

Abstract
Centrosome overduplication or amplification has been observed in many human cancers and in premalignant tissue, but the mechanisms leading to such centrosome aberrations are not fully understood. We previously showed that abnormal mitotic cells with supernumerary centrosomes increase with replicative senescence in human fibroblasts, especially in a polyploid subpopulation. This study examines localization of p53 protein at centrosomes in mitotic cells, which is often observed in association with DNA damage response, to investigate a possible association between p53 localization and numerical centrosome aberrations induced by cellular senescence. Cultures at later passages or the 4th day after exposure to H2O2 showed increased frequencies of mitotic cells with supernumerary centrosomes, especially in a polyploid subpopulation. Immunohistochemical analysis frequently showed p53-positive foci in mitotic cells, and some were localized at centrosomes. The number of p53-positive foci in mitotic cells and its localization to centrosomes increased with replicative and premature senescence. Supernumerary centrosomes showed higher frequencies of p53 localization compared to normally duplicated centrosomes. Centrosome-associated p53 protein was phosphorylated at Ser15. These data suggest a possible association between localization of p53 protein and numerical centrosome aberrations in replicatively or prematurely senescent cells.
1. Introduction
Centrosome overduplication or centrosome amplification has been observed in many human cancers and in premalignant tissue [1–5]. Centrosome aberrations have been linked to genomic instability during tumour progression, because centrosomes dysfunction may cause abnormal spindle assembly and chromosome missegregation, leading to chromosome aneuploidy [1, 3, 5]. Some studies showed that genetic manipulations that cause centrosome amplification in vivo can induce tumor development [6, 7]. The mechanisms leading to centrosome aberrations have been studied extensively but still are not fully understood. However, DNA damage-induced centrosome aberrations such as inactivation, amplification, or fragmentation have been reported. For example, inhibitors of DNA replication such as aphidicolin or hydroxyurea cause centrosome inactivation or centrosome splitting, leading to spindle defects [8–10]. Deficiency of Rad51 recombinase, which is essential for DNA repair, causes supernumerary centrosomes [11]. Radiation causes centrosome amplification in tumour cells and in p53-inactivated or ATR-deficient human cells [12–14]. Several studies have suggested that centrosomal abnormalities resulting from DNA damage response cause mitotic errors and cell death, thus preventing the propagation of damaged cells that might be transformed into malignant cells. However, it is still not clear whether centrosome aberrations are part of the defence mechanism that inhibits carcinogenesis or undesirable pathological phenomena.
Our previous study showed that abnormal mitotic cells with supernumerary (>2/cell) centrosomes increase with replicative senescence in human fibroblasts, especially in a polyploid subpopulation [15]. Moreover, such numerical centrosome aberrations correlated significantly with chromosome misalignment in metaphase cells, suggesting that chromosomal instability with aging might be attributable to centrosome aberrations that are induced with cellular aging.
Recent studies have shown that several proteins involved in the DNA damage response, such as BRCA1, ATM, and p53, localize at centrosomes and regulate cell cycle or centrosome duplication [16–19]. P53 localizes at centrosomes during mitosis, and Ser15 phosphorylation of p53 by ATM, which is a DNA damage response, is required for this localization [20]. Centrosomally localized p53 regulates centrosome duplication in a manner independent of its transactivation function [17]. This study investigated a possible association between p53 localization and numerical centrosome aberrations in senescent cells by examining the localization of p53 protein at centrosomes in mitotic cells from young and near-senescent human fibroblasts. In particular, the effect of polyploidization during cellular senescence on centrosome aberrations and centrosomal localization of p53 was investigated.
2. Materials and Methods
TIG-1 normal human fibroblast cells (21 population doubling levels) were obtained from the Health Science Resources Bank (Tokyo, Japan) and grown in minimum essential medium with α modification (MEM-α; Sigma-Aldrich Co., St. Louis, MO, USA) supplemented with heat-inactivated 10% (v/v) fetal bovine serum. Cells were incubated in a 5% (v/v) CO2 atmosphere at 37°C and passaged every 3 or 4 days, so that cells never exceeded subconfluent density. For immunofluorescence studies, cells were seeded onto sterile glass slides, placed into culture dishes, and incubated for 2 days.
Replicative senescence was achieved by serial passaging until cells stopped proliferation. Premature senescence was induced by brief exposure of the cells to 200 μM H2O2 diluted in Hanks Balanced Salt Solution. The senescent phenotype was evaluated based on the doubling time by counting the cell number at each passage. In some experiments, expression of senescence-associated β-galactosidase (SA-β-gal) was examined using a Senescence Detection Kit (BioVision, CA, USA) to confirm the phenotype. To examine cell cycle profiles, DNA content of the cells was analyzed using CycleTEST Plus (Becton Dickinson Bioscience, USA) and a flow cytometer (FACSCanto II, Becton Dickinson Bioscience, USA).
To detect the localization of centrosomes and p53 protein, cells were fixed with 2% (v/v) formaldehyde, permeabilized with 0.25% (v/v) Triton X-100, and incubated for 1 h at room temperature with specific primary antibodies diluted in PBS containing 2% (w/v) BSA. Primary antibodies were mouse anti-α-tubulin (clone DM1A, 1 : 500 dilution, Sigma, T9025), rabbit anticentrin-2 (N-17, 1 : 800 dilution, Santa Cruz Biotechnology, SC-27793-R), mouse anti-p53 (clone DO-7, 1 : 100 dilution, Santa Cruz Biotechnology, SC-47698), mouse anti-p53 phosphorylated (pSer15), (clone 261352, 1 : 100 dilution, R&D Systems, MAB 1839), and rabbit anti-γ-H2AX (pSer139) (1 : 200 dilution, R&D Systems, AF2288). For fluorescent microscopy, cells treated with primary antibodies were incubated with Alexa Fluor 488-conjugated goat antimouse IgG (Molecular Probes, 1 : 500 dilution) and Alexa Fluor 555-conjugated goat antirabbit IgG (Molecular Probes, 1 : 500 dilution) for 1 h at room temperature. Cells were treated with RNase (1 mg/mL) for 30 min, and cell nuclei were counterstained with 5 μg/mL DAPI. Multiple slides were made from the same culture because the number of mitotic cells decreases as cells approach senescence. The specificity of the primary antibodies was confirmed by comparing it to the reaction with isotype control antibodies. In some experiments, another monoclonal antibody for p53, mouse anti-p53, clone DO-1 (Santa Cruz Biotechnology, SC-126), was also used to confirm the specificity of the reaction to p53 protein. Mouse monoclonal anti-p53 antibodies clone DO-7 and DO-1 are widely used and specificity of the antibodies has been established [21].
Cell cycle profiles were obtained by the use of a laser scanning cytometer (LSC-2, Olympus, Japan). DNA content was measured by DAPI fluorescence using violet (405 nm) laser excitation and a blue channel filter (460–485 nm), then subpopulations of interest were recalled, and localization of chromosomes, centrosomes, and p53 protein in mitotic cells was observed by DAPI, Alexa Fluor 555, and Alexa Fluor 488 fluorescence using appropriate filter sets, respectively. Centrosome number per mitotic cell and frequencies of centrosomal localization of p53 in mitotic cells were analyzed by preparing multiple slides from the same culture to obtain as many mitotic cells as possible.
3. Results
TIG-1 human fibroblasts showed growth retardation after 70 population doubling levels (PDLs) and entered a state of replicative senescence after 80 PDLs. Growth-arrested cells showed typical senescent phenotypes, such as enlarged size and β-galactosidase expression (Figure 1(a)). Cells exposed to 200 μM H2O2 showed the same phenotypes at the 4th day after exposure, indicating that premature senescence was induced by oxidative damage. Cell cycle analysis showed increased frequencies of polyploid cells (with 8C DNA) concomitant with replicative senescence and premature senescence (Figures 1(a), 1(b), and 1(c)).
Figure 1.
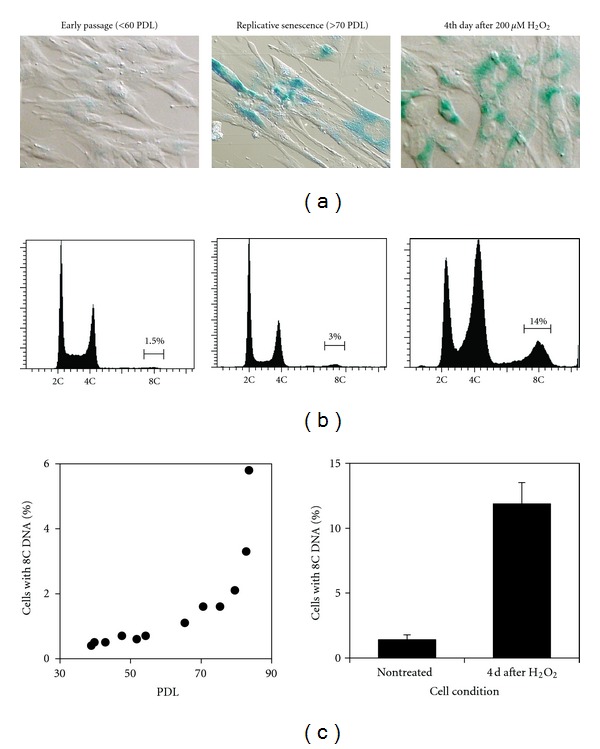
Polyploidization associated with cellular senescence in TIG-1 cells. (a) Differential interference contrast images overlaid with colouring reaction by senescence-associated β-galactosidase. (b) DNA histograms obtained by flow cytometer. (c) Change in frequencies of polyploid cells (with 8C DNA) by population doubling levels in serially passaged cells (left panel) and in cells exposed to 200 μM H2O2 (right panel).
Most diploid mitotic cells at early passages had two centrosomes, each of which consists of two centrioles recognized by anticentrin-2 antibody, with bipolar spindles. More than 60% of the polyploid mitotic cells (8C DNA or more) at early passages also had two centrosomes, although all other polyploid cells had more than two centrosomes (Figures 2(a) and 2(b)). In contrast, cells at later passages or the 4th day after exposure to H2O2 showed increased frequencies of mitotic cells with supernumerary (>2/cell) centrosomes, especially in a polyploid subpopulation (Figures 2(a) and 2(b)). These cells often showed incomplete spindles and misaligned chromosomes (Figure 2(a)). Most of the extra centrosomes with incomplete spindles also had two centrioles similar to centrosomes with bipolar spindles.
Figure 2.
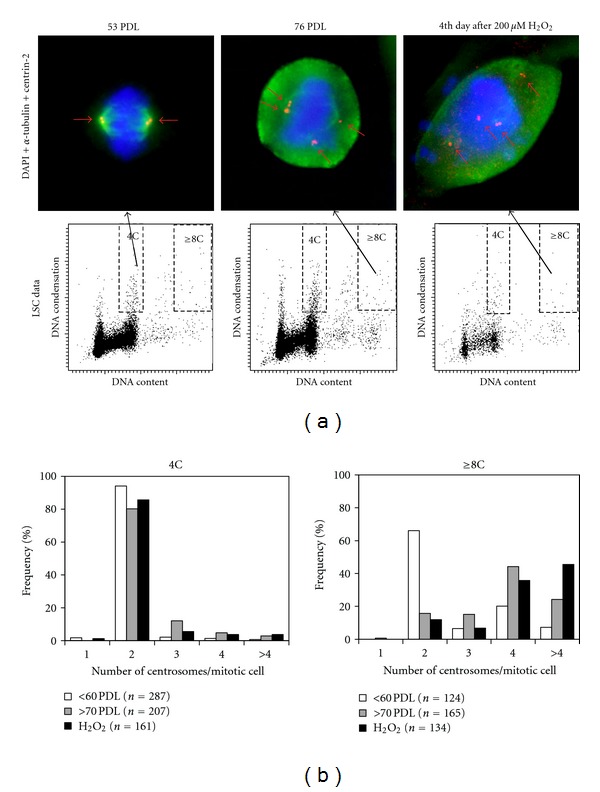
Numerical centrosome aberrations associated with cellular senescence. (a) Upper left panel shows a representative mitotic cell with normal morphology in a diploid subpopulation from early passage, and upper middle and upper right panels show abnormal mitotic cells with supernumerary centrosomes in a polyploid subpopulation from late passage and the 4th day after exposure to H2O2. Small red arrows indicate localization of centrosomes. Chromosomes, centrosomes, and mitotic spindles are labelled with DAPI (blue), anticentrin2 antibody (red), and anti-α-tubulin antibody (green), respectively. Lower panels show corresponding LSC data. (b) Histograms showing frequencies of mitotic cells with different centrosome numbers in a diploid subpopulation (4C) and a polyploid subpopulation (≥8C).
Immunohistochemical analysis showed that p53-positive foci were frequently present in mitotic and in interphase cells. Some foci were localized at centrosomes, and others were on the chromosomes (Figure 3(a)). P53 at centrosomes was always recognized as a single focus and appears to localize in the PCM (pericentriolar material) or on one of two centrioles in a centrosome. The mean number of p53-positive foci per cell was higher in the polyploid subpopulation compared to the diploid subpopulation (Figure 3(b)). Mitotic cells at the 4th day after exposure to H2O2 showed an increased number of p53-positive foci per cell in both subpopulations compared to untreated early passage cells (Figure 3(b)). The frequency of centrosomes with associated p53 protein was about 10% in untreated early passage cells regardless of DNA ploidy, while mitotic cells at later passages showed increased frequencies of p53-associated centrosomes, especially in diploid subpopulations (Figure 3(c)). Interestingly, supernumerary centrosomes showed higher frequencies of p53 localization compared to normally duplicated (2 centrosomes/cell) centrosomes, especially in diploid subpopulations irrespective of cell condition (Figure 3(d)). Centrosome-associated p53 foci were consistently recognized by antibody against p53 phosphorylated at Ser15 (Figure 4), while p53 foci on chromosomes were not phosphorylated at Ser15. These p53 foci on chromosomes were observed more often in the polyploid subpopulation compared to the diploid subpopulation, and they were often colocalized with phosphorylated histone H2AX (γ-H2AX) foci (data not shown).
Figure 3.
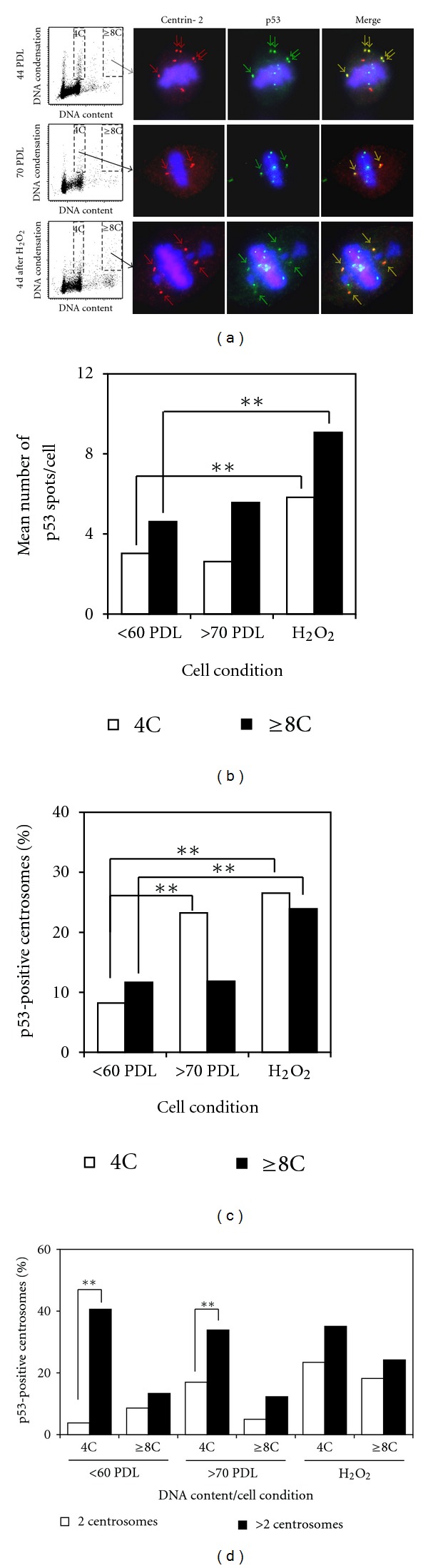
Localization of p53 protein in mitotic cells. (a) Representative mitotic cells showing localization of centrosomes and p53-positive foci from early passages (upper panels), late passages (middle panels), and the 4th day after exposure to H2O2 (lower panels). Arrows indicate centrosomes with p53 proteins. Chromosomes (blue) and centrosomes (red) are labelled as in Figure 2, and p53 protein is labelled with anti-p53 antibody (green). (b) Mean numbers of p53-positive foci in mitotic cells. Each column is the mean of at least 50 mitotic cells from 4 to 6 experiments. **P < 0.01 with unpaired t-test. (c) Mean frequencies of p53-positive centrosomes. Each column is the frequency calculated using pooled data from 4 to 6 experiments. **P < 0.01 with Chi-square test. (d) Mean frequencies of p53-positive centrosomes according to centrosome numbers per cell. Each column is the frequency calculated using pooled data from 4 to 6 experiments. **P < 0.01 with Chi-square test.
Figure 4.
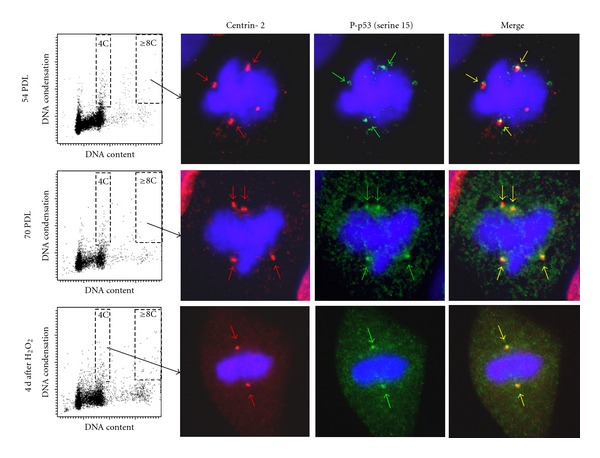
Localization of p53 phosphorylated at Ser15. Representative mitotic cells showing centrosomal localization of phosphorylated p53 (pSer 15) from early passages (upper panels), late passages (middle panels), and the 4th day after exposure to H2O2 (lower panels). Chromosomes (blue) and centrosomes (red) are labelled as in Figure 2, and phosphorylated p53 (pSer 15) is labelled with antiphosphorylated p53 (pSer 15) antibody (green).
4. Discussion
This study showed that mitotic cells with supernumerary (>2/cell) centrosomes increase with cellular aging induced by exposure to H2O2 as well as by serial passaging, especially in a polyploid subpopulation. Because cellular senescence is defined as permanent growth arrest by telomere attrition (replicative senescence) or various stresses (premature senescence) [22–24], the changes associated with cellular senescence in mitotic cells may have occurred in a near-senescent state. Recent studies have shown that cellular senescence is triggered by the DNA damage response induced by telomere attrition or oxidative DNA damage [23–26]. Therefore, cells in a near-senescent state may have extensive DNA damage that causes centrosome aberrations. Supernumerary centrosomes (>2 centrosomes/cell) could be formed due to several different mechanisms including overduplication in diploid cells, reduplication in the course of polyploidization, centrosome fragmentation, or centriole separation. However, supernumerary centrosomes observed in this study are not considered to be formed mainly by centrosome fragmentation or centriole separation, because most of the extra centrosomes consisted of two centrioles as recognized by anticentrin-2 antibody. The point of note is the relationship between numerical centrosome aberrations and DNA ploidy. Because centrosomes are duplicated in synchrony with DNA duplication, endoreduplication of DNA in polyploid cells may result in multiple centrosomes [5, 27]. However, more than 60% of polyploid mitotic cells at early passages had only two centrosomes, while the majority of polyploid mitotic cells formed in association with cellular senescence had more than two centrosomes (Figure 2(b)). In this point, we have recently shown that proliferative tetraploid cells established from normal human fibroblasts had only two centrosomes with bipolar spindles and grew at the same rate as diploid parent cells [28]. Therefore, mechanisms leading to the formation of bipolar spindles with two centrosomes in polyploid cells should exist. Mechanisms other than polyploidization may also be responsible for the numerical centrosome aberrations induced by cellular aging. Centrosome overduplication during cell cycle arrest by DNA damage response should also be considered as a possible mechanism for numerical aberrations of the centrosomes, as suggested by other investigators [11, 29, 30].
The present study shows that p53 protein localizes at centrosomes as shown by others [16, 17, 20, 31, 32], although its role is still not clear. The most significant finding of the present study is that the centrosomal localization of p53 correlated with numerical centrosome aberrations. Supernumerary centrosomes showed consistently higher frequencies of p53 localization compared to normally duplicated centrosomes, although statistical significance was observed only in diploid subpopulations. This correlation suggests that the centrosomal localization of p53 in human fibroblasts may be significantly associated with numerical centrosome aberrations. Moreover, Ser15 phosphorylation of p53 localized at centrosomes suggests that this may be associated with the DNA damage response, because p53 is known to be phosphorylated at Ser15 by ATM in DNA damage response after radiation [33, 34]. Although the significance of p53 localization at supernumerary centrosomes is not clear, it is possible that this localization may be promoted by DNA damage response. However, results of this study are somewhat inconsistent with other studies. The frequency of centrosomes with p53 protein in untreated cells was relatively lower compared to those reported in other studies. But the cells used in other studies were mouse cells in which human p53 genes were introduced [17, 32] or a lymphoblastoid cell line [16, 20, 35]. Therefore, the difference in the frequency of p53 positive centrosomes in untreated cells between this study and other studies might be cell- or species-specific. But the validity of this assumption is not certain, because the frequency of p53 localization to centrosomes in normal human fibroblasts has not been reported so far. It is also possible that sensitivity of the immunofluorescence procedure employed in this study was not sufficient for the complete detection of p53 protein on centrosomes. The majority of signals showing p53 protein on centrosomes were relatively weak.
Another point that needs to be considered is the role of p53 phosphorylation in p53 localization at centrosomes during mitosis. The studies using a lymphoblastoid cell line have shown that ATM-dependent phosphorylation of p53 at serine 15 is required for centrosomal localization of p53, and this phosphorylated p53 is rapidly dephosphorylated upon association with centrosomes [16, 20]. They showed that treatment of cells with serine-phosphatase inhibitor NaF before immunostaining is necessary to detect Ser 15 phosphorylated endogenous p53. In our experiments, however, centrosome-associated p53 foci seemed to be consistently phosphorylated at Ser15 in the absence of phosphatase inhibitors irrespective of DNA ploidy or whether centrosomes were normally duplicated or overduplicated. Although the reason for this discrepancy is not clear, it is possible that ATM-dependent Ser 15 phosphorylation of p53 in DNA damage response promoted localization of p53 at supernumerary centrosomes. Furthermore, we speculate that p53 protein localized at supernumerary centrosomes inactivates their function as the microtubule organizing centres, although this speculation lacks supporting data. Some studies published recently have shown that p53 is necessary to ensure bipolar mitosis and maintain chromosomal stability of tetraploid cells, which would otherwise cause multipolar mitosis due to supernumerary centrosomes [36, 37]. These observations regarding p53 function in maintaining chromosomal stability favor the above hypothesis.
The centrosome function in regulation of cell cycle and mitosis is increasingly being recognized, and many proteins implicated in such regulation have been shown to localize at centrosomes [19, 38–43]. They include cell cycle regulators such as cyclin-dependent kinases or cyclins, mitotic regulators such as Aurora kinases or TACC (transforming acidic coiled-coil), and mitotic checkpoint kinases such as BubR1 or Mad2. Expression and localization of those regulators may be affected directly or indirectly by p53 activation in DNA damage response, leading to control of cell cycle and mitosis. On the other hand, deregulation of those factors may activate p53 and p21 as well, causing premature cellular senescence to minimize chromosomal instability [44, 45]. Functions of centrosomes in regulating cell cycle and mitosis are not fully understood, however, and the significance of p53 localization at centrosomes should be defined more clearly in the near future.
Genetic instability of polyploid cells, which could lead to the formation of aneuploid cells, is well known [27, 46, 47], and an increase with age of polyploid cells in vivo and in vitro has been reported [48–51]. Therefore, age-associated chromosomal instability and tumorigenesis might be attributable to an increased frequency of polyploid cells with age. Results of the present study reveal that numerical centrosome aberration in polyploid cells is significantly associated with cellular aging induced by oxidative stress as well as repeated DNA replication. This association may lead to age-associated chromosomal instability and tumorigenesis. In addition, p53 protein activated during the DNA damage response is suggested to be associated with supernumerary centrosomes during mitosis, which may contribute to maintain chromosomal stability.
References
- 1.Krämer A, Schweizer S, Neben K, et al. Centrosome aberrations as a possible mechanism for chromosomal instability in non-Hodgkin’s lymphoma. Leukemia. 2003;17(11):2207–2213. doi: 10.1038/sj.leu.2403142. [DOI] [PubMed] [Google Scholar]
- 2.Pihan GA, Wallace J, Zhou Y, Doxsey SJ. Centrosome abnormalities and chromosome instability occur together in pre-invasive carcinomas. Cancer Research. 2003;63(6):1398–1404. [PubMed] [Google Scholar]
- 3.Fukasawa K. Centrosome amplification, chromosome instability and cancer development. Cancer Letters. 2005;230(1):6–19. doi: 10.1016/j.canlet.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 4.Giehl M, Fabarius A, Frank O, et al. Centrosome aberrations in chronic myeloid leukemia correlate with stage of disease and chromosomal instability. Leukemia. 2005;19(7):1192–1197. doi: 10.1038/sj.leu.2403779. [DOI] [PubMed] [Google Scholar]
- 5.Nigg EA. Origins and consequences of centrosome aberrations in human cancers. International Journal of Cancer. 2006;119(12):2717–2723. doi: 10.1002/ijc.22245. [DOI] [PubMed] [Google Scholar]
- 6.Ko MA, Rosario CO, Hudson JW, et al. Plk4 haploinsufficiency causes mitotic infidelity and carcinogenesis. Nature Genetics. 2005;37(8):883–888. doi: 10.1038/ng1605. [DOI] [PubMed] [Google Scholar]
- 7.Basto R, Brunk K, Vinadogrova T, et al. Centrosome amplification can initiate tumorigenesis in flies. Cell. 2008;133(6):1032–1042. doi: 10.1016/j.cell.2008.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sibon OCM, Kelkar A, Lemstra W, Theurkauf WE. DNA-replication/DNA-damage-dependent centrosome inactivation in Drosophila embryos. Nature Cell Biology. 2000;2(2):90–95. doi: 10.1038/35000041. [DOI] [PubMed] [Google Scholar]
- 9.Hut HMJ, Lemstra W, Blaauw EH, Van Cappellen GWA, Kampinga HH, Sibon OCM. Centrosomes split in the presence of impaired DNA integrity during mitosis. Molecular Biology of the Cell. 2003;14(5):1993–2004. doi: 10.1091/mbc.E02-08-0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sibon OCM. Centrosomes as DNA damage regulators. Nature Genetics. 2003;34(1):6–7. doi: 10.1038/ng0503-6. [DOI] [PubMed] [Google Scholar]
- 11.Dodson H, Bourke E, Jeffers LJ, et al. Centrosome amplification induced by DNA damage occurs during a prolonged G2 phase and involves ATM. EMBO Journal. 2004;23(19):3864–3873. doi: 10.1038/sj.emboj.7600393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sato N, Mizumoto K, Nakamura M, et al. A possible role for centrosome overduplication in radiation-induced cell death. Oncogene. 2000;19(46):5281–5290. doi: 10.1038/sj.onc.1203902. [DOI] [PubMed] [Google Scholar]
- 13.Kawamura K, Morita N, Domiki C, et al. Induction of centrosome amplification in p53 siRNA-treated human fibroblast cells by radiation exposure. Cancer Science. 2006;97(4):252–258. doi: 10.1111/j.1349-7006.2006.00168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Löffler H, Bochtler T, Fritz B, et al. DNA damage-induced accumulation of centrosomal Chk1 contributes to its checkpoint function. Cell Cycle. 2007;6(20):2541–2548. doi: 10.4161/cc.6.20.4810. [DOI] [PubMed] [Google Scholar]
- 15.Ohshima S, Seyama A. Cellular aging and centrosome aberrations. Annals of the New York Academy of Sciences. 2010;1197:108–117. doi: 10.1111/j.1749-6632.2009.05396.x. [DOI] [PubMed] [Google Scholar]
- 16.Oricchio E, Saladino C, Iacovelli S, Soddu S, Cundari E. ATM is activated by default in mitosis, localizes at centrosomes and monitors mitotic spindle integrity. Cell Cycle. 2006;5(1):88–92. doi: 10.4161/cc.5.1.2269. [DOI] [PubMed] [Google Scholar]
- 17.Shinmura K, Bennett RA, Tarapore P, Fukasawa K. Direct evidence for the role of centrosomally localized p53 in the regulation of centrosome duplication. Oncogene. 2007;26(20):2939–2944. doi: 10.1038/sj.onc.1210085. [DOI] [PubMed] [Google Scholar]
- 18.Zhang S, Hemmerich P, Grosse F. Centrosomal localization of DNA damage checkpoint proteins. Journal of Cellular Biochemistry. 2007;101(2):451–465. doi: 10.1002/jcb.21195. [DOI] [PubMed] [Google Scholar]
- 19.Shimada M, Komatsu K. Emerging connection between centrosome and DNA repair machinery. Journal of Radiation Research. 2009;50(4):295–301. doi: 10.1269/jrr.09039. [DOI] [PubMed] [Google Scholar]
- 20.Tritarelli A, Oricchio E, Ciciarello M, et al. p53 localization at centrosomes during mitosis and postmitotic checkpoint are ATM-dependent and require Serine 15 phosphorylation. Molecular Biology of the Cell. 2004;15(8):3751–3757. doi: 10.1091/mbc.E03-12-0900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bonsing BA, Corver WE, Gorsira MCB, et al. Specificity of seven monoclonal antibodies against p53 evaluated with western blotting, immunohistochemistry, confocal laser scanning microscopy, and flow cytometry. Cytometry. 1997;28(1):11–24. [PubMed] [Google Scholar]
- 22.Ben-Porath I, Weinberg RA. When cells get stressed: an integrative view of cellular senescence. Journal of Clinical Investigation. 2004;113(1):8–13. doi: 10.1172/JCI200420663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW. DNA repair, genome stability, and aging. Cell. 2005;120(4):497–512. doi: 10.1016/j.cell.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 24.Chen J-H, Hales CN, Ozanne SE. DNA damage, cellular senescence and organismal ageing: Causal or correlative? Nucleic Acids Research. 2007;35(22):7417–7428. doi: 10.1093/nar/gkm681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takai H, Smogorzewska A, De Lange T. DNA damage foci at dysfunctional telomeres. Current Biology. 2003;13(17):1549–1556. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 26.Di Micco R, Fumagalli M, Cicalese A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444(7119):638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 27.Storchova Z, Pellman D. From polyploidy to aneuploidy, genome instability and cancer. Nature Reviews Molecular Cell Biology. 2004;5(1):45–54. doi: 10.1038/nrm1276. [DOI] [PubMed] [Google Scholar]
- 28.Ohshima S, Seyama A. Formation of bipolar spindles with two centrosomes in tetraploid cells established from normal human fibroblasts. Human Cell. 2012;25(3):78–85. doi: 10.1007/s13577-012-0046-3. [DOI] [PubMed] [Google Scholar]
- 29.Kuriyama R, Terada Y, Lee KS, Wang CLC. Centrosome replication in hydroxyurea-arrested CHO cells expressing GFP-tagged centrin 2. Journal of Cell Science. 2007;120(14):2444–2453. doi: 10.1242/jcs.008938. [DOI] [PubMed] [Google Scholar]
- 30.Prosser SL, Straatman KR, Fry AM. Molecular dissection of the centrosome overduplication pathway in S-phase-arrested cells. Molecular and Cellular Biology. 2009;29(7):1760–1773. doi: 10.1128/MCB.01124-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morris VB, Brammall J, Noble J, Reddel R. p53 Localizes to the centrosomes and spindles of mitotic cells in the embryonic chick epiblast, human cell lines, and a human primary culture: an immunofluorescence study. Experimental Cell Research. 2000;256(1):122–130. doi: 10.1006/excr.2000.4800. [DOI] [PubMed] [Google Scholar]
- 32.Tarapore P, Tokuyama Y, Horn HF, Fukasawa K. Difference in the centrosome duplication regulatory activity among p53 ’hot spot’ mutants: potential role of Ser 315 phosphorylation-dependent centrosome binding of p53. Oncogene. 2001;20(47):6851–6863. doi: 10.1038/sj.onc.1204848. [DOI] [PubMed] [Google Scholar]
- 33.Caspari T. Checkpoints: how to activate p53. Current Biology. 2000;10(8):R315–R317. doi: 10.1016/s0960-9822(00)00439-5. [DOI] [PubMed] [Google Scholar]
- 34.Kruse J-P, Gu W. Modes of p53 Regulation. Cell. 2009;137(4):609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ciciarello M, Mangiacasale R, Casenghi M, et al. p53 displacement from centrosomes and p53-mediated G1 arrest following transient inhibition of the mitotic spindle. Journal of Biological Chemistry. 2001;276(22):19205–19213. doi: 10.1074/jbc.M009528200. [DOI] [PubMed] [Google Scholar]
- 36.Ho CC, Hau PM, Marxer M, Poon RY. The requirement of p53 for maintaining chromosomal stability during tetraploidization. Oncotarget. 2010;1(7):583–595. doi: 10.18632/oncotarget.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yi Q, Zhao X, Huang Y, et al. P53 dependent centrosome clustering prevents multipolar mitosis in tetraploid cells. PLoS One. 2011;6(11) doi: 10.1371/journal.pone.0027304.Article numbere27304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oikawa T, Okuda M, Ma Z, et al. Transcriptional control of BubR1 by p53 and suppression of centrosome amplification by BubR1. Molecular and Cellular Biology. 2005;25(10):4046–4061. doi: 10.1128/MCB.25.10.4046-4061.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Löffler H, Lukas J, Bartek J, Krämer A. Structure meets function—centrosomes, genome maintenance and the DNA damage response. Experimental Cell Research. 2006;312(14):2633–2640. doi: 10.1016/j.yexcr.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 40.Barr AR, Gergely F. Aurora-A: the maker and breaker of spindle poles. Journal of Cell Science. 2007;120(17):2987–2996. doi: 10.1242/jcs.013136. [DOI] [PubMed] [Google Scholar]
- 41.Schatten H. The mammalian centrosome and its functional significance. Histochemistry and Cell Biology. 2008;129(6):667–686. doi: 10.1007/s00418-008-0427-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y, Ji P, Liu J, Broaddus RR, Xue F, Zhang W. Centrosome-associated regulators of the G2/M checkpoint as targets for cancer therapy. Molecular Cancer. 2009;8(article no. 8) doi: 10.1186/1476-4598-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zyss D, Gergely F. Centrosome function in cancer: guilty or innocent? Trends in Cell Biology. 2009;19(7):334–346. doi: 10.1016/j.tcb.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 44.Lentini L, Barra V, Schillaci T, Di Leonardo A. MAD2 depletion triggers premature cellular senescence in human primary fibroblasts by activating a P53 pathway preventing aneuploid cells propagation. Journal of Cellular Physiology. 2012;227(9):3324–3332. doi: 10.1002/jcp.24030. [DOI] [PubMed] [Google Scholar]
- 45.Schmidt S, Schneider L, Essmann F, et al. The centrosomal protein TACC3 controls paclitaxel sensitivity by modulating a premature senescence program. Oncogene. 2010;29(46):6184–6192. doi: 10.1038/onc.2010.354. [DOI] [PubMed] [Google Scholar]
- 46.Duesberg P, Fabarius A, Hehlmann R. Aneuploidy, the primary cause of the multilateral genomic instability of neoplastic and preneoplastic cells. IUBMB Life. 2004;56(2):65–81. doi: 10.1080/15216540410001667902. [DOI] [PubMed] [Google Scholar]
- 47.Ganem NJ, Storchova Z, Pellman D. Tetraploidy, aneuploidy and cancer. Current Opinion in Genetics and Development. 2007;17(2):157–162. doi: 10.1016/j.gde.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 48.Fujisawa H, Nishikawa T, Zhu BH, et al. Accelerated aging of dermal fibroblast-like cells from the senescence- accelerated mouse (SAM): acceleration of changes in DNA ploidy associated with in vitro cellular aging. Journals of Gerontology. Series A. 1998;53(1):B11–B17. doi: 10.1093/gerona/53a.1.b11. [DOI] [PubMed] [Google Scholar]
- 49.Wagner M, Hampel B, Bernhard D, Hala M, Zwerschke W, Jansen-Dürr P. Replicative senescence of human endothelial cells in vitro involves G1 arrest, polyploidization and senescence-associated apoptosis. Experimental Gerontology. 2001;36(8):1327–1347. doi: 10.1016/s0531-5565(01)00105-x. [DOI] [PubMed] [Google Scholar]
- 50.Jones MR, Ravid K. Vascular smooth muscle polyploidization as a biomarker for aging and its impact on differential gene expression. Journal of Biological Chemistry. 2004;279(7):5306–5313. doi: 10.1074/jbc.M308406200. [DOI] [PubMed] [Google Scholar]
- 51.Walen KH. Bipolar genome reductional division of human near-senescent, polyploid fibroblast cells. Cancer Genetics and Cytogenetics. 2007;173(1):43–50. doi: 10.1016/j.cancergencyto.2006.09.013. [DOI] [PubMed] [Google Scholar]