

Abstract
Recent evidence supports the hypothesis that the oligomers formed by the β-amyloid peptide early in its aggregation process are neurotoxic and may feature in Alzheimer’s disease. Although the mechanism underlying this neurotoxicity remains unclear, interactions of these oligomers with neuronal membranes are believed to be involved. Identifying the neurotoxic species is challenging because β-amyloid peptides form oligomers at very low physiological concentrations (nM), and these oligomers are highly heterogeneous and metastable. Here, we report the use of single-molecule imaging techniques to study the interactions between β-amyloid (1–40) peptides and supported synthetic model anionic lipid membranes. The evolution of the β-amyloid species on the membranes was monitored for up to several days, and the results indicate an initial tight, uniform, binding of β-amyloid (1–40) peptides to the lipid membranes, followed by oligomer formation in the membrane. At these low concentrations, the behavior at early times during the formation of small oligomers is interpreted qualitatively in terms of the two-state model proposed by H. W. Huang for the interaction between amphipathic peptides and membranes. However, the rate of oligomer formation in the membrane and their size are highly dependent on the concentrations of β-amyloid (1–40) peptides in aqueous solution, suggesting two different pathways of oligomer formation, which lead to drastically different species in the membrane and a departure from the two-state model as the concentration increases.
Introduction
The formation of senile plaques in brain tissue is a hallmark of Alzheimer’s disease (AD) (1–3). It has been found that β-amyloid peptides, which are 39–43 amino acid residues in length, are the major component of the plaques (4–6) and are able to self-assemble into highly ordered fibrillar structures in aqueous solution (7,8). There is evidence that β-amyloid (1–40) peptide is more concentrated in the cerebrovascular plaques, whereas β-amyloid (1–42) peptide is more abundant in the neuritic plaques (9). Historical research shows that the composition of β-amyloid deposits in AD brains can also vary considerably from patient to patient (10–12).
The presence of β-amyloid fibrils in brains of AD patients has led to the amyloid hypothesis, which proposed that these fibrils are toxic to neuronal cells and are a causative factor in AD (13,14). However, recent experimental evidence has indicated that early β-amyloid oligomers, rather than the mature fibrils, are likely the neurotoxic species (15–20). Indeed, there is evidence that β-amyloid oligomers, as small as dimers, are capable of disrupting synaptic plasticity and memory (19,20). One hypothesis to explain the neurotoxicity of the β-amyloid oligomers is that these peptide aggregates form ion-conducting pores in neuronal membranes, and that the resulting imbalance of calcium ions across the membranes can either lead to direct cell death or trigger the signaling for apoptosis (21). Small oligomeric structures, which consist of a minimum of 4∼6 monomeric units, are likely involved in the formation of membrane pores (17,22). Moreover, anionic membranes have been found to be especially prone to the pore formation process (23–28), although a few studies have reached contradictory conclusions (29–31). Some recent studies have also suggested other possible mechanisms for β-amyloid-induced calcium permeability, such as by β-amyloid peptide interacting with GM1 ganglioside (32), or by β-amyloid oligomers affecting glutamatergic receptors (33,34).
Despite the growing attention to early β-amyloid oligomers, the study of these potentially pore-forming species has proved challenging, because they are heterogeneous, dynamic, and exist at extremely low concentrations in the brain. It has been suggested that the lack of a clear description of the toxic β-amyloid oligomer has hindered our understanding and interpretation of the controversial results in Alzheimer’s research (35,36). Single-molecule techniques have been shown to be very promising tools for studying these small oligomeric structures, because individual β-amyloid oligomers can be detected and identified at very low peptide concentrations.
In this work, we used single-molecule imaging techniques to study the interactions between β-amyloid (1–40) and a supported anionic lipid membrane to gain deeper understanding of the mechanism of β-amyloid neurotoxicity. In our experiments, the evolution of β-amyloid species in anionic lipid membranes was monitored for up to several days. The results indicate an initial tight, uniform, binding of β-amyloid (1–40) peptide to the lipid membrane, followed by oligomer formation in the membrane. This result is qualitatively compared to the two-state model proposed for peptide membrane interactions (37,38). The results of our single-molecule experiments suggest there are likely two different pathways of oligomer formation in the membrane, both yielding a wide range of oligomer species. A significant fraction of the oligomers formed via one of these pathways is composed of smaller species, which have also been observed by high-resolution atomic force microscopy imaging in previous studies (17,22).
Materials and Methods
β-amyloid (1–40) preparation
The HiLyte Fluor 488 β-amyloid (1–40) (AnaSpec, San Jose, CA) was stored frozen at −20°C. The peptide was dissolved in 2% ammonium hydroxide (J. T. Baker, Phillipsburg, NJ) at 1 mg/ml, and then aliquotted into small samples, lyophilized, and stored at −20°C. To make a fresh β-amyloid (1–40) solution, the lyophilized powder was directly dissolved into 10 mM sodium phosphate, 100 mM sodium chloride, pH7.4 buffer. The same buffer was used in all the experiments conducted in this work. In addition, only freshly dissolved β-amyloid (1–40) peptide was used, at a concentration of 100 nM, and these samples contain ∼70% monomers as shown by single-molecule measurements. Because the oligomers exist in a dynamic equilibrium with monomeric peptide and oligomerization is a high order reaction and hence highly concentration dependent, it appears highly plausible that at lower peptide concentrations as used in out experiments, i.e., 2 nM, the oligomeric fraction is markedly lower and the peptide is very predominantly monomeric.
Total internal reflection fluorescence microscopy
Fluorescence emission from HiLyte Fluor 488 β-amyloid (1–40) peptide was collected by a custom-made inverted total internal reflection fluorescence (TIRF) microscope (39–41). Both β-amyloid peptide and lipid membrane were imaged in the salient environment (10 mM sodium phosphate, 100 mM sodium chloride, pH7.4), and at room temperature (22°C). The microscope was equipped with a 1.45NA 60X oil-immersion objective (Olympus, Center Valley, PA). An Ar+-ion laser (I-90, Coherent, Santa Clara, CA) operating at 488.0 nm was used as the excitation source and delivered a laser power of 500 μW at the sample plane. The fluorescence signal was filtered by a 520/60 nm band-pass filter (Chroma, Rockingham, VT), and recorded by an electron-multiplying CCD camera (Andor, South Windsor, CT) with an exposure time of 50 ms/frame. Due to the evanescent wave in the TIRF geometry, only fluorophores that are very close to the glass/water interface (∼150nm) are excited and detected.
Supported anionic lipid membrane preparation
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC, molecular weight 760.08) and 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (POPG, molecular weight 770.99) (both from Avanti Polar Lipids, Alabaster, AL) were stored at −20°C. In all of our experiments, a mixture of POPC (neutral/zwitterionic) and POPG (negative) with a molar ratio of 1:1 was used to form the supported anionic membranes on coverglasses (Fisher Scientific, Pittsburgh, PA) that had been precleaned with MilliQ (Millipore, Billerica, MA) water rinsing followed by kiln baking at ∼500°C for 2 h.
To form an anionic lipid membrane on the coverglass, small unilamellar vesicles (SUVs) were first made (42,43). 0.5 mg POPC and 0.5 mg POPG were codissolved in chloroform, and then dried under gaseous nitrogen in a fume hood (a molar ratio of 1:1 was achieved due to the close molecular weight of POPC and POPG). A further removal of residual chloroform was achieved by vacuum drying of the sample overnight. Before the preparation of SUVs, the dried lipids were hydrated in buffer (10 mM sodium phosphate, 100 mM sodium chloride, pH 7.4) for 2 h at room temperature, during which the sample was vortexed a few times to completely resuspend the lipids. SUVs were then formed by sonication of the lipid suspension in an ice water bath for 2–5 min until the suspension became clear. Finally, the supported lipid membranes were spontaneously assembled by incubating the freshly prepared SUVs on a clean coverglass surface overnight (43). After the formation of the supported lipid membrane, the unbound SUVs were gently washed off with buffer. 100% surface coverage of the supported bilayer was confirmed under the microscope.
Application of fluorescence recovery after photobleaching (FRAP) to determine membrane fluidity
FRAP experiments were used to measure the mobility of the lipid molecules and the membrane-bound β-amyloid (1–40) peptide (44,45). It should be noted that, for measuring the mobility of lipid molecules, 0.5% nitro-2-1,3-benzoxadiazol-4-yl-POPC (NBD-POPC; Avanti Polar Lipids) was mixed with POPC and POPG to allow fluorescence photobleaching and recovery. In a typical FRAP experiment, a circular area of membrane of ∼45 μm in diameter was photobleached by high intensity laser illumination for a short duration (5 s). (Because the laser intensity is higher than the saturation level of the dye molecules, the uneven illumination still yielded a relatively flat photobleaching profile, which satisfies the assumption in using the following analysis method for extracting diffusion coefficient.) The fluorescence recovery in the photobleached region was then recorded using much weaker illumination intensity (<1/2500 of the photobleaching intensity). Because all the free lipid molecules (or β-amyloid (1–40) peptide) in solution had been washed off extensively, the fluorescence recovery was purely due to the lateral diffusion of the lipid molecules (or the membrane-bound β-amyloid (1–40) peptide) in the membrane. This mobility measurement of the lipid molecules was also used to confirm the integrity of the lipid membranes.
To extract diffusion coefficients from FRAP experiments, the fluorescence recovery curves were fit to the following equation (45) using Mathematica: . In this equation, is the integrated fluorescence intensity over the photobleached region: and are the modified Bessel functions of the first kind . And is the characteristic diffusion time, with w being the radius of the photobleached region, and D being the diffusion coefficient.
β-amyloid (1–40) interaction with lipid membrane
After POPC/POPG lipid membrane was formed on clean coverglass substrate, freshly prepared HiLyte Fluor 488 β-amyloid (1–40) peptide was added to the desired concentration by gentle pipetting. The sample holder was then sealed with parafilm to prevent evaporation of the solution. The incubation of β-amyloid (1–40) peptide with lipid membrane was kept at room temperature (22°C).
Results
It was critical to confirm that the fluorescence labeling of β-amyloid with HiLyte Fluor 488 did not alter the properties of the peptide. NMR studies of β-amyloid peptides structure indicate that the N-terminus of the peptide, where the fluorophore is attached, is flexible, suggesting the peptides should be minimally perturbed by the labeling (8). Membrane conductivity experiments indeed confirmed that both labeled and unlabeled β-amyloid (1–40) peptides permeabilize lipid membranes with similar efficiency (46). In addition, transmission electron microscopy imaging showed that both labeled and unlabeled peptides form fibrillar structures with almost identical morphologies (47).
To study β-amyloid (1–40) peptide interactions with lipid membranes, supported anionic membranes were formed on clean coverglass. HiLyte Fluor 488 β-amyloid (1–40) peptide solution was added and a TIRF microscope was used to monitor the interactions between the peptide and the membrane. Depending on the concentration of the added β-amyloid (1–40), the peptide-membrane interactions were monitored for a period of a few hours, with high peptide concentration, ∼100 nM, to over 6 days, with low peptide concentration, ∼2 nM.
The results of these experiments reveal that the interactions between the peptide and membrane can be divided into two stages where an initial tight and uniform binding of β-amyloid (1–40) peptide to the anionic membrane is followed by the formation of membrane-embedded β-amyloid (1–40) oligomers.
Uniform binding
Following the addition of freshly dissolved HiLyte Fluor 488 β-amyloid (1–40) to the anionic membrane, a uniform binding of the peptide to the membrane took place, as revealed by an elevated and featureless fluorescence across the entire field of view of ∼120 μm in diameter (see Fig. 1 b, and for comparison, a background image with supported membrane only is shown in Fig. 1 a. The variation in the intensity across the field in both images corresponds to the TIRF illumination gradient). The duration of the uniform binding phase (i.e., before discrete structures develop) ranged from <2 h (for high peptide concentration in solution, ∼100 nM) to a few days (for low peptide concentration in solution, ∼2 nM).
Figure 1.
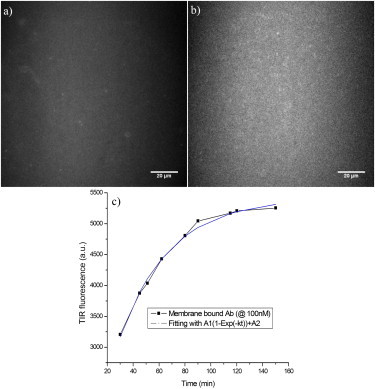
(a) TIRF image of background fluorescence (taken with supported POPC/POPG lipid membrane only). (b) TIRF image of HiLyte Fluor 488 β-amyloid (1–40) peptide bound to POPC/POPG lipid membrane. The concentration of β-amyloid (1–40) peptide in solution is 100 nM. (c) The kinetics of the same concentration of β-amyloid (1–40) peptide binding to the membrane. The data could be fit to a single exponential very well, indicating a first-order binding kinetics.
Binding kinetics
The initial, uniform, binding of the peptide to the membrane was found to be slow, as indicated by the slow increase in the observed mean fluorescence intensity from the membrane-bound β-amyloid (1–40) (Fig. 1 c). This binding is well fit by a single exponential, suggesting the process obeys first-order kinetics.
The following model is proposed for the binding process:
We can further simplify this model based on the following two observations made in our experiments.
-
1.
koff is extremely small. In our experiments, after the aqueous phase containing unbound β-amyloid (1–40) was washed off extensively and replaced by peptide-free buffer, the fluorescence intensity of the membrane-bound peptide did not noticeably decrease even after 130 h. This sets the upper limit of koff at below 2 × 10−6s−1.
-
2.
β-amyloid (1–40) saturates the membrane at very low peptide/phospholipid ratios. Crude measures of β-amyloid (1–40) concentration, via the fluorescence intensity from the solution, were done both before and after the peptide bound to the membrane. No measurable decrease was detected; indicating the majority of the peptide remained in solution. Furthermore, a higher concentration of solution peptide up to 100 nM did not increase the binding of β-amyloid (1–40) to the membrane, as indicated by the lack of change in the fluorescence intensity from the membrane-bound peptide regardless of aqueous peptide concentration between 2 and 100 nM. These observations suggest that the fraction of peptide that bound to the membrane was extremely small ([Solution Aβ] ≈ [Solution Aβ]0 ≫ [Membrane Bound Aβ]). A more direct estimate of the bound peptide, based on the density of the membrane-bound β-amyloid (1–40) peptide confirms this conclusion and is provided in the Discussion.
Combining the previous two observations, the binding of β-amyloid (1–40) to the lipid membrane can be presented as a simplified irreversible process:
Therefore, the binding process shown in Fig. 1 c is described by a single exponential curve A1(1-exp(−kon[Added Aβ] × t)) + A2 (A1 represents the fluorescence intensity when the membrane-bound β-amyloid peptide saturates, and A2 is the fluorescence baseline; i.e., autofluorescence from the membrane). kon= (4.3 ± 0.4) × 103 s−1M−1 was derived from the curve fitting. Various concentrations (20, 50, and 100 nM) of β-amyloid (1–40) were tested, and a consistent kon was found. Data from 100 nM peptide are shown in Fig. 1 c, and data from other concentrations can be found in Fig. S1 in the Supporting Material. With the estimated upper limit for koff (<2 × 10−6s−1), an upper limit for the dissociation constant of the peptide from the membrane can be calculated: Kd = koff /kon <470 pM. This extremely low Kd reveals a very tight binding of β-amyloid (1–40) to the anionic membrane, albeit saturating at a very low peptide/lipid ratio. This Kd is much lower than the value reported in our previous study (23) in that the previous Kd was determined at much higher peptide concentrations (micromolar) where a second binding process involving a much higher peptide/lipid ratio occurs, which is very different from the tight binding at subnanomolar peptide concentrations reported in the current study.
The membrane-bound β-amyloid (1–40) is very mobile
The mobility of lipid molecules as well as of the membrane-bound β-amyloid (1–40) peptide in the membrane was measured by FRAP. A circular region in the membrane was photobleached, and the fluorescence recovery via lateral diffusion was then recorded. Fig. 2, a–d, show four typical FRAP images. To measure the mobility of the lipid molecules, the membrane was made using lipids containing 0.5% NBD-POPC thereby allowing for fluorescence imaging. The diffusion coefficient was derived by fitting of the recovery curve to the model described in Methods. Fig. 2 e shows the recovery curve of the membrane-bound β-amyloid (1–40) peptide and the corresponding curve fitting.
Figure 2.
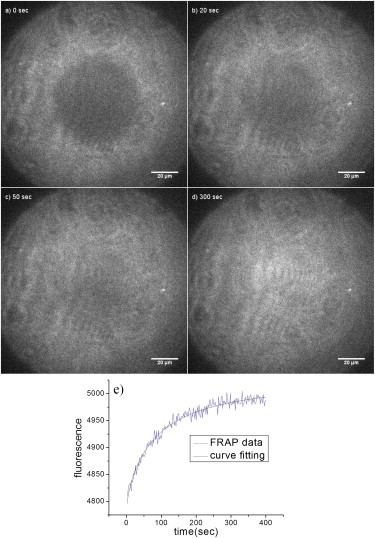
FRAP of a POPC/POPG lipid bilayer. The bilayer was mixed with 0.5% NBD-POPC for fluorescence photobleaching and recovery. Snapshot images at 0 s, 20 s, 50 s, and 300 s are shown in a–d. (e) The fluorescence recovery curve was fit to the equation to extract the diffusion coefficient.
FRAP measurements of the lipid molecules and the membrane-bound β-amyloid (1–40) peptide yielded comparable diffusion coefficients: 1.6 ± 0.1 10−8cm2/s and 2.3 ± 0.2 10−8cm2/s, respectively. This shows that the membrane-bound β-amyloid (1–40) is very mobile, with a diffusion coefficient similar to that of the lipid molecules.
Oligomer formation
Fig. 3 a shows the membrane after incubation with 2 nM of β-amyloid (1–40) peptide for 20 h and then washed off showing no oligomer formation. Prolonged incubation of the membrane-bound peptide led to slow oligomer formation (Fig. 3, b–d). These oligomers appeared as bright spots in the fluorescence images. Inspection of these images revealed that the uniformly bound peptide remained attached to the membrane during oligomer formation, as indicated by the presence of elevated fluorescence backgrounds in all the images. Contrary to the uniformly bound peptide, which showed good mobility in the membrane, the oligomers appeared to be immobilized based on time lapse observation up to 30 min.
Figure 3.

(a) Fluorescence image of the membrane-bound β-amyloid (1–40) peptide, after incubation with 2 nM of β-amyloid (1–40) peptide for 20 h and then washed off, before oligomers formation was observed. (b–d) Fluorescence images showing oligomer formations in the membrane with different concentrations of β-amyloid (1–40) peptide in solution: (b) oligomers formed with 0 nM β-amyloid (1–40) peptides in solution for 130 h; (c) oligomers formed with 2 nM β-amyloid (1–40) peptides in solution for 130 h; (d) oligomers formed with 100 nM β-amyloid (1–40) peptides in solution for 3 h.
Peptide concentration dependence of oligomer formation
Our experiments reveal that the rate of formation of membrane-bound oligomers depends on the concentration of the β-amyloid (1–40) peptide in solution. Experiments using three peptide concentrations (0, 2, and 100 nM in solution) suggested that oligomer formation most likely occurs via two different mechanisms. It should be noted that preincubation of the lipid membrane with β-amyloid (1–40) was performed in all the experiments, as oligomer formation was observed to occur only after a uniform binding of the peptide to the membrane (even for the 0 nM experiments, 2 nM of peptide was incubated with lipid membranes for 20 h before the peptide was washed off extensively, Fig. 3 a).
Oligomer formation with peptide-free solution
When the solution β-amyloid (1–40) was washed off (after 20 h incubation with the membrane), the imaging of the membranes after 130 h revealed the presence of oligomers (Fig. 3 b). There were 34 ± 4.4 oligomers observed per field of view after the 130 h incubation (error bar calculation based on 10 fields of view, and each field of view is ∼120 μm in diameter).
Because there was no β-amyloid (1–40) available in solution, these oligomers clearly formed within (or on) the membrane from β-amyloid (1–40) peptide that was bound to the membrane during the preincubation.
Oligomer formation with low concentration (2 nM) of peptide in solution
When the preincubated membrane with 2 nM β-amyloid (1–40) peptide in solution was followed for 130 h, the oligomer formation (see in Fig. 3 c) was very similar to that seen in the absence of solution peptide (Fig. 3 b). The observed number of oligomers per field of view was 41 ± 6.9 in this case, only slightly higher than that observed with no peptide in solution (error bar calculation based on nine fields of view). The fluorescence intensities of the oligomers in both experiments were very similar as well, suggesting similar sizes as discussed in more detail below. We conclude that although the excess free β-amyloid (1–40) in solution can serve as a reservoir for oligomer formation, the oligomerization under these conditions is still mainly due to membrane-bound peptide in a similar mechanism to that of oligomer formation in the absence of solution peptide.
Oligomer formation with high concentration (100 nM) of peptide in solution
Monitoring the membrane with 100 nM β-amyloid (1–40) in solution for only 2.5 h revealed a dramatically increased level of oligomer formation (Fig. 3 d). Though the incubation time was much shorter, the density and the fluorescence intensities of the oligomers formed in the membrane were much higher than those in the previous two experiments. 132 ± 17 oligomers per field of view were observed after the short incubation (error bar calculation based on eight fields of view). It should be noted that, the gain of the electron-multiplying CCD camera was lowered in these experiments, which accounts for the darker background seen in Fig. 3 d. This much faster oligomer formation suggests that these oligomers were likely formed via a different mechanism.
Comparison of oligomer species
Although fluorescence intensities of individual membrane-bound oligomers can be used to assess their sizes, the illumination gradient caused by both the Gaussian profile of the laser beam and the TIRF geometry makes the absolute intensities of the oligomers’ fluorescence a poor measure of their sizes. Thus, normalized fluorescence intensities were used to compare the different oligomer species.
As had been observed in the experiments, the uniformly bound β-amyloid (1–40) remained associated with the membrane during oligomer formation appearing as an elevated background. This background could be served as a reference for the normalization of the oligomer fluorescence intensities. Based on the rationale that smaller regions, such as 9 × 9 pixels in our images, are more likely to have approximately uniform illumination, the fluorescence intensity of a single pixel can be normalized to its averaged local background. Consequently, an oligomer can be represented as the integration of the normalized pixel intensities within its diffraction-limited area, giving a more accurate estimate of oligomer size. Thus, in the postnormalized images, the background regions that represent the mobile, uniformly bound peptide have normalized intensities of around unity, whereas the oligomers have normalized intensities well above unity.
By normalizing the fluorescence intensities, the oligomeric species in a given experiment can be compared. Moreover, based on our finding that the saturation level of membrane-bound β-amyloid (1–40) is the same regardless of peptide concentration in solution, the sizes of the oligomers in different experiments can also be compared through normalization. The background fluorescence in Fig. 3, c and d, showed very similar intensities, though Fig. 3 d appears to be darker due to different contrast and gain settings. Applying this approach we found that at low concentration of solution peptide only small oligomers were formed, with normalized intensities <12 (as shown in Fig. 4 a, which depicts the data of Fig. 3 c in normalized fluorescence intensities). In contrast, in the presence of a high concentration of solution peptide, much larger oligomers were formed, as indicated by much higher (up to around 100) normalized intensities (shown in Fig. 4 b, which presents the data of Fig. 3 d in normalized fluorescence intensities).
Figure 4.
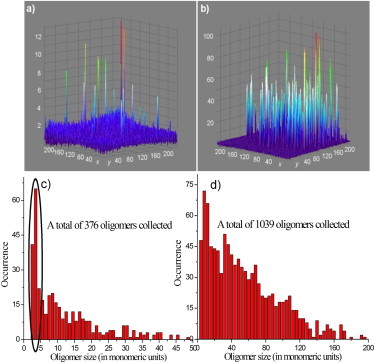
Comparison of the β-amyloid (1–40) oligomers formed at low concentration (2 nM) in a and c, and high concentration (100 nM) in b and d. In a and b, three-dimensional images of oligomers in the membranes are presented, with z axis indicating the normalized fluorescence intensity, and x and y axes representing the pixel numbers of the image. Please see the text for detailed explanation of normalized fluorescence intensity. In c and d, histograms of β-amyloid (1–40) oligomers are compared in oligomeric units. It should be noted that, a significant fraction (34.0%) of the oligomers formed at low peptide concentration, as highlighted in c, are smaller species (trimers/tetramers).
Determination of oligomer species
We showed that the oligomer species are correlated with their normalized fluorescence intensities. However, this normalization did not give a real estimation of oligomer size. On the other hand, single molecule photobleaching has been used to determine in a direct way the number of subunits in molecular assemblies (48,49). By counting the number of steps in photobleaching trajectories, the sizes of spin-coated β-amyloid (1–40) oligomers have been unambiguously determined (47).
We note that the usual photobleaching methodology (47–49) did not yield clear results in the current study due to the presence of mobile β-amyloid (1–40) peptide that contributes to the photobleaching trajectory. However, the individual photobleaching events that can often be identified in the trajectories provided an accurate means of estimating the size of monomeric units of β-amyloid peptide, because a discrete photobleaching step corresponds to the loss of fluorescence from a single β-amyloid peptide molecule (a selection of photobleaching events can be found in Fig. S2, a–f). By comparing the photobleaching fluorescence loss with its local background, we can further calibrate the normalized fluorescence intensities. With the background fluorescence being represented as unity in the normalization process, the ratio of background fluorescence over photobleaching loss yields the number of monomeric β-amyloid units per normalized fluorescence unity. A total of 121 single-step photobleaching events were selected, and an average of 2.5 ± 0.9 monomeric units per normalized fluorescence unit was calculated from the calibration (distribution of monomeric units/normalized fluorescence unit from the selected photobleaching events can be found in Fig. S2 g).
Based on the calibration of the normalized fluorescence unit, the oligomer sizes could be determined with a reasonable degree of confidence (oligomer size = normalized intensity × 2.5). Fig. 4, c and d, show the histograms of oligomeric species formed under different experimental conditions: low β-amyloid peptide concentration (2 nM) in solution in (c), and high peptide concentration (100 nM) in (d). The comparison of the histograms not only shows a large difference in size between the oligomers formed at different peptide concentrations, but also indicates that even under the same experimental condition; there is a large variation in the oligomeric species. Furthermore, among the oligomers formed at low peptide concentration, a significant fraction (34.0%) contains smaller species, as highlighted in Fig. 4 c. These smaller species are estimated to be mainly trimers and tetramers. In fact, high-resolution atomic force microscopy imaging has also revealed similar small oligomer species in membranes (17,22). These species may be the smallest stable oligomers in the membrane and, therefore, could be the origin of pore formation and neurotoxicity. It should be noted that, due to possible fluorophore self-quenching, the sizes of these oligomers may be underestimated, especially for the large species. However, we also note that for HiLyte Fluor 488 β-amyloid (1–40) oligomers sizes up to 20, self-quenching as determined by fluorescence lifetime was not observed.
Discussion
Though there is extensive evidence to support the role of membrane-associated β-amyloid oligomers in AD, the molecular mechanism by which these structures form and the origin of their neurotoxicity remain poorly understood (15–20). More detailed studies of peptide membrane interactions are therefore critical for understanding the underlying mechanisms. Because β-amyloid peptides are found in extremely low physiological concentration and can form heterogeneous and dynamic oligomeric species, these mechanistic studies are challenging for traditional techniques. In our research, single-molecule fluorescence imaging techniques are used to study the peptide membrane interactions.
Our results show that the initial interactions between β-amyloid (1–40) and anionic membranes lead to a very tight and uniform binding of the peptide to the membrane (Fig. 1 b), but at a very low peptide/lipid ratio, followed by the slow formation of a variety of oligomer species in the membrane (Fig. 3, b–d).
A two-state model for peptide membrane interaction
A two-state model has been proposed in the literature for peptide membrane interaction (37,38). Though originally developed specifically for antimicrobial peptides, the model is general and applicable for other peptide membrane interactions.
In this model, membrane-bound peptides exist in two different states: the membrane-bound monomeric state and the oligomeric state. It has been shown that these two states are directly related to the peptide/lipid ratio, which can be defined as the number of membrane-bound peptide over the number of lipid molecules. At low peptide/lipid ratio, the membrane-bound peptide is in the monomeric state, and is at the interface between the hydrophilic headgroups and hydrophobic regions of the lipid, with an orientation parallel to the membrane surface (37,38). When the peptide/lipid ratio is above the threshold value, the peptide oligomerizes in the membrane and assumes a perpendicular orientation relative to the membrane surface.
Though the long timescales and decreasing signal/noise did not allow us to identify the threshold value for the peptide/lipid ratio (<10−5), our results for β-amyloid (1–40) interacting with anionic membranes show behavior that at low concentrations is generally compatible with the previous model. During the initial incubation of β-amyloid with the membrane, where earlier studies have shown β-amyloid assumes a α-helical structure (46), saturation occurs at a very low peptide/lipid ratio but with very high affinity and all the membrane-bound peptide is highly mobile and uniformly distributed in the membrane. Further incubation of the membrane-bound β-amyloid (1–40) leads to the appearance of oligomers in the membrane. The immobility of these oligomers indicates that their constituent peptides are inserted into the membrane. This transition from a mobile monomeric state to an immobile oligomeric one is in accordance with the two-state model. Other studies of molecular tracking show that the largest mobile species is a dimer (C. Chang, D. G. Steel, and A. Gafni, unpublished). In addition, our previous study using circular dichroism (46) has revealed that the small membrane-bound peptide oligomers adopt a mostly helical structure, whereas as larger oligomers develop there is a shift to beta-structure, which can also be evidenced by thioflavin T binding. Previous NMR studies of β-amyloid (1–40) in micellar environment have also shown that the peptide’s C-terminus adopts a helical structure, with the length of the peptide (∼4 nm) spanning the membrane (50). This is comparable with the thickness of a lipid bilayer. Therefore, these perpendicularly oriented oligomers are likely to be anchored by the underlying glass substrate, explaining their immobility. As we discuss below, at higher concentration, our results deviate considerably from this model.
Membrane-bound β-amyloid (1–40) peptide
Our results show that during the initial, uniform, binding phase of β-amyloid (1–40), the fraction of the solution-peptide that binds to the membrane is small, and that membrane saturation occurs at an extremely low peptide/lipid ratio (below 10−5, see discussion below). Because the membrane-bound β-amyloid (1–40) peptide is mobile and evenly distributed, they appear as elevated fluorescence backgrounds in the images (Fig. 1 b). The data show (see Results) that they remain attached to the membrane during oligomer formation.
This elevated fluorescence background has a normalized fluorescence intensity of unity, which suggests the density of the membrane-bound β-amyloid (1–40) peptide to be 2.5 ± 0.9 monomeric units per pixel, or 8.8 ± 3.2 monomeric units/μm2 via the calibration of the normalized fluorescence. The average surface area of a lipid molecule in the membrane is ∼70Å2 (51,52), making for ∼1.43 × 106 lipid molecules/μm2. This reveals that β-amyloid (1–40) membrane concentration at saturation is extremely low, with peptide/lipid ratio of ∼10−5−10−6. This low ratio supports our experimental observation that the fraction of solution β-amyloid (1–40) that binds to the membrane is extremely small. Thus, it can be calculated from this low peptide/lipid ratio that, even with 2 nM peptide in solution, the majority of the peptide (99.8%) was still in solution after the membrane binding sites were saturated.
High affinity binding of β-amyloid (1–40) peptide to black lipid membranes, with similar peptide density, has been observed (46). Murphy et. al. (53) have also reported a similar binding of β-amyloid (1–40) peptide to lipid vesicles using surface plasmon resonance. Isothermal titration calorimetry experiments conducted in our group also suggest a very tight binding of the peptide to the membrane at low peptide/lipid ratio conditions (data not shown). Clearly, this extremely tight and yet extremely low density binding of the peptide to the membrane is puzzling and a satisfactory molecular mechanism to explain it remains to be developed. It should be noted that selective β-amyloid binding to contaminants or impurities in the lipid is unlikely to explain the tight binding, because similar peptide binding densities have been observed on different model membranes with various lipid compositions (46,53).
Pathways of oligomer formation
It was shown under Results that oligomer formation in the membrane did not start until well after a uniform binding of β-amyloid (1–40) peptide to the anionic membrane took place. The concentration-dependent rate of oligomer formation suggests there are likely two different pathways for this process.
Upon washing off solution β-amyloid (1–40), oligomer formation in the membrane was still observed, albeit slowly. This indicates the membrane-bound peptide undergoes a transition from the monomeric state to the oligomeric state. It was also shown in Results that, with low peptide concentration in solution, the in-membrane transition pathway still dominates oligomer formation.
On the basis of the peptide surface density and the diffusion coefficient of the membrane-bound mobile peptide (assuming it to be monomeric), we calculated the collision frequency of the mobile peptide in the membrane using the equation (54,55), where Φ is the collision frequency per unit area, C is the density of the peptide (8.8 × 108/cm2), D is the two-dimensional diffusion coefficient (2.3 × 10−8cm2/s), and a is the collision radius, which can be approximated as the cross-section diameter of β-amyloid (1–40) oligomers. From the sizes of the oligomeric pore reported in the literature (17,22), a is on the order of ten nanometers. This calculation yields ∼7×1010 collisions/cm2/s for the monomeric peptide diffusing in the membrane, giving ∼90 collisions/s for each monomer. It is thus clear that the pathway leading to oligomer formation by membrane-bound peptide is far from a diffusion-controlled process, because the collision frequency between the mobile peptide molecules is much higher than the number of oligomers observed on our measurement of (0.7 oligomers/cm2/s). It is possible that the oligomerization of the membrane-bound peptide may require some specific orientations or more subunits during peptide collision in the membrane. It is also possible that the actual collision frequency of membrane-bound peptide is much less than the previous estimate. The two-state model suggests that the membrane will undergo a local thinning/deformation upon peptide adsorption. Because the local membrane thinning/deformation is repulsive due to a high energy cost for overlapping these areas, this may create barriers around the membrane-bound peptide molecules, and prevent them from colliding with each other to form stable oligomers (37,38).
In the presence of a high peptide concentration in solution, however, the rate of oligomer formation in the membrane was found to be much higher. Because the surface density of the membrane-bound mobile peptide was the same for both high and low peptide concentrations in solution (in Fig. 3, c and d), the rate of oligomer formation via the membrane-peptide transition pathway has to be the same as well. This suggests that oligomer formation on the membrane via bound peptide diffusion cannot account for the much faster oligomer formation seen at high solution peptide. Clearly, a second pathway of oligomer formation has to exist, which likely involves either a direct insertion of solution peptide into a membrane-bound preexisting oligomer, or a rapid replenishing of membrane-bound monomeric peptides that have been incorporated into oligomeric species. Indeed, oligomer formation at a low solution peptide concentration of 2 nM did not show any contribution from the direct insertion pathway, although the latter pathway dominated at 100 nM solution peptide. The fast forming oligomers may further serve as nuclei for subsequent binding of β-amyloid peptide to the membrane, leading to more severe membrane permeabilization, or even initiating β-amyloid fibril formation. Indeed, mesh-like β-amyloid deposits were observed after prolonged incubation of 100 nM β-amyloid (1–40) peptide with POPC/POPG lipid membrane for 20 h (data can be found in Fig. S3). Therefore, the studies of these smaller species and their membrane permeabilization may provide new, to our knowledge, insights into the molecular mechanisms underlying the failure of cellular homeostasis in AD.
Fast-forming amyloid (1–40) have recently also been reported to develop during a short (∼10 min) incubation of 50 nM peptide with SHSY5Y neuroblastoma cells (56). As found in the current work, no oligomers were detected in solution revealing that the cell membrane also greatly facilitates the rate of oligomerization, and supports the further growth of the oligomers because cell-bound oligomers were found to be significantly larger than those formed on the surface of the glass slide. It should be noted that the rate of on-cell oligomerization was much higher than that here reported, with oligomers appearing within minutes. Whether this is due solely to intrinsic differences between the model membrane and the much more complex cell membrane or to differences in experimental conditions (the cells were kept in growth medium, which contains a variety of metal ions, nutrients, and proteins that are known to facilitate oligomer formation) is of interest and is the subject of current studies.
Conclusions
Although the membrane-disruption by bound β-amyloid oligomer hypothesis has been supported by recent evidence, there is a lack of understanding of the underlying mechanism at the molecular level. Here, we used single-molecule studies to follow the interactions between β-amyloid (1–40) and anionic bilayer lipid membrane, and the subsequent oligomer formation. Our results lead us to conclude that the interaction between β-amyloid (1–40) and anionic membrane starts with an extremely tight binding of monomeric peptide, and that further incubation leads to oligomer formation in the membrane. The sizes of the formed oligomers are dependent on the concentration of β-amyloid (1–40) peptide in solution. Specifically, at low nanomolar concentrations of β-amyloid (1–40), a range that is of physiological significance, a large fraction of the formed oligomers exist as small species (trimer/tetramer), suggesting a possible mechanism for the origin of pore-forming oligomers. Single-molecule imaging is uniquely suited to the identification of early β-amyloid oligomers on biological membranes, their evolution, and membrane interactions. The approaches described in this work, when applied to live cell membranes, should provide a more detailed understanding of the molecular mechanism of AD.
Acknowledgments
This research was supported by National Institutes of Health (NIH) grant R21 AG027370 and by a Senior Scholar Award from the Ellison Medical Foundation.
Supporting Material
References
- 1.Alzheimer A., Stelzmann R.A., Murtagh F.R. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 1995;8:429–431. doi: 10.1002/ca.980080612. [DOI] [PubMed] [Google Scholar]
- 2.Kidd M. Paired helical filaments in electron microscopy of Alzheimer’s disease. Nature. 1963;197:192–193. doi: 10.1038/197192b0. [DOI] [PubMed] [Google Scholar]
- 3.Terry R.D., Gonatas N.K., Weiss M. Ultrastructural studies in Alzheimer’s presenile dementia. Am. J. Pathol. 1964;44:269–297. [PMC free article] [PubMed] [Google Scholar]
- 4.Glenner G.G., Wong C.W. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 5.Selkoe D.J., Abraham C.R., Duffy L.K. Isolation of low-molecular-weight proteins from amyloid plaque fibers in Alzheimer’s disease. J. Neurochem. 1986;46:1820–1834. doi: 10.1111/j.1471-4159.1986.tb08501.x. [DOI] [PubMed] [Google Scholar]
- 6.Masters C.L., Simms G., Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Serpell L.C., Smith J.M. Direct visualisation of the beta-sheet structure of synthetic Alzheimer’s amyloid. J. Mol. Biol. 2000;299:225–231. doi: 10.1006/jmbi.2000.3650. [DOI] [PubMed] [Google Scholar]
- 8.Lührs T., Ritter C., Riek R. 3D structure of Alzheimer’s amyloid-beta(1–42) fibrils. Proc. Natl. Acad. Sci. USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lue L.F., Kuo Y.M., Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am. J. Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gravina S.A., Ho L., Younkin S.G. Amyloid beta protein (A beta) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at A beta 40 or A beta 42(43) J. Biol. Chem. 1995;270:7013–7016. doi: 10.1074/jbc.270.13.7013. [DOI] [PubMed] [Google Scholar]
- 11.Mori H., Takio K., Selkoe D.J. Mass spectrometry of purified amyloid beta protein in Alzheimer’s disease. J. Biol. Chem. 1992;267:17082–17086. [PubMed] [Google Scholar]
- 12.Roher A.E., Lowenson J.D., Ball M.J. beta-Amyloid-(1–42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc. Natl. Acad. Sci. USA. 1993;90:10836–10840. doi: 10.1073/pnas.90.22.10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hardy J., Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 14.Mudher A., Lovestone S. Alzheimer’s disease-do tauists and baptists finally shake hands? Trends Neurosci. 2002;25:22–26. doi: 10.1016/s0166-2236(00)02031-2. [DOI] [PubMed] [Google Scholar]
- 15.Demuro A., Mina E., Glabe C.G. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 2005;280:17294–17300. doi: 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- 16.Kourie J.I., Culverson A.L., Laohachai K.N. Heterogeneous amyloid-formed ion channels as a common cytotoxic mechanism: implications for therapeutic strategies against amyloidosis. Cell Biochem. Biophys. 2002;36:191–207. doi: 10.1385/CBB:36:2-3:191. [DOI] [PubMed] [Google Scholar]
- 17.Lin H., Bhatia R., Lal R. Amyloid beta protein forms ion channels: implications for Alzheimer’s disease pathophysiology. FASEB J. 2001;15:2433–2444. doi: 10.1096/fj.01-0377com. [DOI] [PubMed] [Google Scholar]
- 18.Arispe N., Pollard H.B., Rojas E. Giant multilevel cation channels formed by Alzheimer disease amyloid beta-protein [A beta P-(1–40)] in bilayer membranes. Proc. Natl. Acad. Sci. USA. 1993;90:10573–10577. doi: 10.1073/pnas.90.22.10573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shankar G.M., Li S., Selkoe D.J. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klyubin I., Betts V., Rowan M.J. Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J. Neurosci. 2008;28:4231–4237. doi: 10.1523/JNEUROSCI.5161-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dong Z., Saikumar P., Venkatachalam M.A. Calcium in cell injury and death. Annu. Rev. Pathol. 2006;1:405–434. doi: 10.1146/annurev.pathol.1.110304.100218. [DOI] [PubMed] [Google Scholar]
- 22.Quist A., Doudevski I., Lal R. Amyloid ion channels: a common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA. 2005;102:10427–10432. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong P.T., Schauerte J.A., Gafni A. Amyloid-beta membrane binding and permeabilization are distinct processes influenced separately by membrane charge and fluidity. J. Mol. Biol. 2009;386:81–96. doi: 10.1016/j.jmb.2008.11.060. [DOI] [PubMed] [Google Scholar]
- 24.Terzi E., Hölzemann G., Seelig J. Alzheimer beta-amyloid peptide 25–35: electrostatic interactions with phospholipid membranes. Biochemistry. 1994;33:7434–7441. doi: 10.1021/bi00189a051. [DOI] [PubMed] [Google Scholar]
- 25.Terzi E., Hölzemann G., Seelig J. Self-association of beta-amyloid peptide (1–40) in solution and binding to lipid membranes. J. Mol. Biol. 1995;252:633–642. doi: 10.1006/jmbi.1995.0525. [DOI] [PubMed] [Google Scholar]
- 26.McLaurin J., Chakrabartty A. Characterization of the interactions of Alzheimer beta-amyloid peptides with phospholipid membranes. Eur. J. Biochem. 1997;245:355–363. doi: 10.1111/j.1432-1033.1997.t01-2-00355.x. [DOI] [PubMed] [Google Scholar]
- 27.Del Mar Martínez-Senac M., Villalaín J., Gómez-Fernández J.C. Structure of the Alzheimer beta-amyloid peptide (25–35) and its interaction with negatively charged phospholipid vesicles. Eur. J. Biochem. 1999;265:744–753. doi: 10.1046/j.1432-1327.1999.00775.x. [DOI] [PubMed] [Google Scholar]
- 28.Bokvist M., Lindström F., Gröbner G. Two types of Alzheimer’s beta-amyloid (1–40) peptide membrane interactions: aggregation preventing transmembrane anchoring versus accelerated surface fibril formation. J. Mol. Biol. 2004;335:1039–1049. doi: 10.1016/j.jmb.2003.11.046. [DOI] [PubMed] [Google Scholar]
- 29.Ege C., Lee K.Y.C. Insertion of Alzheimer’s A beta 40 peptide into lipid monolayers. Biophys. J. 2004;87:1732–1740. doi: 10.1529/biophysj.104.043265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ikeda K., Matsuzaki K. Driving force of binding of amyloid beta-protein to lipid bilayers. Biochem. Biophys. Res. Commun. 2008;370:525–529. doi: 10.1016/j.bbrc.2008.03.130. [DOI] [PubMed] [Google Scholar]
- 31.Kremer J.J., Sklansky D.J., Murphy R.M. Profile of changes in lipid bilayer structure caused by beta-amyloid peptide. Biochemistry. 2001;40:8563–8571. doi: 10.1021/bi010417x. [DOI] [PubMed] [Google Scholar]
- 32.Yanagisawa M., Ariga T., Yu R.K. Cytotoxic effects of GM1 ganglioside and amyloid b-peptide on mouse embryonic neural stem cells. ASN Neuro. 2010;2:49–56. doi: 10.1042/AN20090063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kelly B.L., Ferreira A. beta-Amyloid-induced dynamin 1 degradation is mediated by N-methyl-D-aspartate receptors in hippocampal neurons. J. Biol. Chem. 2006;281:28079–28089. doi: 10.1074/jbc.M605081200. [DOI] [PubMed] [Google Scholar]
- 34.Pellistri F., Bucciantini M., Stefani M. Nonspecific interaction of prefibrillar amyloid aggregates with glutamatergic receptors results in Ca2+ increase in primary neuronal cells. J. Biol. Chem. 2008;283:29950–29960. doi: 10.1074/jbc.M803992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benilova I., Karran E., De Strooper B. The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat. Neurosci. 2012;15:349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- 36.Campioni S., Mannini B., Chiti F. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 2010;6:140–147. doi: 10.1038/nchembio.283. [DOI] [PubMed] [Google Scholar]
- 37.Huang H.W. Action of antimicrobial peptides: two-state model. Biochemistry. 2000;39:8347–8352. doi: 10.1021/bi000946l. [DOI] [PubMed] [Google Scholar]
- 38.Huang H.W. Molecular mechanism of antimicrobial peptides: the origin of cooperativity. Biochim. Biophys. Acta. 2006;1758:1292–1302. doi: 10.1016/j.bbamem.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 39.Axelrod D., Burghardt T.P., Thompson N.L. Total internal reflection fluorescence. Annu. Rev. Biophys. Bioeng. 1984;13:247–268. doi: 10.1146/annurev.bb.13.060184.001335. [DOI] [PubMed] [Google Scholar]
- 40.Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Traffic. 2001;2:764–774. doi: 10.1034/j.1600-0854.2001.21104.x. [DOI] [PubMed] [Google Scholar]
- 41.Moerner W.E., Fromm D.P. Methods of single-molecule fluorescence spectroscopy and microscopy. Rev. Sci. Instrum. 2003;74:3597–3619. [Google Scholar]
- 42.Morrissey Lab Protocol for Preparing Phospholipid Vesicles (SUV) by Sonication. Avanti Polar Lipids. http://avantilipids.com/images/PDF/MorrisseyLabProtocolForPrepSuvBySonication.pdf. Accessed 2008.
- 43.Cremer P.S., Boxer S.G. Formation and spreading of lipid bilayers on planar glass supports. J. Phys. Chem. 1999;103:2554–2559. [Google Scholar]
- 44.Axelrod D., Koppel D.E., Webb W.W. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys. J. 1976;16:1055–1069. doi: 10.1016/S0006-3495(76)85755-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soumpasis D.M. Theoretical analysis of fluorescence photobleaching recovery experiments. Biophys. J. 1983;41:95–97. doi: 10.1016/S0006-3495(83)84410-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schauerte J.A., Wong P.T., Gafni A. Simultaneous single-molecule fluorescence and conductivity studies reveal distinct classes of Abeta species on lipid bilayers. Biochemistry. 2010;49:3031–3039. doi: 10.1021/bi901444w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ding H., Wong P.T., Steel D.G. Determination of the oligomer size of amyloidogenic protein β-amyloid(1–40) by single-molecule spectroscopy. Biophys. J. 2009;97:912–921. doi: 10.1016/j.bpj.2009.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coles M., Bicknell W., Craik D.J. Solution structure of amyloid beta-peptide(1–40) in a water-micelle environment. Is the membrane-spanning domain where we think it is? Biochemistry. 1998;37:11064–11077. doi: 10.1021/bi972979f. [DOI] [PubMed] [Google Scholar]
- 49.Shu D., Zhang H., Guo P. Counting of six pRNAs of phi29 DNA-packaging motor with customized single-molecule dual-view system. EMBO J. 2007;26:527–537. doi: 10.1038/sj.emboj.7601506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ulbrich M.H., Isacoff E.Y. Subunit counting in membrane-bound proteins. Nat. Methods. 2007;4:319–321. doi: 10.1038/NMETH1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lantzsch G., Binder H., Heerklotz H. Surface area per molecule in lipid/C12En membranes as seen by fluorescence resonance energy transfer. J. Fluoresc. 1994;4:339–343. doi: 10.1007/BF01881452. [DOI] [PubMed] [Google Scholar]
- 52.Burke L.I., Patil G.S., Cornwell D.G. Surface areas of naturally occurring lipid classes and the quantitative microdetermination of lipids. J. Lipid Res. 1973;14:9–15. [PubMed] [Google Scholar]
- 53.Kremer J.J., Murphy R.M. Kinetics of adsorption of beta-amyloid peptide Abeta(1–40) to lipid bilayers. J. Biochem. Biophys. Methods. 2003;57:159–169. doi: 10.1016/s0165-022x(03)00103-9. [DOI] [PubMed] [Google Scholar]
- 54.Hardt S.L. Rates of diffusion controlled reactions in one, two and three dimensions. Biophys. Chem. 1979;10:239–243. doi: 10.1016/0301-4622(79)85012-7. [DOI] [PubMed] [Google Scholar]
- 55.Gupte S., Wu E.S., Hackenbrock C.R. Relationship between lateral diffusion, collision frequency, and electron transfer of mitochondrial inner membrane oxidation-reduction components. Proc. Natl. Acad. Sci. USA. 1984;81:2606–2610. doi: 10.1073/pnas.81.9.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Johnson R.D., Schauerte J.A., Steel D.G. Direct observation of single amyloid-β(1–40) oligomers on live cells: binding and growth at physiological concentrations. PLoS ONE. 2011;6:e23970. doi: 10.1371/journal.pone.0023970. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.