

Abstract
Several voltage-gated channels share a proline-valine-proline (proline hinge) sequence motif at the intracellular side of S6. We studied the proline hinge in Kv1.4 channels, which inactivate via two mechanisms: N- and C-type. We mutated the second proline to glycine or alanine: P558A, P558G. These mutations were studied in the presence/absence of the N-terminal to separate the effects of the interaction between the proline hinge and N- and C-type inactivation. Both S6 mutations slowed or removed N- and C-type inactivation, and altered recovery from inactivation. P558G slowed activation and N- and C-type inactivation by nearly an order of magnitude. Sensitivity to extracellular acidosis and intracellular quinidine binding remained, suggesting that transmembrane communication in N- and C-type inactivation was preserved, consistent with our previous findings of major structural rearrangements involving S6 during C-type inactivation. P558A was very disruptive: activation was slowed by more than an order of magnitude, and no inactivation was observed. These results are consistent with our hypothesis that the proline hinge and intracellular S6 movement play a significant role in inactivation and recovery. Computer modeling suggests that both P558G and P558A mutations modify early voltage-dependent steps and make a final voltage-insensitive step that is rate limiting at positive potentials.
Introduction
Voltage-gated potassium (K) channels comprise a large and diverse class of ion channels that play a variety of physiological roles in an assortment of tissues. In this study we examined the gating of Kv1.4 (encoded by KCNA4), which is a member of the Shaker-related family of voltage-gated K+ channels. Depolarization produces a rapidly inactivating current through Kv1.4 channels, which is thought to be the molecular basis of the α-subunit of the channel underlying the transient outward current seen in the endocardium of several mammalian species (1), as well as to underlie an A type current in brain (2) and smooth muscle (3). Kv channels are thought to have many common structural properties, with a putative structure of six-transmembrane spanning segments, including the charged voltage-sensing S4 segment. Four subunits combine to form the minimal pore-forming unit. The S6 regions of the Kv families are similar, and Kv1, Kv2, Kv3, and Kv4 have a proline-valine-proline (PVP) motif, or proline hinge, in the inner portion of the S6 segment, which is well established to play a role in the activation gating of this general class of channels (4–6). This region is also thought to form the receptor for binding of the small lipophilic N-terminal domain of some α-subunits (such as Kv1.4 and Shaker) and ancillary subunits involved in rapid N-type inactivation. More recently, this region was shown to be critical for closure of the pore region through slow or C-type inactivation, and S6 was shown to be critical for determining the coupling between N- and C-type inactivation (7–9).
Kv1 channels open rapidly in response to a depolarizing stimulus, but in the presence of a continued stimulus, they move into an inactivated state. Kv1.4 channels have two distinct forms of inactivation: N-type and C-type. N-type inactivation is well described and is the result of the lipophilic ball of the N-terminal binding to the open channel, which results in occlusion of the pore (10–15). C-type inactivation is less well defined and involves rearrangement of the extracellular face of the channel (16), a substantial closure of the inner pore (9,17), and changes in the selectivity filter (18,19). C-type inactivation is sensitive to extracellular permeant ions (20–22), extracellular acidosis (8,23) and [TEA] (20,24–26), intracellular quinidine binding (7,27), and intracellular osmotic pressure changes (17), as well as to mutations on the extracellular face of the channel near the mouth of the pore and on the intracellular side of the pore (8,28,29).
The three gating processes of activation and N- and C-type inactivation are hypothesized to be coupled. Activation is thought to be primary, with both N- and C-type inactivation proceeding once a sufficient degree of activation is achieved. All three processes involve conformational changes at the intracellular mouth of the pore. However, multiple hypotheses concerning the nature of C-type inactivation and its interaction with activation and N-type inactivation have been proposed. C-type inactivation has been viewed as a relatively limited change in extracellular domains, a change linked to deep pore residues, or a large-scale change of the entire pore (9,22,30). These mechanisms are not necessarily mutually exclusive, and there is considerable debate as to which mechanisms dominate under physiological conditions.
We have proposed a scheme for C-type inactivation in which C-type inactivation is coupled to activation and involves large-scale movement of S6 from an activated state, and closure (or partial closure) of the intracellular mouth of the pore, which is similar in magnitude to the movement that accompanies opening of the channel during the activation transition (31). This leads to testable predictions on inactivation behavior due to mutations at the proline hinge, which are not readily predicted from other hypothesized mechanisms of C-type inactivation. In particular, our paradigm of structural changes in gating predicts that when the hinge prolines are mutated, activation will be affected and 1), N-and C-type inactivation will show parallel shifts in steady-state inactivation that mirror the changes in activation; 2), a reduction in the activation rate will slow or inhibit both N- and C-type inactivation; and 3), there will be a reduction in the rate and degree of steady-state inactivation for N-and C-type inactivation. We show that these predicted differences are indeed observed when mutations are made in the proline hinge region. The effects of the P558A hinge mutation on activation are sensitive to the presence of the N-terminal ball, suggesting that the modified S6 may result in a closed conformation that is distinct from the natural closed conformation.
Materials and Methods
Oocyte preparation
Xenopus laevis (Xenopus Express) were cared for according to standards approved by the Institutional Animal Care and Use Committee of the University at Buffalo. The frogs were anesthetized in tricaine (1 g/l; Sigma, St. Louis, MO). Oocytes were digested in (mM) 82.5 NaCl, 2 KCl, 1 MgCl2, 5 HEPES; pH 7.4, 1 mg ml−1 collagenase, type I (Sigma) and shaken for 1.5–2 h. Defolliculated oocytes (stages V–VI) were injected with <50 ng mRNA (Nanoject; Drummond Scientific). We used mRNA for Kv1.4 originally isolated in ferret heart (32). ΔN had amino acids 2–146 deleted. The following mutant constructs were created (QuikChange; Stratagene) and screened: 556A, 556D, 556G, 556S, 558A, 558D, 558G, and 558S. Only 558G and 558A gave measureable currents.
Voltage clamp
Oocytes were voltage-clamped at room temperature with a two-electrode clamp (CA-1B; Dagan). Microelectrodes (resistances: 0.5–1.5 MΩ) were fabricated from 1.5 mm o.d. borosilicate glass tubing (TW150-4; WPI) using a two-stage puller and filled with 3 M KCl. The control extracellular solution (ND96) contained (mM) 96 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, 10 HEPES (pH 7.4). The 98 mM K+ solution contained (mM) 98 KCl, 1 MgCl2, 1.8 CaCl2, 10 HEPES (pH 7.4).
Data were digitized and analyzed using pCLAMP (Axon). Further analysis was performed using Clampfit (Axon), Excel (Microsoft), and Origin (OriginLab). Data were filtered at 2 kHz. Data are shown as the mean ± SE. Confidence levels were calculated using Student’s paired t-test. The sensitivities of measurements of activation and inactivation to the pulse length duration are shown in Fig. S1, Fig. S2, Fig. S3, Fig. S4, and Table S1 of the Supporting Material. We define N-type inactivation as the binding of the N-terminal domain to the activated channel after a depolarizing pulse. We define C-type inactivation as the channels entering a nonconducting state after activation by a depolarizing pulse by a mechanism other than N-type. When inactivation and activation were well separated kinetically, we measured the rate of development of inactivation as the rate of decay of current following an activating positive step.
Mathematical modeling
We used a mathematical model of Kv1.4 with four closed states (an activated, nonconducting, preopen state; an open state; and both C- and N-type inactivation) to interpret several of our experimental results:
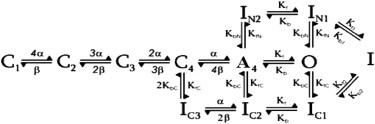
The derivation of this model is shown in Fig. S5 and Fig. S6.
Results
Current-voltage relationships
We studied the effects of P558A and P558G, i.e., with either a glycine or an alanine substituted for the second proline at 558 in Kv1.4 (see Fig. 1). Other mutations at 556 and 558 did not result in measurable currents (see Materials and Methods). Sensitivity to mutation at these positions suggests that the prolines in the putative hinge region play a critical role in channel function. The only mutations that resulted in functional currents were G and A. N-type inactivation of Kv1.4 channels can be eliminated by removal of amino acids 2–146 from the N-terminal, forming a ΔN construct. We therefore characterized the four channel constructs with these two mutations (Kv1.4[P558A], Kv1.4[P558A]ΔN, Kv1.4[P558G], and Kv1.4[P558G]ΔN) to examine the effects on both N- and C-type inactivation.
Figure 1.
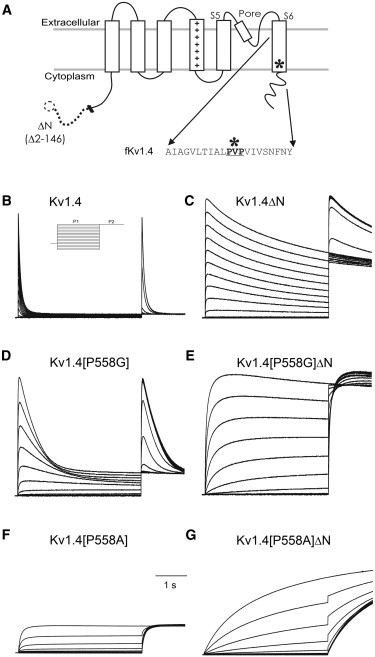
Mutations in Kv1.4. (A) Topological cartoon. Kv1.4 is thought be a tetramer of four subunits, each of which has six transmembrane segments, with a voltage sensor in S4. The position of the proline hinge (∗) and the sequence of the amino acids around it are shown. In the ΔN construct, amino acids 2–146 are removed from N-terminal lipophilic ball responsible for N-type inactivation. Representative traces from a two-pulse protocol on channels expressed in Xenopus oocytes. P1 (4 s) was to potentials from −100 and +50 mV, P2 (1 s) was to +50 mV. Holding potential: −90 mV. (B) Kv1.4 (inset: voltage protocol). (C) Kv1.4ΔN. (D) Kv1.4[P558G]. (E) Kv1.4[P558G]ΔN. (F) Kv1.4[P558A]. (G) Kv1.4[P558A]ΔN.
Proline hinge mutations resulted in severely modified gating properties (Fig. 1). In these traces, an initial 4 s depolarization was applied from the holding potential, −90 mV, to a range of potentials between −100 and +50 mV in 10 mV steps. This was followed by a 1 s pulse at +50 mV before returning to the holding potential of −90 mV. Both Kv1.4[P558G] and Kv1.4[P558A] inactivate relatively slowly. Even though the N-terminal is still present in both the Kv1.4[P558G] and Kv1.4[P558A] channels, the presence of the proline mutation slows (see also Fig. S7) or eliminates rapid N-type inactivation. In addition, activation of Kv1.4[P558A] is dramatically slowed. In the Kv1.4[P558G]ΔN and Kv1.4[P558A]ΔN constructs, which do not have the N-terminal ball, activation is slowed, and C-type inactivation is slowed or eliminated entirely. This qualitative finding shows the predicted link between slowing of activation and slowing/decreasing of both N- and C-type inactivation.
Peak I-V relationships are shown in Fig. 2. Relative to Kv1.4, the threshold for activation of both Kv1.4[P558G] and Kv1.4[P558A] are at more positive potentials. Similarly, relative to Kv1.4ΔN, the threshold for activation of both Kv1.4[P558G]ΔN and Kv1.4[P558A]ΔN are at more positive potentials. Fig. 2, C and D, show peak I-V relationships for Kv1.4[P558A] versus Kv1.4[P558A]ΔN, and Kv1.4[P558G] versus Kv1.4[P558G]ΔN, respectively, plotted on the same graph for easy comparison. Alteration of the proline hinge clearly shifts the threshold of activation more positive for both mutations, and the shift is independent of the presence of the N-terminal.
Figure 2.
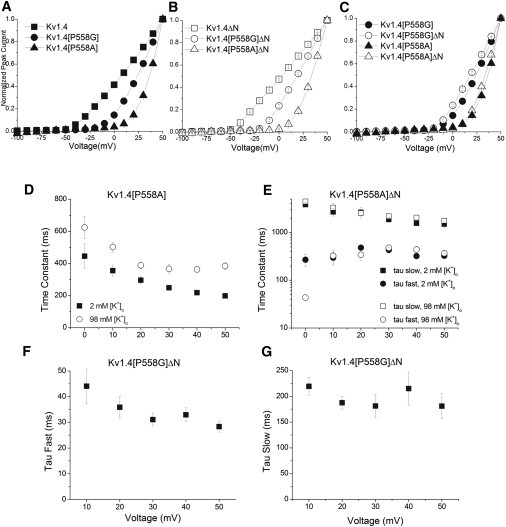
Activation. Peak current elicited by the P1 pulse from a two-pulse protocol (see Fig. 1, inset) plotted versus V to generate I-V relationships for (A) Kv1.4 (■, n = 9), Kv1.4[P558G] (●, n = 23) and Kv1.4[P558A] (▴, n = 6); and (B) Kv1.4ΔN (□, n = 6), Kv1.4[P558G]ΔN (○, n = 16), and Kv1.4[P558A]ΔN (Δ, n = 17) (error bars smaller than symbols). (C) Replotted for easy comparison are Kv1.4[P558G] (●), Kv1.4[P558G]ΔN (○), Kv1.4[P558A] (▴), and Kv1.4[P558A]ΔN (Δ) activation. Activation of Kv1.4[P558A] was well fit by a single exponential and Kv1.4[P558A]ΔN was well fit by a double exponential. (D) Time constants for Kv1.4[P558A] with 2 (■, n = 7) and 98 mM (○, n = 7) [K+]o. (E) Kv1.4[P558A]ΔN fast and slow time constants in 2 mM [K+]o (fast: ●, slow: ■, n = 7) and 98 mM [K+]o (fast: ○, slow: □, n = 7). (F and G) Fast (F) and slow (G) time constants of activation of Kv1.4[P558G]ΔN.
Activation rate
Kv1.4 activates very rapidly. However, mutation of the P558 to either glycine (Fig. 1, D and E) or alanine (Fig. 1, F and G) slows activation. Activation of Kv1.4[P558A]ΔN channels is not complete, even at the end of a 4 s pulse (Fig. 1 G) and any sigmoidicity of activation is completely abolished. This change suggests that the activation process is rate limited by a single slow step (see Fig. S8).This may be similar to the cooperative step described for C-type inactivation (33) which involves a coordinated movement of all four subunits, as opposed to four independent activation gates as in the Hodgkin-Huxley formalism. Fig. 2, D and E shows the rate of activation as a function of voltage for Kv1.4[P558A] and Kv1.4[P558A]ΔN. Changing the extracellular solution from 2 to 98 mM [K+]o did not affect the activation rate of Kv1.4[P558A]ΔN, but slowed activation of Kv1.4[P558A]. The apparent increase in activation rate is unlikely to reflect a mechanism that is similar to that described for the rapid binding of the Kvβ1.2 N-terminal domain (34) (see Fig. S9), but may involve binding to and stabilizing a preactivated state. Fig. 2, F and G, show the fast and slow time constants of activation of Kv1.4[P558G]ΔN. Both components are voltage insensitive, indicating that both the proline hinge mutations cause a voltage-insensitive step to become rate limiting in channel activation.
Isochronal activation
Isochronal activation of Kv1.4[P558G] and Kv1.4[P558G]ΔN was measured relative to Kv1.4 and Kv1.4ΔN (Fig. 3). The relative magnitude of the peak tail current on the step to −50 mV (see inset in Fig. 5) was used to determine the degree of activation, given in Table 1. There was no significant difference between V1/2 for either Kv1.4 and Kv1.4ΔN (p > 0.8) or Kv1.4[P558G] and Kv1.4[P558G]ΔN (p > 0.5), but there were significant differences in V1/2 for Kv1.4 versus Kv1.4[P558G] and Kv1.4ΔN versus Kv1.4[P558G]ΔN (both p ≪ 0.001). For both the N-terminal-intact and N-terminal-deleted constructs, the P558G mutation shifted the value of V1/2 by ∼66 mV. This shift in V1/2 and decrease in slope factor is qualitatively similar to findings in Kv1.5 channels (4). There is a significant decrease in the slope factor between Kv1.4 and Kv1.4[P558G] and Kv1.4ΔN versus Kv1.4[P558G]ΔN (both p ≪ 0.001), but no difference between Kv1.4 and Kv1.4ΔN (p > 0.8) or Kv1.4[P558G] and Kv1.4[P558G]ΔN (p > 0.4). The change in V1/2 and slope were therefore the same across constructs for both the N-terminal-intact and N-terminal-deleted constructs, despite the effects of the N-terminal domain on the apparent activation rate of the P558G mutants. Again, this is consistent with the idea that a rapid overlapping inactivation mechanism is responsible for the apparent slowing of activation by the N-terminal domain in the P558G mutants. We propose that the decreased slope and shift of V1/2 is consistent with the idea that a late rate-limiting cooperative step in the activation of mutant channels becomes dominant and increases the total energy needed to open the channel distributed across multiple transitions (see Supporting Material).
Figure 3.
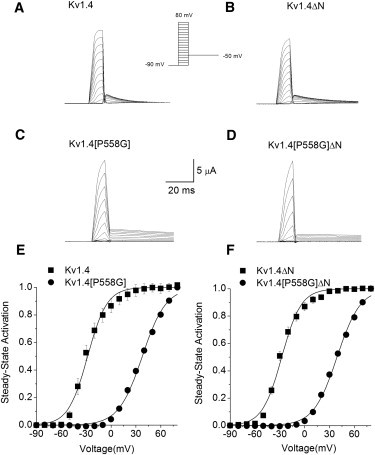
Isochronal activation. A single 10 ms depolarizing pulse from the holding potential of −90 mV was applied in 10 mV steps to potentials from −100 to +80 mV, followed by a step to −50 mV. Representative traces are shown from (A) Kv1.4, (B) Kv1.4ΔN, (C) Kv1.4[P558G], and (D) Kv1.4[P558G]ΔN. Isochronal activation calculated as the peak current measured on repolarization to −50 mV for (E) Kv1.4 (■, n = 4) and Kv1.4[P558G] (●, n = 10, error bars smaller than symbols), and (F) Kv1.4ΔN (■, n = 5) and Kv1.4[P558G]ΔN (●, n = 9, error bars smaller than symbols). Solid lines are Boltzmann fits to data 1/(1+exp((V−V1/2)/k)).
Figure 5.
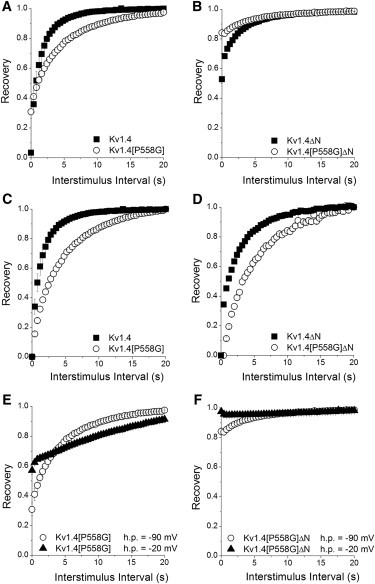
Recovery from inactivation was measured using a standard variable-interval protocol. An initial 5 s pulse (P1) from –90 to +50 mV was followed by a P2 pulse after an interval of 0–20 s in 400 ms intervals. The ratio of peak currents (Ipeak,P2/Ipeak,P1) is plotted versus pulse interval. Recovery is shown for (A) Kv1.4 (■, n = 8) and Kv1.4[P558G] (○, n = 12), and (B) Kv1.4ΔN (■, n = 6) and Kv1.4[P558G]ΔN (○, n = 10). Changing the interpulse holding potential from −90 to −20 mV altered the recovery rate. (C and D) Data from panels A and B, respectively, double normalized for easy comparison of relative recovery rates. (E) Recovery of Kv1.4[P558G] at a holding potential of −90 mV (○, n = 12) and –20 mV (▴, n = 5). (F) Recovery of Kv1.4[P558G]]ΔN at a holding potential of −90 mV (○, n = 10) and –20 mV (▴, n = 5).
Table 1.
Summary of gating parameters
| Kv1.4 | Kv1.4[P558G] | Kv1.4 ΔN | Kv1.4 [P558G]ΔN | |
|---|---|---|---|---|
| Activation | ||||
| Vhalf activation | −28.2 ± 2.5 mV∗ | 37.4 ± 1.4 mV∗ | −27.5 ± 1.6 mV† | 38.9 ± 1.3 mV† |
| Slope factor | 11.5 ± 1.0‡ | 14.2 ± 0.4‡ | 11.8 ± 1.0§ | 13.9 ± 0.2§ |
| Inactivation | ||||
| Vhalf inactivation | −51.8 ± 1.0 mV¶ | 2.3 ± 1.1 mV¶ | −45.5 ± 0.8 mV|| | 2.9 ± 2.1 mV|| |
| Slope factor | 3.2 ± 0.1∗∗ | 8.0 ± 0.5∗∗ | 3.6 ± 0.2†† | 11.3 ± 0.6†† |
| Recovery from inactivation | ||||
| τ | 4.5 ± 0.3 s | 5.0 ± 0.4 s | ||
Superscripts denote comparison between the same measurements of WT and N-terminal deletion constructs with their respective P558G mutant constructs (e.g., ∗compares Vhalf activation of Kv1.4 and Kv1.4[P558G]). All pairs were statistically significantly different (∗,†,‡,§.¶,||,∗∗,†† p ≪ 0.001). Data are mean ± SE.
Steady-state inactivation
We calculated the steady-state inactivation from experiments using the voltage protocol shown in Fig. 1. The pseudo-steady-state inactivation relationships are shown in Fig. 4, and the data are summarized in Table 1. Consistent with the idea that both N- and C-type inactivation are coupled to activation and opening of the intracellular gate, both of the V1/2 values of steady-state inactivation are shifted positively by ∼50 mV in a roughly parallel fashion. This is similar to the change in steady-state activation seen in the mutant channels. The presence of the mutation significantly altered the slopes of the steady-state inactivation relationship for both the N-terminal-attached and -deleted constructs compared with Kv1.4 and Kv1.4ΔN (in both cases, p ≪ 0.001). Furthermore, the slope factors for both Kv1.4[P558G] and Kv1.4[P558G]ΔN (i.e., with the mutation but in the presence or absence of the N-terminal) processes are nearly identical. This shallow slope for steady-state inactivation is qualitatively similar to the shallower slope of the activation process, but has quantitative differences that may reflect the complexity of activation and the coupling process. A mutant cycle analysis (see Supporting Material) also suggests a complex process involving direct interactions between the N-terminal and the proline hinge.
Figure 4.
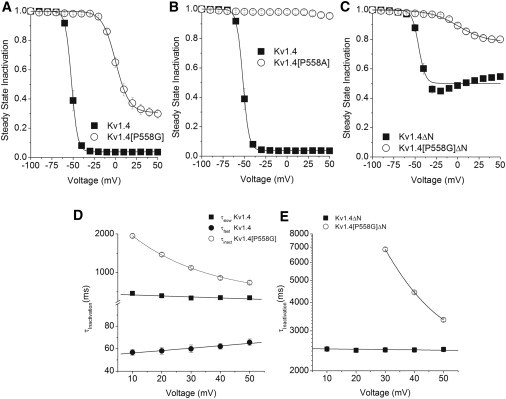
Inactivation. Pseudo steady-state inactivation relationships were determined from a two-pulse protocol (see Fig. 2) by calculating the ratio of the peak current in the P2 pulse to the maximum value of the peak P1 current. Steady-state inactivation relationships are shown for (A) Kv1.4 (■, n = 9) and Kv1.4[P558G] (○, n = 23), (B) Kv1.4 (■, n = 9) and Kv1.4[P558A] (○, n = 6), and (C) Kv1.4ΔN (■, n = 6) and Kv1.4[P558G]ΔN (○, n = 16). Solid lines are Boltzmann fits to the data: 1/{1+exp((V−V1/2)/k)}. P558G steady-state inactivation was relatively insensitive to pulse length (see Supporting Material). The voltage dependence of inactivation time constants is shown. (D) Kv1.4 was best fit with two exponentials, τfast (●) and τslow (■) (n = 8, solid lines are linear fits), and Kv1.4[P558G] was fit by a single exponential (○, n = 9, solid line is exponential fit of τ). (E) Kv1.4ΔN (■, n = 8, solid line is linear fit) and Kv1.4[P558G]ΔN (○, n = 14, solid line is exponential fit of τ) were best fit with single exponentials. Error bars are smaller than symbols.
Inactivation rate
Inactivation of Kv1.4 was well fit with two exponentials. Of the functional channels used in this study, only the proline-to-glycine mutation exhibited inactivation. Kv1.4[P558G] inactivation was well fit by a single exponential and exhibited a strong V dependency, with the rate of inactivation slowing by a factor of 3 over the range of +20 to +50 mV, as shown in Fig. 4, presumably due to overlap with the highly shifted steady-state activation (31). Kv1.4ΔN had no significant difference in the range of 10–50 mV (analysis of variation, n = 8, p > 0.7). Although Kv1.4[P558G]ΔN showed little inactivation, the inactivation measured was strongly voltage dependent.
Recovery from inactivation
Recovery from inactivation in Kv1.4 channels is governed by recovery from C-type inactivation (22). We measured recovery using a standard gapped-pulse protocol with depolarizations from −90 to +50 mV separated by a variable interstimulus interval. The ratio of the magnitude of the first and second pulse peak currents was used as a measure of the degree of recovery from inactivation. Fig. 5 shows the fraction of current elicited by the second pulse as a fraction of the peak current in the first pulse, plotted versus interstimulus interval. The slowing of the rate of recovery between Kv1.4 and Kv1.4[P558G] was virtually identical to the slowing between Kv1.4ΔN and Kv1.4[P558G]ΔN, i.e., the slowing of recovery of inactivation was similar regardless of whether the forward mechanism of inactivation was mediated by the N-type or C-type mechanism (Fig. 5, C and D). There was no significant difference between recovery of Kv1.4[P558G] and Kv1.4[P558G] ΔN (Table 1, p > 0.28). This suggests that the proline hinge mutation did not disrupt the coupling between N- and C-type inactivation. However, voltage-dependent recovery from C-type inactivation was made slower by the proline hinge mutation over a wide range of potentials, despite its location on the intracellular side of S6 (Fig. 5, E and F).
Effect of extracellular acidosis
One indicator of transmembrane communication from the intracellular to the extracellular side is sensitivity to a changes in extracellular pH transduced by an extracellular histidine at position 508 near the mouth of the kv1.4 and in homologous positions in related channels (8,23,35). Extracellular acidosis in wild-type channels tends to accelerate the development of both N- and C-type inactivation. Similarly, acidosis tends to slow recovery from inactivation. These kinetic effects occur in parallel for both N- and C-type inactivation. As shown in Fig. 6, the P558G mutation retains this characteristic. Acidosis increases both the rate and extent of inactivation in the N-terminal-intact and -deleted constructs, and clearly slows the recovery from inactivation. Kv1.4[P558A]ΔN shows a marked slowing in the development of activation.
Figure 6.
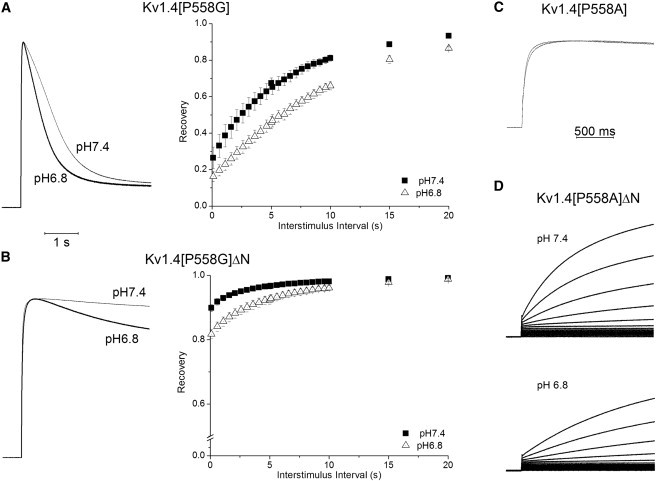
Effects of pH. (A) i: Representative traces show that acidosis speeds Kv1.4[P558G] inactivation. ii: Recovery of Kv1.4[P558G] is slowed by acidosis (same protocol as in Fig. 5). (B) i: Representative traces show that acidosis speeds Kv1.4[P558G]ΔN inactivation. ii: Recovery of Kv1.4[P558G]ΔN is also slowed by acidosis, in similarity to Kv1.4[P558G], suggesting that the common recovery through a C-type state is intact in these mutants. Both constructs show increases in the rate and extent of development of inactivation. (C) Acidosis has little effect on Kv1.4[P558A]. (D) Acidosis slows development of slow Kv1.4[P558A]ΔN activation.
Effect of quinidine
We verified that coupling between lipophilic binding in the intracellular pore is coupled to the development of C-type inactivation by using the quaternary ammonium compound quinidine, which is often employed as a surrogate for N-type inactivation (7,27). The overlap of the binding site for quinidine and the N-terminal was demonstrated by the observation that for both P558G and P558A, removal of the N-terminus resulted in an increase in the apparent affinity for quinidine similar to that seen for WT Kv1.4 (Fig. 7 E). This is consistent with competition between quinidine and the N-terminus for binding to an intracellular site. This competition is not altered by the proline hinge mutation, despite a small increase in affinity for quinidine. Furthermore, as expected, the inactivation rate of P558G was increased by quinidine, and showed a recovery rate consistent with a C-type inactivation mechanism.
Figure 7.
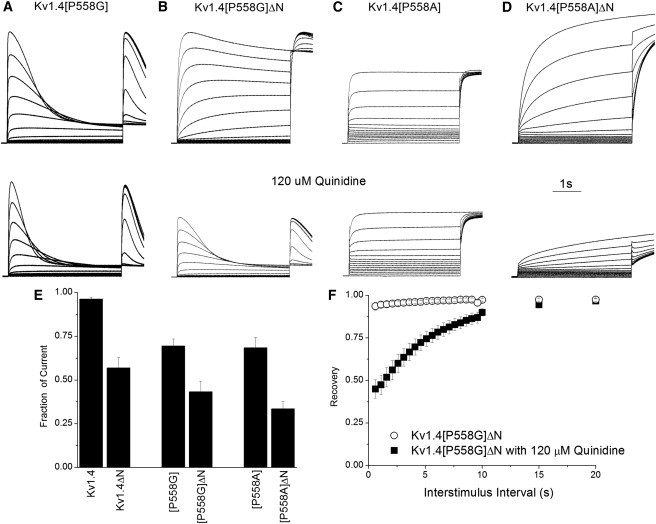
Effect of quinidine. Representative traces show control currents (top row) and the effect of 120 μM quinidine (bottom row, same scale as in the top row for each mutant) on (A) Kv1.4[P558G], (B) Kv1.4[P558G]ΔN, (C) Kv1.4[P558A], and (D) Kv1.4[P558A]ΔN. (E) Effects of the N-terminus on inhibition by quinidine. The fraction of the maximum current during the P1 trace that remains in 120 μM quinidine is shown for each construct. Note that current inhibition is increased by removal of the N-terminus for all constructs. (F) Recovery from inactivation of Kv1.4[P558G]ΔN in the presence and absence of 120 μM quinidine.τ
The P558A kinetics was also sensitive to quinidine, with slowing of activation (Fig. 7, C and D). Slowing was attenuated by the presence of the N-terminal, again consistent with competition between N-terminal and quinidine binding, and a role for N-terminal binding to a distinct preactivated state. The fact that quinidine does not augment the apparent rate of activation argues against the notion that it works via a similar mechanism in the Kvβ1.2 N-terminal domain (34). Instead, we propose that binding of the N-terminal stabilizes the channel in a modified preactivated closed state similar to the one from which C-type inactivation proceeds in Kv1.4 (31), but which does not inactivate with the P558A mutation. Thus, the mutation may prevent the channel from returning to a more stable and slower resting conformation. Quinidine, which is much smaller than the N-terminal, may be less effective in stabilizing this putative preopen state.
Discussion
Kv1.4 channels undergo a series of conformational changes in response to voltage. Activation is the primary voltage-dependent event after depolarization. The voltage dependence of activation requires that a net charge must move in the transmembrane electrical field to produce the physical work necessary to move the channel out of the stable closed conformation. The S4 transmembrane domain in K+ channels contains a highly conserved structure, with every third position occupied by a lysine or arginine that is positively charged in the physiological range of pH. At the resting membrane potential, many of these positive charges are on the intracellular face of the membrane. Upon membrane depolarization, these residues move through the membrane due to the increased electrical driving force and reach the extracellular face (36,37). The transmembrane movement of S4 initiates large-scale conformational changes that result in an open, conducting pore. Although there is a long history of experiments showing a prominent role for the S4-S5 linker in coupling voltage-dependent movement of S4 to opening of the core domains, this linkage became much clearer with the elucidation of the crystal structure of the core domains of Kv1.2 (38). Movement of S5 subsequent to movement of S4 may convey energy and move the lower portion of S6 into an open or conducting conformation.
There are three general pictures of intracellular pore opening. Flexibility and orientation of S6 plays a prominent role in lining the pore in the open model of KcsA, MthK, and Kv1.2 channels (38–40). In all of these structural models, activation involves a rotational movement of S6 as the channel moves from the closed to the open state. However, the putative ending conformation is very different for the KcsA, Kv1.2, and MthK channels. Kv1.2 is much more widely splayed open than KcsA, and both the length and the physical orientation of the putative rigid α-helices are different in all three crystal structures. This suggests that there is tremendous physical diversity in the open-channel conformation, even in related channels, and that there is some tolerance for rigidity in the S6 domain.
In Kv1.4 channels, the considerable flexibility in the intracellular gate is thought to arise from a disruption of α-helical structure at the proline hinge (i.e., a PVP sequence), although glycine may also play a role (41). Proline hinge motifs are found in a diverse range of proteins. If a proline is present within an α-helix structure, the proline will be unable to form hydrogen bonds with the carbonyl oxygens of the residues on the upper and lower turns of the helix, and the helical structure will not be supported. A single proline is sufficient to introduce flexibility in an α-helical structure, and when two or more prolines are found in close proximity, they tend to break the regular structure of the α-helix. In the case of Kv1 channels, a kink is introduced into the helix by the PVP sequence on the intracellular side of S6, which confers a site of conformational flexibility. This flexibility is critical for the rapid activation properties of Kv channels and allows rapid opening of the S6 gate.
Opening of the S6 gate is not the only requirement for channel opening and conducting. Other portions of the channel must also be in the open and conducting conformation. The selectivity filter region has a very specific relationship between structural arrangement and function. There is clear evidence that permeant ions interact with the pore to stabilize the open state (42). In addition, there is thought to be an extracellular gate in or near the selectivity region in some channels (18,19). Thus, there are multiple sites that play a potential role in the regulation of channel conduction, and some of these are also involved in the various forms of inactivation.
Inactivation is usually thought of as the process by which a channel becomes nonconducting in the presence of a continued stimulus. Early studies in Shaker splice variants showed at least two mechanisms of inactivation: N-type and C-type (43). The better-understood inactivation mechanism is N-type, which occurs on the order of approximately milliseconds to approximately tens of milliseconds. N-type inactivation is mediated by a tethered ball mechanism in which a lipophilic segment of ∼20 amino acids in the N-terminus of the channel binds at the intracellular pore mouth (11). C-type inactivation is broadly defined as an inactivation mechanism that is intrinsic to the core domain and is not mediated by an N-type mechanism. Regions critical for C-type inactivation include the extracellular side of S6 and a specific residue in the extracellular H5-S6 loop equivalent to position 449 in Shaker B (for a review, see Rasmusson et al. (22). C-type inactivation is slowed by increasing external permeant ion concentrations and by the application of extracellular, but not intracellular, TEA+ (22). Thus, C-type inactivation was determined to involve closure of the extracellular pore mouth and cooperative interactions between multiple subunits (33).
C-type inactivation in Kv1.4 channels has also been shown to involve large-scale rearrangements of the core α-subunits. These rearrangements have been hypothesized to be responsible for the allosteric component of coupling between N-terminal (or drug) binding at the intracellular mouth of the pore (7,8,27). This coupling causes the rapid development of a C-type inactivated state, which in turn dominates the properties of recovery. In the forward direction, after a depolarizing pulse, activation orients the channel so that both N- and C-type inactivation can proceed. This coupling of inactivation to activation has been proposed to involve the exposure of an S6 binding site for N-type inactivation (7). Similarly, we have proposed that after activation, C-type inactivation involves a closure (or partial closure) of the intracellular mouth of the pore that is similar in magnitude to opening during activation (17).
If activation and N- and C-type inactivation are dependent on the orientation and movement of the intracellular side of S6, the rates and completeness of all three will be strongly affected by reduced flexibility at the pore hinge region. As expected, our experiments with the mutation of P558 yielded the following results: 1) Shifts in the voltage dependence of steady-state activation paralleled changes in inactivation. Both N- and C-type inactivation showed virtually identical shifts in both steady-state V1/2 and slope factor. These changes in inactivation mirrored the changes in V1/2 and the reduced slope factor for activation. 2) Slowing of activation correlated with slowing of both types of inactivation. The extreme slowing of activation in the P558A constructs effectively abolished both forms of inactivation. 3) Flexibility in the hinge region allowed easier opening and closing of the S6 domain, and more flexibility in binding and orienting the N-terminus. The idea that flexibility (rather than size or hydrophobicity) is key in C-type inactivation is bolstered by the insensitivity of C-type inactivation and recovery rates to a V to I mutation at the PVP hinge in Kv1.1 channels (44). Our data for the proline hinge mutations show the expected reductions in the rate and degree of steady-state inactivation for N- and C-type inactivation.
The modified gating induced by the P558G and P558A mutations retained some important aspects of the model of C-type inactivation and its coupling to N-type inactivation involving movement of S6. The effects of pH on the modified gating characteristics of the P558G and P558A are consistent with the established mechanisms of pH regulation on N- and C-type inactivation (8,23) in that acidosis speeded development and slowed recovery in P558G. In P558A, it slowed activation in the N-terminal-deleted construct.
Recovery from inactivation is the opposite process from inactivation. Recovery from C-type inactivation is predicted to require some rearrangement of the intracellular vestibule (9,17). This suggests that the rate of recovery should be slower in the less-flexible P558G mutants, as observed in our results. This slowing of C-type inactivation has implications for N-type inactivation. In Kv1.4, one of the key indicators of coupling between N- and C-type inactivation is that recovery occurs at the same rate regardless of the presence or absence of the N-terminal. As noted in our results, recovery from inactivation in the P558G mutants occurs at the same slowed rate in the presence or absence of the N-terminal domain. This suggests that although the properties of N- and C-type inactivation have been altered, the coupling between them remains intact.
Our data and data from other studies (9,22) are difficult to reconcile with a purely K-exit model of N- and C-type coupling (45). The K-exit model was based on experiments using completely permeant-ion-free solutions. Little noted in those studies was the fact that there are two components: a permeant ion component and an allosteric/chemical component (N-terminal or drug binding) (45,46). Many subsequent studies focused on the permeant-ion-binding mechanism but neglected the allosteric chemical component. In the presence of physiological K concentrations, the relative roles of these components are unclear. However, under physiological conditions, there is consistent evidence that C-type inactivation involves major conformational changes (8,9,17,22,47) remote from the selectivity filter portion of the permeation pathway. These conformational changes likely contribute to the chemical component.
Cuello et al. (47) also proposed functional coupling between activation and inactivation gates in K+ channels. The proposed model of the gating cycle for KcsA channels is defined by four kinetic states with activation and selectivity filter gates that can exist in either conductive or nonconductive conformations. The two gates were proposed to be coupled such that opening of the activation gates stabilizes the nonconductive conformation of the filter, and inactivating the filter stabilizes the activation gate in its closed conformation. This is a clear indication that there are strong energetic interactions between the filter gate and the orientation of the S6 domain. The residues that were proposed by Cuello et al. (47) to transduce the movement of S6 to the filter are dependent on the orientation of the relatively rigid S6, and energetic compatibility with a closed conformation of the activation gate or a similar conformation. This is the same as our proposed mechanism, with a slight difference in the ordering of events, i.e., in our model, entering an activated closed state promotes C-type inactivation as opposed to an open activated state. Small differences in the hydrophobicity of the side chains in S6 may determine which transition is most likely, and the interaction with the S1–S4 domains may also affect the coupling between processes.
The accumulation of physical evidence indicating that large-scale conformational changes during C-type inactivation involve movement of S6 began with the observation that the V561A (Kv1.4 numbering) mutation at the C-terminal end of S6 in Kv1.1 associated with episodic ataxia strongly influenced C-type inactivation (48). This mutation also changed the sensitivity of C-type inactivation in Kv1.4 mutant channels, such that elevated [K+]o increased the rate of C-type inactivation instead of slowing it (8). Our own earlier work measuring changes in aqueous volume with applied osmotic pressure demonstrated that the change in pore volume associated with C-type inactivation was similar to that associated with activation (17). The work of Cuello et al. (47) supports an energetic interaction with conformational states of S6 related to the activation gate (49,50). In addition, data from other channel types (51,52) show that inactivation in Ca2+ and Na+ channels may also be dependent on a functional proline hinge and the intracellular conformational changes in S6.
In conclusion, we have examined the role of the proline hinge of Kv1.4 channels in N- and C-type inactivation. Flexibility in the hinge region was seen to be critical for both N- and C-type inactivation, as well as for activation. These findings confirm a role for large-scale movement of this region in C-type inactivation and in formation of a binding site for N-type inactivation.
Supporting Material
References
- 1.Barry D.M., Nerbonne J.M. Myocardial potassium channels: electrophysiological and molecular diversity. Annu. Rev. Physiol. 1996;58:363–394. doi: 10.1146/annurev.ph.58.030196.002051. [DOI] [PubMed] [Google Scholar]
- 2.Sheng M., Tsaur M.L., Jan L.Y. Subcellular segregation of two A-type K+ channel proteins in rat central neurons. Neuron. 1992;9:271–284. doi: 10.1016/0896-6273(92)90166-b. [DOI] [PubMed] [Google Scholar]
- 3.Korovkina V.P., England S.K. Molecular diversity of vascular potassium channel isoforms. Clin. Exp. Pharmacol. Physiol. 2002;29:317–323. doi: 10.1046/j.1440-1681.2002.03651.x. [DOI] [PubMed] [Google Scholar]
- 4.Labro A.J., Raes A.L., Snyders D.J. Gating of Shaker-type channels requires the flexibility of S6 caused by prolines. J. Biol. Chem. 2003;278:50724–50731. doi: 10.1074/jbc.M306097200. [DOI] [PubMed] [Google Scholar]
- 5.del Camino D., Holmgren M., Yellen G. Blocker protection in the pore of a voltage-gated K+ channel and its structural implications. Nature. 2000;403:321–325. doi: 10.1038/35002099. [DOI] [PubMed] [Google Scholar]
- 6.Hackos D.H., Chang T.H., Swartz K.J. Scanning the intracellular S6 activation gate in the Shaker K+ channel. J. Gen. Physiol. 2002;119:521–532. doi: 10.1085/jgp.20028569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bett G.C., Rasmusson R.L. Inactivation and recovery in Kv1.4 K+ channels: lipophilic interactions at the intracellular mouth of the pore. J. Physiol. 2004;556:109–120. doi: 10.1113/jphysiol.2003.055012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li X., Bett G.C., Rasmusson R.L. Regulation of N- and C-type inactivation of Kv1.4 by pHo and K+: evidence for transmembrane communication. Am. J. Physiol. Heart Circ. Physiol. 2003;284:H71–H80. doi: 10.1152/ajpheart.00392.2002. [DOI] [PubMed] [Google Scholar]
- 9.Kurata H.T., Fedida D. A structural interpretation of voltage-gated potassium channel inactivation. Prog. Biophys. Mol. Biol. 2006;92:185–208. doi: 10.1016/j.pbiomolbio.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 10.Holmgren M., Jurman M.E., Yellen G. N-type inactivation and the S4-S5 region of the Shaker K+ channel. J. Gen. Physiol. 1996;108:195–206. doi: 10.1085/jgp.108.3.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoshi T., Zagotta W.N., Aldrich R.W. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- 12.Jerng H.H., Covarrubias M. K+ channel inactivation mediated by the concerted action of the cytoplasmic N- and C-terminal domains. Biophys. J. 1997;72:163–174. doi: 10.1016/S0006-3495(97)78655-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopez G.A., Jan Y.N., Jan L.Y. Evidence that the S6 segment of the Shaker voltage-gated K+ channel comprises part of the pore. Nature. 1994;367:179–182. doi: 10.1038/367179a0. [DOI] [PubMed] [Google Scholar]
- 14.Zagotta W.N., Hoshi T., Aldrich R.W. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science. 1990;250:568–571. doi: 10.1126/science.2122520. [DOI] [PubMed] [Google Scholar]
- 15.Isacoff E.Y., Jan Y.N., Jan L.Y. Putative receptor for the cytoplasmic inactivation gate in the Shaker K+ channel. Nature. 1991;353:86–90. doi: 10.1038/353086a0. [DOI] [PubMed] [Google Scholar]
- 16.Liu Y., Jurman M.E., Yellen G. Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron. 1996;16:859–867. doi: 10.1016/s0896-6273(00)80106-3. [DOI] [PubMed] [Google Scholar]
- 17.Jiang X., Bett G.C., Rasmusson R.L. C-type inactivation involves a significant decrease in the intracellular aqueous pore volume of Kv1.4 K+ channels expressed in Xenopus oocytes. J. Physiol. 2003;549:683–695. doi: 10.1113/jphysiol.2002.034660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ogielska E.M., Aldrich R.W. Functional consequences of a decreased potassium affinity in a potassium channel pore. Ion interactions and C-type inactivation. J. Gen. Physiol. 1999;113:347–358. doi: 10.1085/jgp.113.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kiss L., Korn S.J. Modulation of C-type inactivation by K+ at the potassium channel selectivity filter. Biophys. J. 1998;74:1840–1849. doi: 10.1016/S0006-3495(98)77894-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoshi T., Zagotta W.N., Aldrich R.W. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- 21.López-Barneo J., Hoshi T., Aldrich R.W. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- 22.Rasmusson R.L., Morales M.J., Strauss H.C. Inactivation of voltage-gated cardiac K+ channels. Circ. Res. 1998;82:739–750. doi: 10.1161/01.res.82.7.739. [DOI] [PubMed] [Google Scholar]
- 23.Claydon T.W., Boyett M.R., Orchard C.H. Inhibition of the K+ channel kv1.4 by acidosis: protonation of an extracellular histidine slows the recovery from N-type inactivation. J. Physiol. 2000;526:253–264. doi: 10.1111/j.1469-7793.2000.00253.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Armstrong C.M. Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. J. Gen. Physiol. 1971;58:413–437. doi: 10.1085/jgp.58.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Choi K.L., Aldrich R.W., Yellen G. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+ channels. Proc. Natl. Acad. Sci. USA. 1991;88:5092–5095. doi: 10.1073/pnas.88.12.5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lipkind G.M., Hanck D.A., Fozzard H.A. A structural motif for the voltage-gated potassium channel pore. Proc. Natl. Acad. Sci. USA. 1995;92:9215–9219. doi: 10.1073/pnas.92.20.9215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang S., Morales M.J., Rasmusson R.L. Kv1.4 channel block by quinidine: evidence for a drug-induced allosteric effect. J. Physiol. 2003;546:387–401. doi: 10.1113/jphysiol.2002.029512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Busch A.E., Hurst R.S., Kavanaugh M.P. Current inactivation involves a histidine residue in the pore of the rat lymphocyte potassium channel RGK5. Biochem. Biophys. Res. Commun. 1991;179:1384–1390. doi: 10.1016/0006-291x(91)91726-s. [DOI] [PubMed] [Google Scholar]
- 29.Mitcheson J., Perry M., Stansfeld P., Sanguinetti M.C., Witchel H., Hancox J. Structural determinants for high-affinity block of hERG potassium channels. Novartis Found. Symp. 2005;266:136–150. discussion 150–138. [PubMed] [Google Scholar]
- 30.Bett G.C., Rasmusson R.L. Modification of K+ channel-drug interactions by ancillary subunits. J. Physiol. 2008;586:929–950. doi: 10.1113/jphysiol.2007.139279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bett G.C., Dinga-Madou I., Rasmusson R.L. A model of the interaction between N-type and C-type inactivation in Kv1.4 channels. Biophys. J. 2011;100:11–21. doi: 10.1016/j.bpj.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Comer M.B., Campbell D.L., Strauss H.C. Cloning and characterization of an Ito-like potassium channel from ferret ventricle. Am. J. Physiol. 1994;267:H1383–H1395. doi: 10.1152/ajpheart.1994.267.4.H1383. [DOI] [PubMed] [Google Scholar]
- 33.Panyi G., Sheng Z., Deutsch C. C-type inactivation of a voltage-gated K+ channel occurs by a cooperative mechanism. Biophys. J. 1995;69:896–903. doi: 10.1016/S0006-3495(95)79963-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Castellino R.C., Morales M.J., Rasmusson R.L. Time- and voltage-dependent modulation of a Kv1.4 channel by a β-subunit (Kv β 3) cloned from ferret ventricle. Am. J. Physiol. 1995;269:H385–H391. doi: 10.1152/ajpheart.1995.269.1.H385. [DOI] [PubMed] [Google Scholar]
- 35.Kehl S.J., Eduljee C., Fedida D. Molecular determinants of the inhibition of human Kv1.5 potassium currents by external protons and Zn(2+) J. Physiol. 2002;541:9–24. doi: 10.1113/jphysiol.2001.014456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larsson H.P., Baker O.S., Isacoff E.Y. Transmembrane movement of the Shaker K+ channel S4. Neuron. 1996;16:387–397. doi: 10.1016/s0896-6273(00)80056-2. [DOI] [PubMed] [Google Scholar]
- 37.Bezanilla F. Voltage sensor movements. J. Gen. Physiol. 2002;120:465–473. doi: 10.1085/jgp.20028660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Long S.B., Campbell E.B., Mackinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 2005;309:903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- 39.Doyle D.A., Morais Cabral J., MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 40.Jiang Y., Lee A., MacKinnon R. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 2002;417:515–522. doi: 10.1038/417515a. [DOI] [PubMed] [Google Scholar]
- 41.Ding S., Ingleby L., Ahern C.A., Horn R. Investigating the putative glycine hinge in Shaker potassium channel. J. Gen. Physiol. 2005;126:213–226. doi: 10.1085/jgp.200509287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Consiglio J.F., Korn S.J. Influence of permeant ions on voltage sensor function in the Kv2.1 potassium channel. J. Gen. Physiol. 2004;123:387–400. doi: 10.1085/jgp.200308976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jan L.Y., Jan Y.N. Structural elements involved in specific K+ channel functions. Annu. Rev. Physiol. 1992;54:537–555. doi: 10.1146/annurev.ph.54.030192.002541. [DOI] [PubMed] [Google Scholar]
- 44.Imbrici P., Grottesi A., Pessia M. Contribution of the central hydrophobic residue in the PXP motif of voltage-dependent K+ channels to S6 flexibility and gating properties. Channels (Austin) 2009;3:39–45. doi: 10.4161/chan.3.1.7548. [DOI] [PubMed] [Google Scholar]
- 45.Baukrowitz T., Yellen G. Use-dependent blockers and exit rate of the last ion from the multi-ion pore of a K+ channel. Science. 1996;271:653–656. doi: 10.1126/science.271.5249.653. [DOI] [PubMed] [Google Scholar]
- 46.Baukrowitz T., Yellen G. Modulation of K+ current by frequency and external [K+]: a tale of two inactivation mechanisms. Neuron. 1995;15:951–960. doi: 10.1016/0896-6273(95)90185-x. [DOI] [PubMed] [Google Scholar]
- 47.Cuello L.G., Jogini V., Perozo E. Structural basis for the coupling between activation and inactivation gates in K(+) channels. Nature. 2010;466:272–275. doi: 10.1038/nature09136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adelman J.P., Bond C.T., Maylie J. Episodic ataxia results from voltage-dependent potassium channels with altered functions. Neuron. 1995;15:1449–1454. doi: 10.1016/0896-6273(95)90022-5. [DOI] [PubMed] [Google Scholar]
- 49.Panyi G., Deutsch C. Probing the cavity of the slow inactivated conformation of Shaker potassium channels. J. Gen. Physiol. 2007;129:403–418. doi: 10.1085/jgp.200709758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Panyi G., Deutsch C. Cross talk between activation and slow inactivation gates of Shaker potassium channels. J. Gen. Physiol. 2006;128:547–559. doi: 10.1085/jgp.200609644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Raybaud A., Dodier Y., Parent L. The role of the GX9GX3G motif in the gating of high voltage-activated Ca2+ channels. J. Biol. Chem. 2006;281:39424–39436. doi: 10.1074/jbc.M607405200. [DOI] [PubMed] [Google Scholar]
- 52.Zhao Y., Yarov-Yarovoy V., Catterall W.A. A gating hinge in Na+ channels: a molecular switch for electrical signaling. Neuron. 2004;41:859–865. doi: 10.1016/s0896-6273(04)00116-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.