slight decrease in the autophagosome flux in CT cybrids and a substantial decline in mitochondria motility. However, it is well established in the literature that starvation induces ‘bulk’ autophagy. A possibility is that those findings could be associated with an intracellular redistribution and specific alterations in the membrane binding ability of cytoplasmic dyneins to organelles (55,56), contributing to their defective transport, but further studies are needed to clarify this point.
Thus, the key role of mitochondria as ATP fuel suppliers for microtubule turnover and microtubule-based motor proteins implies that inherent mitochondrial dysfunction and/or related defects in their distribution in sPD cybrids will have a profound effect on the autophagy-related vesicular trafficking and, as a consequence, autophagy turnover maintenance and function. Interestingly, alteration in microtubules associated with deficient axoplasmic flow, altered mitochondrial turnover and loss of synaptic connectivity was observed in vulnerable neurons in AD (57).
In conjunction with altered mitochondrial function and motility, tubulin cytoskeleton alterations may contribute to the accumulation of protein aggregates and autophagic vesicles, and/or associated substrates, as observed in our sPD cybrids model. The levels of p62 and α-synuclein oligomerization profiles were concomitantly altered in these cells. These findings strongly support the hypothesis that aggregate-prone proteins, such as α-synuclein, initially interact with p62, and then aggregation of the protein complex occurs in a p62-dependent manner but the aggregates are not efficiently degraded by autophagy. Our data show that the levels of autophagic activity and its efficiency in the clearance of specific substrates and/or aggregated α-synuclein are dependent on the metabolic status of the cell.
Under these circumstances, the resulting accumulation of α-synuclein oligomers, other toxic protein products and/or damaged organelles, such as dysfunctional mitochondria, will obviously be detrimental for cell functioning and survival. Intracellular accumulation of toxic protein oligomers might in turn contribute to further failure of the autophagic system in cells, as aggregates might physically block vesicular trafficking by secondarily interacting with or sequestrating key elements involved in this process. Interestingly, when neuronal cybrids are differentiated, the resultant neurites exhibit several characteristics of blocked axons, such as α-synuclein aggregates reminiscent of LBs (58) and accumulation of vesicles resembling nonfused autophagic structures. Moreover, an inefficient degradation of dysfunctional mitochondria due to a decreased autophagic activity will uphold a positive feedback loop that will further propagate the neurodegenerative pathway in PD (59). Accordingly, long-term accumulation of autophagic compartments within cytosol can also be harmful as autophagosomes can became leaky, and if enzyme leakage occurs after fusion, it can lead to lysosomal breakdown and release of lysosomal hydrolases, which frequently activates cell death (12).
In summary, data reported in this work support our hypothesis that a mitochondrial dysfunction induces an alteration in microtubule assembly that hinders autophagy turnover, which, in turn, potentiates the accumulation of α-synuclein oligomers and, finally, prompts apoptosis.
Although it is predictable that autophagy may be a new therapeutic target in PD, the results obtained in this study indicate that therapeutic modulation in the context of sPD should be aimed at improving autophagy intermediates ‘backing up’. For example, targeting trafficking proteins (60), and proteins involved in promoting autophagosome–lysosome fusion, such as those regulating endosomal fusion and multivesicular bodies (61,62), will likely improve the efficiency of autophagosome maturation into autophagolysosomes for subsequent substrate degradation.
MATERIALS AND METHODS
Chemicals, antibodies and cell media
Ammonium chloride was from Merck KGaA (Darmstadt, Germany). 3-MA, leupeptin, MPP+, rapamycin, poli-d-lysine, poli-l-lysine, 5-fluoro-2′-deoxyuridine (FUdR), urd, cytosine β-d-arabinofuranoside (araC), paclitaxel (taxol) and nocodazole were from Sigma Chemical Co (St Louis, MO, USA). The colorimetric substrate acetyl-Asp-Glu-Val-Asp p-nitroaniline (Ac-DEVD-pNA) was from CalbiochemTM, Merck KGaA. For western blotting analysis, the following antibodies were used and the working dilutions are given in brackets: rabbit monoclonal antibody (mAb) anti-α-tubulin [clone 11H10 (1:1000)], rabbit polyclonal antibody (pAb) anti-Bcl-2 (1:1000), rabbit pAb anti-LC3B (1:1000), rabbit pAb anti-SQSTM1/p62 (1:1000), rabbit pAb anti-COX IV (1:1000) (all were from Cell Signaling Technology, Inc., Danvers, MA, USA). Mouse mAb anti-Beclin-1 (1:1000) was from BD Biosciences (San Diego, CA, USA). Mouse mAb anti-Hsp60 (1:1000) was from Millipore (Billerica, MA, USA). Rabbit pAb anti-VDAC/Porin (1:800) was from Abcam (Cambridge, UK). Mouse mAb anti-α-synuclein [clone LB509 (1:100)] was from Invitrogen Corporation (Camarillo, CA, USA). Rabbit pAb anti-TOM20 (1:200) was from Santa Cruz Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Alkaline phosphatase-conjugated secondary antibodies (1:15 000) were from GE Healthcare UK Limited (Buckinghamshire, UK). For immunocytochemistry, the following antibodies were used: rabbit pAb anti-LC3B (1:200), rabbit mAb anti-LC3B (clone D11) XP® (1:400) and mouse mAb anti-β3-tubulin [clone TU-20 (1:200)] (all were from Cell Signaling Technology, Inc.). Mouse mAb anti-LAMP-1 [clone H4A3 (1:100)] was from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA, USA). Rabbit pAb anti-TOM20 (1:200) was from Santa Cruz Biotechnology, Inc. Alexa Fluor® 488-conjugated goat anti-rabbit and Alexa Fluor® 594-conjugated goat anti-mouse antibodies were from Molecular Probes (Eugene, OR, USA). For immunoprecipitation, the following antibodies were used: rabbit pAb anti-Bcl-2 (1:1000) and rabbit pAb anti-Beclin-1 (1:100) from Cell Signaling Technology, Inc.
Neurobasal medium, cell lines' culture media, dialyzed, non-dialyzed and heat-inactivated fetal bovine serum (FBS), streptomycin, penicillin and B-27 supplement were from GibcoTM, Invitrogen Corporation.
NT2 Rho0 cell growth medium consisted of OPTIMEM supplemented with 10% heat-inactivated FBS, 200 μg/ml sodium pyruvate, 150 μg/ml uridine and 10 000 U/ml penicillin and 10 µg/ml streptomycin. Cybrid cell-line selection medium consisted of Dulbecco's modified Eagle's medium, supplemented with 10% dialyzed FBS, 100 IU/ml penicillin and 50 μg/ml streptomycin. Cybrid cell-line expansion medium consisted of OPTIMEM supplemented with 10% non-dialyzed FBS and 10 000 U/ml penicillin and 10 µg/ml streptomycin. Cybrid cell lines and NT2 cell-line growth medium consisted of OPTIMEM medium supplemented with 10% heat-inactivated FBS, 10 000 U/ml penicillin and 10 µg/ml streptomycin.
Cell-line culture and treatments
Human NT2 (Ntera2/D1) cells, a neuronally committed human teratocarcinoma cell line (63–65), were purchased from Stratagene Cloning Systems (La Jolla, CA, USA) and were cultured as described previously (46). NT2 Rho0 cells were kindly provided by R.H.S. (University of Kansas School of Medicine, Kansas City, Kansas, USA) and were cultured as indicated elsewhere (4,66). Cybrid cell lines were routinely cultured in growth medium as described previously (67).
Cell treatment conditions included normal nutrient (serum+) and serum starvation (hereafter simply ‘starvation’) by culturing cells in serum-free medium (serum−) for 6 h before harvesting or fixation. Compounds included MPP+ prepared as 10 mm stocks in water immediately before use and added to the medium at 1 mm final concentration. Where indicated, 20 mm ammonium chloride and 100 μm leupeptin, and 10 mm 3-MA or 0.5 μm rapamycin were added in the culture medium in the last 4 h of MPP+-treated or fed/starved cells. To modulate microtubule dynamic instability, 5 nm taxol and 1 µm nocodozole were used in normal nutrient conditions. For all experimental procedures, controls were performed in the absence of those agents.
Human subjects
sPD patients and healthy individuals were all recruited after approval by the University of Kansas School of Medicine Institutional Review Board. Individuals in the PD group were followed regularly in a tertiary referral movement disorders clinic at the Kansas University Medical Center and met criteria commonly used to diagnose PD in clinical and research settings (68). None of the patients were believed to have alternative diagnoses, degeneration of related systems, drug-induced parkinsonism or any other serious medical illness. Enrollment was also contingent on the absence of a diagnosis for another neurodegenerative disease.
The CT subjects were participants of a longitudinal ‘normal aging’ cohort that is characterized serially by the Brain Aging Project at the University of Kansas School of Medicine. These CT subjects have not been diagnosed with a neurodegenerative or pre-neurodegenerative disease condition. The age of the PD subjects who participated in this study was 64 ± 12.8 years, and for the CT subjects it was 74.3 ± 5.5 years.
After informed consent was given, sPD (n= 9) and age-matched CT (n= 5) subjects underwent a 10 ml phlebotomy using tubes containing acid-citrate-dextrose, as an anticoagulant, to provide the platelets needed for cell fusions.
Generation of cybrid cell lines
Cybrid approach consists of the transfer of sPD or healthy subjects' platelet mitochondria to mtDNA-depleted recipient cells (Rho0 cells), generating hybrid cell lines (cybrids). The resulting cybrid cell lines express the nuclear genes of the recipient Rho0 cell line and the mitochondrial genes of the platelet donor.
To generate cybrid cell lines for this study, we used a clonal stock of human teratocarcinoma cells containing no mtDNA (NT2 Rho0 cell line) created in NT2 cells by long-term exposure to 5 µg/ml ethidium bromide to deplete mtDNA selectively (4). Platelets (which contain mtDNA but not nDNA) from PD subjects are known to have reduced complex I activity relative to CT subjects' (3). We used platelet mitochondria to generate cybrid cell lines from both sPD and disease-free CT subjects. Previously, platelets were isolated from the individual blood samples and then were fused with NT2 Rho0 cells by co-incubation in polyethylenoglycol as described previously (66). The resulting cybrids were plated on T75 flasks, maintained for 1 week in Rho0 growth medium and then switched to cybrid selection medium for 6 weeks. NT2 Rh0 cells lack intact mtDNA, do not possess a functional mitochondrial electron chain and are auxotrophic for pyruvate and uridine. Maintaining cells in selection medium removes Rh0 cells that have not repopulated their mtDNA with platelet mtDNA. ‘Mock fusions’, in which NT2 Rho0 cells were not co-incubated with platelets, were performed in parallel with the proper fusions. During the selection period, all cells from the mock fusions died. After selection was complete, the cybrids were changed to cybrid expansion medium. Flasks were maintained in this medium at 37°C, 5% CO2 for 24 h prior to harvesting.
We used nine sPD and five CT cybrid cell lines for key experiments, such as those addressing the autophagic flux and the initiation steps of the autophagic pathway. In the remaining experiments, we used the three sPD and three CT cells lines showing the greatest differences in terms of mitochondrial bioenergetics as characterized previously (16).
mtDNA screening
Total DNA was extracted from three sPD and three CT cybrids by using standard methods and quantified by UV spectrophotometry (λ = 260 nm). Automated sequencing analysis was used, according to the manufacturer's instructions (3130 ABI Prism sequencing system), with BigDye® Terminator Ready Reaction Mix v3.1 (Applied Biosystems), for the investigation of the seven mtDNA genes coding for complex I subunits, corresponding to ND1, ND2, ND3, ND4L, ND4, ND5 and ND6, allowing the screening of confirmed pathogenic mutations, reported mutations, polymorphisms and novel sequence variations in these genes, according to MITOMAP (www.mitomap.org). All sequences were analyzed using Sequencing Analysis v5.4 and SeqScape v.2.5 software, by comparison with reference sequence obtained from GenBank database.
In silico analysis was performed for unpublished sequence variations found, using PolyPhen-2® and ClustalW2® for evolutionary conservation analysis among species.
Cybrid cell-line neuronal differentiation
Cybrid lines were differentiated to form process-bearing neuronal cells according to the procedure of Paquet-Durand et al. (69). Briefly, cells were allowed to form free floating conglomerates in non-tissue culture plastic, 10 cm diameter dishes in growth medium supplemented with 10 µm all-trans retinoic acid, renewed every 2 days. After 8 days in culture, the conglomerates were seeded in T75 cell culture flasks and cultures were kept for 10 days to completely generate a large number of cell aggregates. A single-cell suspension was then derived from the conglomerates by trypsinization and transferred to T175 culture flasks. Cells were cultured for 4 days to expand the culture and to further obtain conditioned medium. After that, cells were selectively trypsinized and transferred again to T75 culture flasks, where they were kept for 2 weeks to eliminate persistent undifferentiated cells by adding mitotic inhibitors (araC 1 μm, FudR 10 μm, urd 10 μm) to the growth medium. Finally, neuron-like cells (1 × 104 per coverslip) were plated onto glass coverslips (16 mm diameter) pre-coated with poly-d-lysine in 50:50 conditioned medium and allowed to improve neurite outgrowth for 4 days for further immunocytochemical experiments.
The enrichment and purity of neuronal-like cultures was evaluated by comparing the number of cells staining positively with antibodies directed against neuronal marker β-tubulin III, with the number of nuclei stained with Hoechst 33342. The numbers of β-tubulin III-positive cells and Hoechst 33342-positive nuclei were counted in 10 randomly selected visual fields. This was performed for two independent experiments.
Isolation of peripheral blood mononuclear cells
Twenty milliliters of venous blood from both sPD and disease-free CT subjects was collected by venipuncture in K2EDTA-containing tubes. Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll-Histopaque 1077 density gradient centrifugation, according to the manufacturer's instruction. Briefly, blood samples were diluted with the same amount of Hanks's balanced salt solution (HBSS), layered on Ficoll-Histopaque 1077 and centrifuged for 30 min at 300g at room temperature. PBMCs were collected from the interface between serum and Ficoll-Histopaque 1077 gradients and washed once with HBSS. PBMC protein extracts were obtained by using 1% Triton X-100 containing hypotonic lysis buffer [25 mm HEPES, pH 7.5, 2 mm MgCl2, 1 mm EDTA and 1 mm EGTA, supplemented with 2 mm DTT, 0.1 mm phenylmethylsulfonylfluoride (PMSF) and a 1:1000 dilution of a protease inhibitor cocktail].
Animals
Experiments involving animals were approved by and performed in accordance with the University of Coimbra Institutional Animal Care and Use Committee guidelines and European Community Council Directive for the Care and Use of Laboratory Animals (86/609/ECC).
Culturing and treatment of primary cortical neurons
Primary neuronal cultures were prepared as described previously (70), with minor modifications. Cerebral cortices were removed from embryonic day 15–16 of Wistar rats and were aseptically dissected and combined in Ca2+- and Mg2+-free Krebs buffer [120 mm NaCl, 4.8 mm KCl, 1.2 mm KH2PO4, 13 mm glucose, 10 mm HEPES (pH 7.4)] and then incubated in Krebs solution supplemented with BSA (0.3 g/l) containing trypsin (0.5 g/l) and DNase I (0.04 g/l) for 10 min at 37°C. Tissue digestion was stopped by the addition of trypsin inhibitor (type II-S; 0.75 g/l) in Krebs buffer containing DNase I (0.04 g/l), followed by a centrifugation at 140 g for 5 min. After washing the pellet once with Krebs buffer, the cells were dissociated mechanically and resuspended in fresh Neurobasal medium supplemented with 2 mm l-glutamine, 2% B-27 supplement, penicillin (100 000 U/l) and streptomycin (100 mg/l). The cells were seeded on poly-l-lysine (0.1 g/l)-coated dishes at a density of 0.33 × 106 cells/cm2 for western blotting. For immunofluorescence studies, neurons were mounted on poly-l-lysine-coated glass coverslips at a density of 0.1 × 106 cells/cm2. The cultures were maintained in serum-free Neurobasal medium supplemented with B-27 at 37°C in a humidified atmosphere of 5% CO2, 95% air for 6 days before treatment in order to allow neuronal differentiation. After 6 days in vitro, cultured neurons were treated with 50 µm MPP+, 24 h before fixation or harvesting. Where indicated, 20 mm ammonium chloride and 20 μm leupeptin, and 10 mm 3-MA or 10 nm rapamycin were added in the culture medium in the last 4 h of MPP+ treatment. The composition of the cultures was determined by immunolabeling in addition to physiological characterization. The majority of cells (90–95%) were positive for neuronal markers (Map2, TuJ1 or NeuN), whereas <10% of cells showed immunolabeling for the astrocytic marker glial fibrillary acidic protein.
Subcellular fractioning proteoextracts
Mitochondria and cytosol-enriched fractions were obtained by using the ProteoExtract Subcellular Proteome Extraction Kit from CalbiochemTM, Merck KGaA. Cells were grown in T75 flasks and treated under various conditions as described earlier for cell lines. Then, cells were harvested and subcellular fractions were prepared according to the manufacturer's specifications. To prepare samples for western blotting, protein concentrations were determined using the PierceTM BCA Protein Assay Kit (Thermo Scientific, Rockford, IL, USA) according to the manufacturer's instructions for plate reader.
Electron microscopy analysis
Cybrids and Rho0 cell suspensions were fixed in 2.5% gluteraldehyde/2% paraformaldehyde in 100 mm sodium cacodylate, pH 7.43 (SC), and post-fixed in 1% osmium tetroxide in SC followed by 1% uranyl acetate. After ethanol dehydration and embedding in LX112 resin (LADD Research Industries), ultrathin sections were stained with uranyl acetate followed by lead citrate. Grids were viewed on a JEOL JEM 1400 transmission electron microscope operated at 80 kV. For the quantification of organelles in cells, 15 different electronmicrographs for each cell line were analyzed. Autophagic vacuoles were identified using previously established criteria (71).
Immunocytochemistry
Cells were grown on glass coverslips (16 mm diameter) in 12-well plates. Following treatment, cells were washed twice with serum-free medium and fixed with 4% paraformaldehyde for 20 min at room temperature. The fixed cells were washed again with PBS, permeabilized with 0.2% Triton X-100 and incubated with 3% BSA, to prevent nonspecific binding, for 30 min. Then, cells were incubated with primary antibody overnight followed by washing with PBS and secondary antibody incubation for 1 h. After washing twice in PBS, nuclei were counterstained with Hoechst 33342. After a final wash, the coverslips were immobilized on a glass slide with mounting medium (DakoCytomation, Dako, Glostrup, Denmark). Negative CTs omitting each primary antibody were performed in each case, and no staining was seen (data not shown). Images were acquired on a Zeiss LSM 510 meta-confocal microscope (63 × 1.4NA plan-apochromat oil immersion lens) by using the Zeiss LSM510 v3.2 software (Carl Zeiss, Inc., Thornwood, NY, USA) and analyzed using Zeiss LSM Image Examiner. LC3 puncta number and size were quantified using the ‘analyze particles’ function of the ImageJ v1.39k (NIH, Bethesda, MD, USA) program after thresholding of images with size settings from 0.2–10 pixel2 and a circularity of 0–1. At least 20 cells were examined for each condition.
To quantify two parameters of mitochondrial morphology, a custom-written ImageJ macro containing plug-ins (72) was used. The cells stained with TOM20 antibody were extracted to grayscale, inverted to show mitochondria-specific fluorescence as black pixels and thresholded to optimally resolve individual mitochondria. This macro traces mitochondrial outlines, using the ‘analyze particles’ function. The area/perimeter ratio was employed as an index of mitochondrial interconnectivity, and inverse roundness was used as a measure of mitochondrial elongation.
Colocalization of two antibodies (LC3/LAMP1) was quantified in thresholded images with the JACoP plug-in of the ImageJ software, according to Bolte and Cordelieres (73).
Live-cell imaging
Cybrid cells were grown on ibidiTreat µ-Slide eight-well plates. Following treatments, cells were washed twice with HBSS, and mitochondria or late autophagosomes and earlier vacuoles in the autophagy pathway were labeled with 100 nm MitoTracker Green (Invitrogen, Carlsbad, CA, USA) for 30 min at 37°C in the dark, according to Du et al. (74) or using the Cyto-ID™ Green Detection Reagent provided in the Cyto-ID™ Autophagy Detection Kit (Enzo, Lausen, Switzerland), respectively, according to the manufacturer's instructions. After a gentle wash, cells were kept in HBSS and were imaged for mitochondrial or autophagic vacuole movements. Time-lapse images were captured under a Zeiss LSM 510 meta-confocal microscope with a stage-based chamber (5% CO2, 37°C). The inverted microscope was driven by the LSM software and images were taken every 2 s for a total of 4 min under 63× magnification (Zeiss Plan-ApoChromat 63×, 1.4NA).
For transport analysis, mitochondria or autophagosomes were considered not mobile if they remained stationary for the entire recording period. Movement was counted only if the displacement was more than the length of the mitochondrion (∼2 μm) or of the autophagosome (∼500 nm).
For each time-lapse movie, mitochondria or autophagic vacuoles were manually tracked and transport parameters were generated using the ImageJ software plug-in Multiple Kymograph, submitted by J. Rietdorf and A. Seitz (European Molecular Biology Laboratory, Heidelberg, Germany). Movement velocity data were determined from the kymographic images and were calculated based on the slope (v = dx/dt) obtained for each profile along the recording time. Each series of images was recorded for at least three randomly selected MitoTracker Green or Cyto-ID™ Green-labeled cells per culture and three independent cultures per condition.
SDS–PAGE and immunoblotting
Individual cell lines were washed with PBS, scraped and lysed on ice in RIPA buffer [150 mm NaCl, 1% NP40, 0.1% SDS, 0.5% DOC, 50 mm Tris–HCl, pH 7.4, supplemented with 0.1 mm PMSF, 2 mm dithiothreitol (DTT) and a 1:1000 dilution of a protease inhibitor cocktail]. Cell suspensions were incubated on ice for 15 min and centrifuged at 20 000g for 15 min. Cleared lysates were assayed for protein concentration using the PierceTM BCA Protein Assay Kit. For each sample, equal amounts of protein (50 µg) were separated under reducing conditions on 12 or 15% SDS–PAGE gels.
For the analysis of α-synuclein oligomerization, a detergent solubility fractionation was performed by using 1% Triton X-100 containing hypotonic lysis buffer (25 mm HEPES, pH 7.5, 2 mm MgCl2, 1 mm EDTA and 1 mm EGTA, supplemented with 2 mm DTT, 0.1 mm PMSF and a 1:1000 dilution of a protease inhibitor cocktail) and incubating cell suspensions for 15 min on ice followed by ultracentrifugation (163 400g, 15 min, 4°C). The supernatant was designated Triton X-100-soluble fraction, and the pellet was redissolved in 1% SDS-containing lysis buffer and sonicated for 10 s (Triton X-100-insoluble fraction). Seventy-five micrograms of each cell lysate was loaded onto 10% SDS–PAGE gels under non-reducing and non-denaturating conditions.
After transfer to ImmobilonTM-P PVDF (polyvinylidene difluoride) membranes (Millipore), the membranes were incubated for 1 h in Tris-buffered saline (TBS) solution containing 0.1% Tween 20 and 5% nonfat milk, followed by an overnight incubation with the respective primary antibody at 4°C with gentle agitation. Membranes were further washed three times with TBS and 0.1% Tween and then incubated with the corresponding secondary antibody for 2 h at room temperature. The membranes were washed again three times and bound antibodies detected using the enhanced chemifluorescence reagent ECF (Amersham Biosciences UK Limited, Buckinghamshire, UK) according to the manufacturer's instructions. Blots were visualized using a VersaDoc imaging system (Bio-Rad, Hercules, CA, USA) and quantified using the Quantity-One software (Bio-Rad).
Immunoprecipitation assay
Cells were scraped and lysed on ice in a non-denaturing lysis buffer [20 mm Tris–HCl (pH 7.0), 100 mm NaCl, 2 mm EDTA, 2 mm EGTA, supplemented with 0.1% SDS, 1% Triton X-100, 2 mm DTT, 0.1 mm PMSF and a 1:1000 dilution of a protease inhibitor cocktail]. Cellular suspensions were centrifuged at 20 000g, 10 min at 4°C and whole lysates were assayed for protein concentration as described earlier. Five hundred micrograms of each sample were precleared with Protein A Sepharose beads (GE Healthcare Bio-Sciences, Uppsala, Sweden) for 1 h, 4°C, and then incubated with primary antibody, overnight at 4°C and with nutation. Protein A-Sepharose beads were then added to samples followed by 2 h incubation. The beads were spun down and washed seven times in washing buffer [1% Triton X-100, 500 mm NaCl, 2 mm EDTA, 2 mm EGTA, 20 mm Tris–HCl (pH 7.0)]. The last supernatant was collected and 25 μl of 2× sample buffer was added. The samples were boiled at 95–100°C for 5 min to denature the protein and to separate it from the protein-A beads. The boiled proteins were centrifuged at 20 000g for 5 min at room temperature and the supernatants collected. Samples were separated by SDS–PAGE and subjected to western blotting as aforementioned. A shift <5 kDa is observed in the molecular weight of Bcl-2 in the IP, which might be due to different salt concentrations between the lysis buffer and the IP buffer.
Evaluation of mitochondrial respiratory chain NADH–ubiquinone oxidoreductase activity
NT2 Rho+ cells growing in 75 cm2 flasks were replated into 10 cm Petri-dishes at the concentration of 3.0 × 105 cells/ml. Cell treatment conditions were as mentioned earlier.
The activity of mitochondrial NADH–ubiquinone oxidoreductase (complex I: EC 1.6.99.3) was determined by using a modified version of the method of Ragan et al. (75). Briefly, after treatments, cells were harvested on ice with a low stringency lysis buffer [20 mm HEPES (pH 7.4), 250 mm sucrose, 1 mm EDTA, 1 mm EGTA]. Then, protein lysates of homogenated cells were prepared using a Teflon-fitted glass hand homogenizer and assayed for protein concentration using the Bio-Rad protein dye assay reagent (Bio-Rad). Complex I activity was measured by following the decrease in NADH absorbance at 340 nm (ɛ = 6.81/mm/cm) that occurs when ubiquinone is reduced to form ubiquinol. The reaction was initiated by adding 50 µm ubiquinone to the reaction mixture, containing 30 µg of protein lysate suspended in potassium phosphate buffer [25 mm K2HPO4, 25 mm KH2PO4 (pH 7.2)], in the presence of 2.5 mg/ml BSA, 4 mm MgCl2 and 1 mm KCN, at 30°C. After 5 min, 10 µm rotenone, a complex I inhibitor, was added and the reaction was monitored for a further 5 min period. Complex I specific activity was expressed in nanomoles per minute per milligram of protein and represents the rotenone sensitive rates.
Caspase-3 activation assay
Caspase-3 activation assays were performed by using the method described by Cregan et al. (76) with minor modifications. After treatments, cells were washed once in ice-cold PBS and harvested on ice with a lysis buffer containing 25 mm HEPES (pH 7.5), 1 mm EDTA, 1 mm EGTA, 2 mm MgCl2, supplemented with 2 mm DTT, 0.1 mm PMSF and a 1:1000 dilution of a protease inhibitor cocktail. The cellular suspension was frozen/thawed three times on liquid nitrogen and centrifuged at 20 000g, 10 min at 4°C. The resulting supernatant was collected and assayed for protein concentration using Bio-Rad protein dye assay reagent (Bio-Rad). To evaluate caspase-3 activation, cell extracts containing 50 μg of protein were incubated in a reaction buffer [25 mm HEPES (pH 7.5), 0.1% (w/v) 3[(3-cholamidopropyl) dimethylammonio]-propanesulfonic acid, 10% (w/v) sucrose, 2 mm DTT] with 100 μm Ac-DEVD-pNA, the colorimetric substrate for caspase-3, for 2 h at 37°C. The enzymatic cleavage of the chromophore pNA from the substrate was detected at 405 nm using a Spectramax Plus 384 spectrophotometer (Molecular Devices, Sunnyvale, CA, USA).
Data analysis
All data were expressed as mean ± SEM of at least three independent experiments. Statistical analyses were performed using GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA). Differences between two data sets were evaluated by two-tailed unpaired Student's t-test. Statistical tests between multiple data sets and conditions were carried out using a one-way or two-way analysis of variance (ANOVA) followed by Bonferroni's post hoc test or the Newman–Keuls multiple comparison test to determine statistical significance, as appropriate. A P-value <0.05 was considered statistically significant.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
ACKNOWLEDGEMENTS
We are grateful to Dr Ana Maria Cuervo for critically reviewing the manuscript and to Drs Rosa Resende, Maria João Santos and João Pratas for technical support.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by the Portuguese Foundation for Science and Technology (FCT-MEC, Portugal), grant PTDC/SAU-NEU/102710/2008 to S.M.C. D.M.A. and A.R.E. are supported by fellowships from FCT-MEC (SFRHD/BD/38743/2007 and SFRH/BPD/75044/2010, respectively). B.P. is supported by the Cellular and Tissue Aging Core funded by the grant NIH/NIA AG038072 to Ana Maria Cuervo. R.H.S. is funded by the grant NIH P30AG035982 to the KU ADC. Funding to pay the Open Access publication charges for this article was provided by PTDC/SAU-NEU/102710/2008 grant.
REFERENCES
- 1.Forno L.S. Neuropathology of Parkinson's disease. J. Neuropathol. Exp. Neurol. 1996;55:259–272. doi: 10.1097/00005072-199603000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Schapira A.H., Cooper J.M., Dexter D., Jenner P., Clark J.B., Marsden C.D. Mitochondrial complex I deficiency in Parkinson's disease. Lancet. 1989;1:1269. doi: 10.1016/s0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- 3.Parker W.D., Jr, Boyson S.J., Parks J.K. Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Ann. Neurol. 1989;26:719–723. doi: 10.1002/ana.410260606. [DOI] [PubMed] [Google Scholar]
- 4.Swerdlow R.H., Parks J.K., Miller S.W., Tuttle J.B., Trimmer P.A., Sheehan J.P., Bennett J.P., Jr, Davis R.E., Parker W.D., Jr Origin and functional consequences of the complex I defect in Parkinson's disease. Ann. Neurol. 1996;40:663–671. doi: 10.1002/ana.410400417. [DOI] [PubMed] [Google Scholar]
- 5.Swerdlow R.H. Does mitochondrial DNA play a role in Parkinson's disease? A review of cybrid and other supportive evidence. Antioxid. Redox Signal. 2012;16:950–964. doi: 10.1089/ars.2011.3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cardoso S.M. The mitochondrial cascade hypothesis for Parkinson's disease. Curr. Pharm. Des. 2011;17:3390–3397. doi: 10.2174/138161211798072508. [DOI] [PubMed] [Google Scholar]
- 7.Esteves A.R., Arduino D.M., Swerdlow R.H., Oliveira C.R., Cardoso S.M. Oxidative stress involvement in alpha-synuclein oligomerization in Parkinsons disease cybrids. Antioxid. Redox Signal. 2009;11:439–448. doi: 10.1089/ars.2008.2247. [DOI] [PubMed] [Google Scholar]
- 8.Trimmer P.A., Keeney P.M., Borland M.K., Simon F.A., Almeida J., Swerdlow R.H., Parks J.P., Parker W.D., Jr, Bennett J.P., Jr Mitochondrial abnormalities in cybrid cell models of sporadic Alzheimer's disease worsen with passage in culture. Neurobiol. Dis. 2004;15:29–39. doi: 10.1016/j.nbd.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 9.Lee J.Y., Koga H., Kawaguchi Y., Tang W., Wong E., Gao Y.S., Pandey U.B., Kaushik S., Tresse E., Lu J., et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010;29:969–980. doi: 10.1038/emboj.2009.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee J.Y., Nagano Y., Taylor J.P., Lim K.L., Yao T.P. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J. Cell Biol. 2010;189:671–679. doi: 10.1083/jcb.201001039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arduino D.M., Esteves A.R., Silva D.F., Martins-Branco D., Santos D., Pimentel D.F., Cardoso S.M. Therapeutic intervention at cellular quality control systems in Alzheimer's and Parkinson's diseases. Curr. Pharm. Des. 2011;17:3446–3459. doi: 10.2174/138161211798072481. [DOI] [PubMed] [Google Scholar]
- 12.Wong E., Cuervo A.M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 2010;13:805–811. doi: 10.1038/nn.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dagda R.K., Zhu J., Kulich S.M., Chu C.T. Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress: implications for Parkinson's disease. Autophagy. 2008;4:770–782. doi: 10.4161/auto.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y., McMillan-Ward E., Kong J., Israels S.J., Gibson S.B. Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J. Cell Sci. 2007;120:4155–4166. doi: 10.1242/jcs.011163. [DOI] [PubMed] [Google Scholar]
- 15.Anglade P., Vyas S., Javoy-Agid F., Herrero M.T., Michel P.P., Marquez J., Mouatt-Prigent A., Ruberg M., Hirsch E.C., Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol. Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- 16.Esteves A.R., Lu J., Rodova M., Onyango I., Lezi E., Dubinsky R., Lyons K.E., Pahwa R., Burns J.M., Cardoso S.M., et al. Mitochondrial respiration and respiration-associated proteins in cell lines created through Parkinson's subject mitochondrial transfer. J. Neurochem. 2010;113:674–682. doi: 10.1111/j.1471-4159.2010.06631.x. [DOI] [PubMed] [Google Scholar]
- 17.Esteves A.R., Domingues A.F., Ferreira I.L., Januario C., Swerdlow R.H., Oliveira C.R., Cardoso S.M. Mitochondrial function in Parkinson's disease cybrids containing an nt2 neuron-like nuclear background. Mitochondrion. 2008;8:219–228. doi: 10.1016/j.mito.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 18.Koopman W.J., Verkaart S., Visch H.J., van der Westhuizen F.H., Murphy M.P., van den Heuvel L.W., Smeitink J.A., Willems P.H. Inhibition of complex I of the electron transport chain causes O2−.-mediated mitochondrial outgrowth. Am. J. Physiol. Cell Physiol. 2005;288:C1440–1450. doi: 10.1152/ajpcell.00607.2004. [DOI] [PubMed] [Google Scholar]
- 19.Koopman W.J., Visch H.J., Verkaart S., van den Heuvel L.W., Smeitink J.A., Willems P.H. Mitochondrial network complexity and pathological decrease in complex I activity are tightly correlated in isolated human complex I deficiency. Am. J. Physiol. Cell Physiol. 2005;289:C881–C890. doi: 10.1152/ajpcell.00104.2005. [DOI] [PubMed] [Google Scholar]
- 20.Ghadially F.N. As you like it, part 3: a critique and historical review of calcification as seen with the electron microscope. Ultrastruct. Pathol. 2001;25:243–267. doi: 10.1080/019131201300343874. [DOI] [PubMed] [Google Scholar]
- 21.Lloreta-Trull J., Serrano S. Biology and pathology of the mitochondrion. Ultrastruct. Pathol. 1998;22:357–367. doi: 10.3109/01913129809103357. [DOI] [PubMed] [Google Scholar]
- 22.Rubinsztein D.C., Cuervo A.M., Ravikumar B., Sarkar S., Korolchuk V., Kaushik S., Klionsky D.J. In search of an ‘autophagomometer’. Autophagy. 2009;5:585–589. doi: 10.4161/auto.5.5.8823. [DOI] [PubMed] [Google Scholar]
- 23.Larm J.A., Vaillant F., Linnane A.W., Lawen A. Up-regulation of the plasma membrane oxidoreductase as a prerequisite for the viability of human Namalwa rho0 cells. J. Biol. Chem. 1994;269:30097–30100. [PubMed] [Google Scholar]
- 24.Morre D.M., Lenaz G., Morre D.J. Surface oxidase and oxidative stress propagation in aging. J. Exp. Biol. 2000;203:1513–1521. doi: 10.1242/jeb.203.10.1513. [DOI] [PubMed] [Google Scholar]
- 25.Beal M.F. Experimental models of Parkinson's disease. Nat. Rev. Neurosci. 2001;2:325–334. doi: 10.1038/35072550. [DOI] [PubMed] [Google Scholar]
- 26.Martinez T.N., Greenamyre J.T. Toxin models of mitochondrial dysfunction in Parkinson's disease. Antioxid. Redox Signal. 2012;16:920–934. doi: 10.1089/ars.2011.4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nicotra A., Parvez S. Apoptotic molecules and MPTP-induced cell death. Neurotoxicol. Teratol. 2002;24:599–605. doi: 10.1016/s0892-0362(02)00213-1. [DOI] [PubMed] [Google Scholar]
- 28.Shibata M., Lu T., Furuya T., Degterev A., Mizushima N., Yoshimori T., MacDonald M., Yankner B., Yuan J. Regulation of intracellular accumulation of mutant Huntingtin by Beclin 1. J. Biol. Chem. 2006;281:14474–14485. doi: 10.1074/jbc.M600364200. [DOI] [PubMed] [Google Scholar]
- 29.Funderburk S.F., Wang Q.J., Yue Z. The Beclin 1-VPS34 complex—at the crossroads of autophagy and beyond. Trends Cell Biol. 2010;20:355–362. doi: 10.1016/j.tcb.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Veech G.A., Dennis J., Keeney P.M., Fall C.P., Swerdlow R.H., Parker W.D., Jr, Bennett J.P., Jr Disrupted mitochondrial electron transport function increases expression of anti-apoptotic bcl-2 and bcl-X(L) proteins in SH-SY5Y neuroblastoma and in Parkinson disease cybrid cells through oxidative stress. J. Neurosci. Res. 2000;61:693–700. doi: 10.1002/1097-4547(20000915)61:6<693::AID-JNR13>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 31.Pattingre S., Tassa A., Qu X., Garuti R., Liang X.H., Mizushima N., Packer M., Schneider M.D., Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 32.Maiuri M.C., Le Toumelin G., Criollo A., Rain J.C., Gautier F., Juin P., Tasdemir E., Pierron G., Troulinaki K., Tavernarakis N., et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Esteves A.R., Arduino D.M., Swerdlow R.H., Oliveira C.R., Cardoso S.M. Microtubule depolymerization potentiates alpha-synuclein oligomerization. Front. Aging Neurosci. 2010;1:5. doi: 10.3389/neuro.24.005.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bains M., Heidenreich K.A. Live-cell imaging of autophagy induction and autophagosome-lysosome fusion in primary cultured neurons. Methods Enzymol. 2009;453:145–158. doi: 10.1016/S0076-6879(08)04007-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee S., Sato Y., Nixon R.A. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer's-like axonal dystrophy. J. Neurosci. 2011;31:7817–7830. doi: 10.1523/JNEUROSCI.6412-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arduino D.M., Esteves A.R., Oliveira C.R., Cardoso S.M. Mitochondrial metabolism modulation: a new therapeutic approach for Parkinson's disease. CNS Neurol. Disord. Drug Targets. 2010;9:105–119. doi: 10.2174/187152710790966687. [DOI] [PubMed] [Google Scholar]
- 37.Strous G.J., Du Maine A., Zijderhand-Bleekemolen J.E., Slot J.W., Schwartz A.L. Effect of lysosomotropic amines on the secretory pathway and on the recycling of the asialoglycoprotein receptor in human hepatoma cells. J. Cell Biol. 1985;101:531–539. doi: 10.1083/jcb.101.2.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Macaskill A.F., Rinholm J.E., Twelvetrees A.E., Arancibia-Carcamo I.L., Muir J., Fransson A., Aspenstrom P., Attwell D., Kittler J.T. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron. 2009;61:541–555. doi: 10.1016/j.neuron.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cuervo A.M., Stefanis L., Fredenburg R., Lansbury P.T., Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 40.Martinez-Vicente M., Talloczy Z., Kaushik S., Massey A.C., Mazzulli J., Mosharov E.V., Hodara R., Fredenburg R., Wu D.C., Follenzi A., et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J. Clin. Invest. 2008;118:777–788. doi: 10.1172/JCI32806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vogiatzi T., Xilouri M., Vekrellis K., Stefanis L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008;283:23542–23556. doi: 10.1074/jbc.M801992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xilouri M., Vogiatzi T., Vekrellis K., Park D., Stefanis L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS One. 2009;4:e5515. doi: 10.1371/journal.pone.0005515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bjorkoy G., Lamark T., Brech A., Outzen H., Perander M., Overvatn A., Stenmark H., Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bartels T., Choi J.G., Selkoe D.J. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arduino D.M., Esteves A.R., Domingues A.F., Pereira C.M., Cardoso S.M., Oliveira C.R. ER-mediated stress induces mitochondrial-dependent caspases activation in NT2 neuron-like cells. BMB Rep. 2009;42:719–724. doi: 10.5483/bmbrep.2009.42.11.719. [DOI] [PubMed] [Google Scholar]
- 46.Arduino D.M., Esteves A.R., Cardoso S.M., Oliveira C.R. Endoplasmic reticulum and mitochondria interplay mediates apoptotic cell death: relevance to Parkinson's disease. Neurochem. Int. 2009;55:341–348. doi: 10.1016/j.neuint.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 47.Plomp P.J., Gordon P.B., Meijer A.J., Hoyvik H., Seglen P.O. Energy dependence of different steps in the autophagic-lysosomal pathway. J. Biol. Chem. 1989;264:6699–6704. [PubMed] [Google Scholar]
- 48.Plomp P.J., Wolvetang E.J., Groen A.K., Meijer A.J., Gordon P.B., Seglen P.O. Energy dependence of autophagic protein degradation in isolated rat hepatocytes. Eur. J. Biochem. 1987;164:197–203. doi: 10.1111/j.1432-1033.1987.tb11011.x. [DOI] [PubMed] [Google Scholar]
- 49.Schellens J.P., Vreeling-Sindelarova H., Plomp P.J., Meijer A.J. Hepatic autophagy and intracellular ATP. A morphometric study. Exp. Cell Res. 1988;177:103–108. doi: 10.1016/0014-4827(88)90028-6. [DOI] [PubMed] [Google Scholar]
- 50.Schellens J.P., Meijer A.J. Energy depletion and autophagy. Cytochemical and biochemical studies in isolated rat hepatocytes. Histochem. J. 1991;23:460–466. doi: 10.1007/BF01041376. [DOI] [PubMed] [Google Scholar]
- 51.Van Laar V.S., Arnold B., Cassady S.J., Chu C.T., Burton E.A., Berman S.B. Bioenergetics of neurons inhibit the translocation response of Parkin following rapid mitochondrial depolarization. Hum. Mol. Genet. 2011;20:927–940. doi: 10.1093/hmg/ddq531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kimura S., Noda T., Yoshimori T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Funct. 2008;33:109–122. doi: 10.1247/csf.08005. [DOI] [PubMed] [Google Scholar]
- 53.Fass E., Shvets E., Degani I., Hirschberg K., Elazar Z. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J. Biol. Chem. 2006;281:36303–36316. doi: 10.1074/jbc.M607031200. [DOI] [PubMed] [Google Scholar]
- 54.Jahreiss L., Menzies F.M., Rubinsztein D.C. The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic. 2008;9:574–587. doi: 10.1111/j.1600-0854.2008.00701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin S.X., Ferro K.L., Collins C.A. Cytoplasmic dynein undergoes intracellular redistribution concomitant with phosphorylation of the heavy chain in response to serum starvation and okadaic acid. J. Cell Biol. 1994;127:1009–1019. doi: 10.1083/jcb.127.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin S.X., Collins C.A. Regulation of the intracellular distribution of cytoplasmic dynein by serum factors and calcium. J. Cell Sci. 1993;105:579–588. doi: 10.1242/jcs.105.2.579. [DOI] [PubMed] [Google Scholar]
- 57.Cash A.D., Aliev G., Siedlak S.L., Nunomura A., Fujioka H., Zhu X., Raina A.K., Vinters H.V., Tabaton M., Johnson A.B., et al. Microtubule reduction in Alzheimer's disease and aging is independent of tau filament formation. Am. J. Pathol. 2003;162:1623–1627. doi: 10.1016/s0002-9440(10)64296-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trimmer P.A., Schwartz K.M., Borland M.K., De Taboada L., Streeter J., Oron U. Reduced axonal transport in Parkinson's disease cybrid neurites is restored by light therapy. Mol. Neurodegener. 2009;4:26. doi: 10.1186/1750-1326-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chu C.T. Introduction. Autophagy dysregulation in neuropathology. Brain Pathol. 2012;22:80–81. doi: 10.1111/j.1750-3639.2011.00547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pandey U.B., Nie Z., Batlevi Y., McCray B.A., Ritson G.P., Nedelsky N.B., Schwartz S.L., DiProspero N.A., Knight M.A., Schuldiner O., et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 61.Jager S., Bucci C., Tanida I., Ueno T., Kominami E., Saftig P., Eskelinen E.L. Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 2004;117:4837–4848. doi: 10.1242/jcs.01370. [DOI] [PubMed] [Google Scholar]
- 62.Filimonenko M., Stuffers S., Raiborg C., Yamamoto A., Malerod L., Fisher E.M., Isaacs A., Brech A., Stenmark H., Simonsen A. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J. Cell Biol. 2007;179:485–500. doi: 10.1083/jcb.200702115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pleasure S.J., Lee V.M. NTera 2 cells: a human cell line which displays characteristics expected of a human committed neuronal progenitor cell. J. Neurosci. Res. 1993;35:585–602. doi: 10.1002/jnr.490350603. [DOI] [PubMed] [Google Scholar]
- 64.Cardoso S.M., Santos S., Swerdlow R.H., Oliveira C.R. Functional mitochondria are required for amyloid beta-mediated neurotoxicity. FASEB J. 2001;15:1439–1441. doi: 10.1096/fj.00-0561fje. [DOI] [PubMed] [Google Scholar]
- 65.Sodja C., Fang H., Dasgupta T., Ribecco M., Walker P.R., Sikorska M. Identification of functional dopamine receptors in human teratocarcinoma NT2 cells. Brain Res. Mol. Brain Res. 2002;99:83–91. doi: 10.1016/s0169-328x(01)00324-2. [DOI] [PubMed] [Google Scholar]
- 66.Swerdlow R.H., Parks J.K., Cassarino D.S., Maguire D.J., Maguire R.S., Bennett J.P., Jr, Davis R.E., Parker W.D., Jr Cybrids in Alzheimer's disease: a cellular model of the disease? Neurology. 1997;49:918–925. doi: 10.1212/wnl.49.4.918. [DOI] [PubMed] [Google Scholar]
- 67.Esteves A.R., Arduino D.M., Swerdlow R.H., Oliveira C.R., Cardoso S.M. Dysfunctional mitochondria uphold calpain activation: contribution to Parkinson's disease pathology. Neurobiol. Dis. 2010;37:723–730. doi: 10.1016/j.nbd.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 68.Litvan I., Bhatia K.P., Burn D.J., Goetz C.G., Lang A.E., McKeith I., Quinn N., Sethi K.D., Shults C., Wenning G.K. Movement Disorders Society Scientific Issues Committee report: SIC Task Force appraisal of clinical diagnostic criteria for Parkinsonian disorders. Mov. Disord. 2003;18:467–486. doi: 10.1002/mds.10459. [DOI] [PubMed] [Google Scholar]
- 69.Paquet-Durand F., Tan S., Bicker G. Turning teratocarcinoma cells into neurons: rapid differentiation of NT-2 cells in floating spheres. Brain Res. Dev. Brain Res. 2003;142:161–167. doi: 10.1016/s0165-3806(03)00065-8. [DOI] [PubMed] [Google Scholar]
- 70.Agostinho P., Oliveira C.R. Involvement of calcineurin in the neurotoxic effects induced by amyloid-beta and prion peptides. Eur. J. Neurosci. 2003;17:1189–1196. doi: 10.1046/j.1460-9568.2003.02546.x. [DOI] [PubMed] [Google Scholar]
- 71.Nixon R.A., Wegiel J., Kumar A., Yu W.H., Peterhoff C., Cataldo A., Cuervo A.M. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005;64:113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 72.Dagda R.K., Cherra S.J., III, Kulich S.M., Tandon A., Park D., Chu C.T. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J. Biol. Chem. 2009;284:13843–13855. doi: 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bolte S., Cordelieres F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 74.Du H., Guo L., Yan S., Sosunov A.A., McKhann G.M., Yan S.S. Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc. Natl Acad. Sci. USA. 2010;107:18670–18675. doi: 10.1073/pnas.1006586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ragan C.I., Wilson M.T., Darley-Usmar V.M., Lowe P.N. Subfractionation of Mitochondria, and isolation of the proteins of oxidative phosphorylation. Mitochondria, a practical approach. London: IRL Press; 1987. [Google Scholar]
- 76.Cregan S.P., MacLaurin J.G., Craig C.G., Robertson G.S., Nicholson D.W., Park D.S., Slack R.S. Bax-dependent caspase-3 activation is a key determinant in p53-induced apoptosis in neurons. J. Neurosci. 1999;19:7860–7869. doi: 10.1523/JNEUROSCI.19-18-07860.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.