

Abstract
The synthesis of efficient water-oxidation catalysts demands insight into the only known, naturally occurring water-oxidation catalyst, the oxygen-evolving complex (OEC) of photosystem II (PSII). Understanding the water oxidation mechanism requires knowledge of where and when substrate water binds to the OEC. Mn catalase in its Mn(III)-Mn(IV) state is a protein model of the OEC’s S2 state. From 17O-labeled water exchanged into the di-μ-oxo di-Mn(III,IV) coordination sphere of Mn catalase, CW Q-band ENDOR spectroscopy revealed two distinctly different 17O signals incorporated in distinctly different time regimes. First, a signal appearing after two hours of 17O exchange was detected with a 13.0 MHz hyperfine coupling. From similarity in the time scale of isotope incorporation and in the 17O μ-oxo hyperfine coupling of the di-μ-oxo di-Mn(III,IV) bipyridine model (Usov, O. M.; Grigoryants, V. M.; Tagore, R.; Brudvig, G. W.; Scholes, C. P. J. Am. Chem. Soc. 2007, 129, 11886-11887), this signal was assigned to μ-oxo oxygen. EPR line broadening was obvious from this 17O μ-oxo species. Earlier exchange proceeded on the minute or faster time scale into a non-μ-oxo position, from which 17O ENDOR showed a smaller 3.8 MHz hyperfine coupling and possible quadrupole splittings, indicating a terminal water of Mn(III). Exchangeable proton/deuteron hyperfine couplings, consistent with terminal water ligation to Mn(III), also appeared. Q-band CW ENDOR from the S2 state of the OEC was obtained following multi-hour 17O exchange, which showed a 17O hyperfine signal with a 11 MHz hyperfine coupling, tentatively assigned as μ-oxo-17O by resemblance to the μ-oxo signals from Mn catalase and the di-μ-oxo di-Mn(III,IV) bipyridine model.
Keywords: ENDOR, Manganese Catalase, OEC, PSII, electronic structure
Introduction
Efficient solar energy-driven water-oxidation catalysts represent a very promising solution to the problem of generating renewable solar fuels,1, 2 cheaply and on a large scale.3–5 A starting point for such catalytic systems is the photosystem II (PSII) oxygen-evolving complex (OEC).6 Composed of four Mn ions and a calcium ion, in addition to a proteinacious ligation environment, the OEC oxidizes in a multistep reaction two water molecules with the energy from four photons of visible light.
Catalytic hypotheses and concomitant model development have been greatly aided by crystallographic structural information on PSII at a resolution down to 1.9 Å.7–9 However, radiation damage has interfered with identification of the substrate water-binding sites. Identification of these sites is key to understanding the mechanism by which waters are activated for O-O bond formation. EPR spectroscopy can provide information on the binding of water/hydroxo/oxo species to the metal ions in the OEC, and these data may correlate with the substrate waters by comparison to the substrate exchange rates measured by mass spectrometry.10–14
The paramagnetic S2 state of the OEC has a characteristic 1.80 T wide, 18+ line CW X-band EPR difference spectrum (S2 minus S1) or ‘multiline signal’, centered at g = 2.15 An early EPR study detected broadening of this signal due to exchange of 17O from 17O-labeled water into the OEC.16 This initial work showed that oxygen from water could interact with the manganese ions in the OEC. More recent studies have provided evidence for 17O that exchanged from 17O-labeled water into the OEC. X-band ESEEM (Electron Spin Echo Envelope Modulation) provided evidence for 17O in the OEC with a hyperfine coupling of ~ 5 MHz.17 This ESEEM signal was assigned as a terminal water ligated by π bonding to Mn(III),17 based on the relation of the 17O hyperfine couplings to those found for terminal water π bonded to low spin ferric cytochrome P450.18 X-band HYSCORE (Hyperfine Sublevel Correlation) spectroscopy measurements on the OEC exchanged with 17O-water showed a single pair of structureless features assigned as a 17O coupling of ≈ 10 MHz.19 However, while our present submission was in revision, an Addition/Correction was published using a 15N substitution to show that the 2008 Su et al.19 nominal 17O HYSCORE signal actually derived from a 14N spectral subtraction artifact.20
Di-μ-oxo Mn(III)-Mn(IV) bipyridine had been investigated as a simple chemical model of the OEC which lacks the complexity of that higher nuclearity cluster because it has μ-oxo oxygens but no terminal waters. Mass spectrometry confirmed that the μ-oxo ligands of this Mn(III)-Mn(IV) bipyridine model complex exchanged from 18O-labeled water.21 CW Q-band ENDOR and X-band EPR line broadening experiments revealed a 17O hyperfine coupling of 12.8 MHz.22 Given the breadth of the ENDOR line shape, we would take this hyperfine coupling as an estimate of the isotropic coupling. Thus, to date, the most reliable 17O μ-oxo hyperfine coupling has been reported for the di-μ-oxo Mn(III)-Mn(IV) bipyridine model, for which mass spectrometry experiments confirm that the μ-oxo oxygen can exchange with isotopically enriched oxygen from water.21
As evidenced by rapid mass spectroscopic detection of 18O2 product, two substrate 18O-waters were found to exchange very quickly into the OEC itself, with product formation times of ~0.01 s and 0.5 s for a fast and a slow site respectively (in the S2 state).10–14 It is not clear where these catalytically relevant, rapidly exchanging water binding sites of the OEC are located. There is resonance Raman evidence for a slower oxygen isotope exchange occurring on the order of tens of minutes into a Raman-detectable μ-oxo group localized in the OEC.23 The mass spectroscopic study on the di-μ-oxo Mn(III)-Mn(IV) bipyridine model which had been dissolved in acetonitrile showed that oxygen ligand exchange into the di-μ-oxo groups from microliter quantities of 18O-water occurred in about 20 min.21 In the same study21 terminal water ligand exchange rates of the di-μ-oxo Mn(III)-Mn(IV) terpy model complex could not be resolved, but the μ-oxo ligand exchange time of 400 s from this terpy model compound was the shortest time measured for di-μ-oxo models.
The rate of water exchange into a di-μ-oxo di-Mn system could be affected by an aqueous environment, proteinacious ligation, seclusion from the bulk phase, and electrostatic effects on pKa, all factors not under control in the organic solvent of the model work of Tagore et al.21, 24 An improved model, also having the potential for terminal water ligation, could have a similar di-μ-oxo di-Mn center imbedded inside a soluble protein, and Mn catalase (MnCat) was chosen for that model.
The location and environmental character of a di-Mn di-μ-oxo center is known in the proteinaceous system of MnCat; the positions of both bridging oxygens, protein ligands, terminal water, and potentially critical second sphere amino acids are crystallographically known, as shown in Figure 1. MnCat is a 29.6 kDa hexamer forming-protein found in Lactobacillus plantarum25 and also Thermus thermophilus.26 It contains a di-Mn catalytic core with solvent derived aquo, hydroxo27 or μ-oxo bridges,28 depending on the oxidation state of the Mn. Under catalytic conditions, the Mn core oscillates between (II,II) and (III,III) oxidation states to disproportionate peroxide in a ping-pong mechanism.29, 30 The enzyme can be oxidized beyond the (III,III) state by treatment with hydrogen peroxide and hydroxylamine25, 31 or KIO4.26, 32, 33 This form of the enzyme, while catalytically inactive, is EPR active,32, 34–36 producing a 16-line signal at g = 2 which is diagnostic of antiferromagnetically coupled Mn(III)-Mn(IV).26, 33 EXAFS of this form of MnCat has also indicated a decrease in the Mn-Mn distance consistent with oxidation of one manganese to Mn(IV),37 and this decrease was consistent with the Mn(III)-Mn(IV) distance found in di-μ-oxo models.38 Advanced paramagnetic resonance studies have already been performed on MnCat in the (III,IV) state,37, 39,40 but the 17O ENDOR signals from potential μ-oxo or terminal water sites of MnCat have not been reported.
Figure 1.
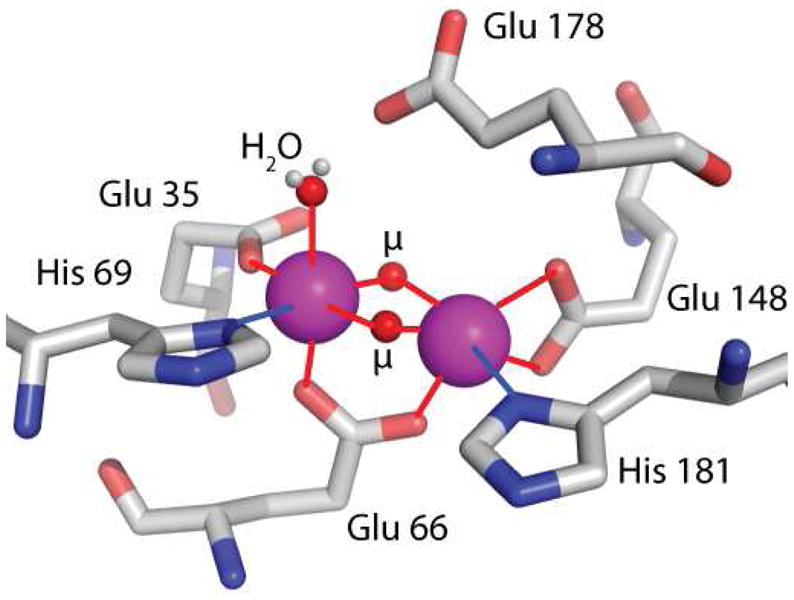
Shown is the local liganding environment of the di-manganese center of MnCat from L. plantarum. The structure is taken from the X-ray crystallographic structure of the Mn(III)-Mn(III) derivative, PDB structure 1JKU.27 Glu178 does not directly ligate the manganese but is within hydrogen bonding distance (2.5 Å) of the liganding water oxygen. The μ labels indicate solvent derived ligands. It is expected that there would be two μ-oxos in the (III,IV) form, and potentially a μ-oxo and μ-OH in the (III,III) state.27
With the goal of learning about the mechanism of water binding and O2 production in PSII, this work is focused on using ENDOR and EPR spectroscopy to characterize the binding and exchange of 17O from 17O-enriched water into both terminal and μ-oxo sites of MnCat. Data for this proteinaceous structural mimic of PSII provides the background to understand water exchange into PSII, its time course, locale, and catalytic relevance.
Materials and Methods
Preparative Methods
MnCat Preparation
MnCat was isolated from L. plantarum according to published procedures.25, 41 The superoxidized Mn(III)-Mn(IV) state of MnCat was prepared with minor modifications as previously described.25, 27, 31 MnCat was oxidized by redox cycling in the presence of hydrogen peroxide and hydroxylamine. Briefly, MnCat (0.1 mM active sites in 50 mM potassium phosphate buffer pH 7, containing 0.1 mM EDTA) was dialyzed against 1 L of 0.1 mM hydroxylamine and 0.1 mM H2O2 at 4 °C for 4 h. The protein was then transferred to 1 L of fresh 0.1 mM H2O2 solution (two changes), and finally, dialyzed overnight against 50 mM potassium phosphate buffer, pH 7, containing 0.1 mM EDTA. The sample, concentrated to 90 mg mL−1 (3 mM active sites), was then combined with a known volume of H216O or H217O for CW X-band EPR experiments. For Q-band ENDOR, H217O and glycerol; H216O and glycerol; or D2O and deuterated glycerol (99% deuterium enrichment, Cambridge Isotope Laboratories, MA) were added. (Glycerol at 50% vol/vol is a glassing agent that prevents aggregation of paramagnetic centers upon freezing.) After addition of reagents, the sample was frozen at 77 K. Exchange time with the added H217O reagent was monitored to differentiate between short exchange periods (1–5 min) and long-term exchange (> 2 hours). H217O was supplied at 90 % enrichment from Cambridge Isotope Laboratories. Exchange of D2O was accomplished in about a minute and the sample frozen; the sample was then unfrozen, deuterated glycerol added, and refrozen in about 5 minutes.
Photosystem II Preparation
Dark-adapted PSII membranes were prepared by the method of Berthold et al.42 The membranes were brought in 2.0 mm i.d., 2.4 mm o.d. quartz EPR-ENDOR tubes to a final concentration of 10–20 mg chl/mL and a ~74 % concentration of exchangeable H217O with a method outlined in Supporting Information, Figure 1S. Following dark adaptation for 24 hours, samples were frozen by plunging in liquid nitrogen. To achieve the S2 state for both the H217O sample and the H216O control, samples were illuminated for 1 min in an acetone bath at 200 K with a xenon arc lamp.
X-band EPR Methods
Mn-Cat cw X band EPR
Samples were stored at 77 K. CW X-band EPR scans were acquired on a Bruker ELEXSYS E500 EPR spectrometer equipped with a SHQ resonator and an Oxford ESR-900 helium-flow cryostat. The 16-line EPR signal was recorded at 7.5 K with the following instrumental parameters: microwave frequency 9.38 GHz, modulation frequency 100 kHz, modulation amplitude 3 G, microwave power 5 mW.
Photosystem II CW X band EPR
Following creation of the S2 state by photoillumination, the EPR line widths of S2 state samples enriched in H217O were compared with those of with non-enriched H216O samples. EPR scans were acquired as for MnCat.
CW Q-band ENDOR Methods
Spectroscopic Methods
CW Q-band (34.1 GHz) ENDOR measurements were performed under dispersion (χ′), rapid passage, field-modulated conditions with a cryogenically tunable TE011 Q-band resonator43 at 2 K as previously reported.22, 44, 45 For study of PSII, S2 samples were transferred from liquid N2 on a specially designed wand into the He-filled ENDOR cavity in the dark within 3 min of illumination. In this method of CW ENDOR, one monitors the radio-frequency (RF)-induced change in the rapid-passage, 100 kHz field modulated dispersion EPR signal as the frequency of the RF field is swept. RF power is pulsed with 100 μsec on/900 μsec off, with a peak power that is generally 20 W. Through previous experience we have determined that strongly coupled protons (coupling ≥ 4 MHz), 14N, and 17O are best resolved with a higher field modulation ≥ 2.5 Gauss p.t.p. (peak-to-peak)22, 44, 45 while weakly coupled nuclei, notably deuterons here, are best resolved with smaller field modulation ≤ 0.5 Gauss p.t.p.44 The frequencies of ENDOR features were determined from the average frequency of spectra taken with increasing and with decreasing frequency sweeps. CW field-modulated ENDOR has higher sensitivity than pulse methods for broad ENDOR signals, such as those of 17O.46 However, pulse methods, not available to us, appear to give better resolution of small detailed couplings, such as those of deuterons.47 We are aware that CW ENDOR, as carried out in pumped liquid helium, can also be subject to baseline artifacts from RF and field modulation-induced heating.
First-Order ENDOR Theory
A nucleus, N, with spin I, e.g., 17O (I = 5/2) or 14N (I = 1), can be described by a Spin Hamiltonian of the form:
| (1) |
Where βn is the nuclear Bohr magneton and Ngn is the nuclear g-value (1gn = 5.585, 2gn = 0.8574, 14gn = 0.4038, 17gn = −0.7572, 1ν = 52.928 MHz, 2ν = 8.124 MHz, 14ν = 3.816 MHz, 17ν = 7.178 MHz at 1.2431 T) for protons, deuterium, 14N, and 17O, respectively. NAxx, NAyy, and NAzz are the components of the hyperfine tensor, and for nuclei with I > ½, NPxx, NPyy, and NPzz, are the components of the quadrupolar tensor, where NPxx + NPyy + NPzz = 0. [NPzz = (3e2qzzNQ/h)/(4I(2I−1)) where e is the electronic charge; h is Planck’s constant; NQ is the nuclear quadrupole coupling constant of a particular nucleus; I is the nuclear spin operator, and qzz is the electric field gradient (EFG) along the “z” direction, a direction which may coincide for 14N or 17O with a bonding direction.] This terminology for Spin Hamiltonian parameters follows that used in our recent publications.44, 45, 48, 49
17O ENDOR Frequencies and Hyperfine Analysis
To first order, the 17O ENDOR frequencies will be:
| (2a) |
| (2b) |
The 17ν+ ENDOR branch has been the branch generally observed by Q-band CW rapid passage ENDOR on biologically relevant complexes,46, 50–52 including the μ-oxo oxygens of the Mn(III)-Mn(IV) bipyridine dimer.22 For those μ-oxo oxygens of the Mn(III)-Mn(IV) bipyridine dimer, the 17ν−ENDOR branch is close to zero frequency because of the near cancellation of |17A/2| ≈ 6.4 MHz and 17ν = 7.18 MHz at 1.243 T). Relevant details of 17O quadrupole and anisotropic hyperfine couplings are provided in the discussion and the Supporting Information.
14N ENDOR Frequencies and Hyperfine Analysis
The first-order expressions for spin 1 14N ENDOR frequencies are:
| (3a) |
| (3b) |
where 14A is the hyperfine coupling, 14P the quadrupolar coupling, and 14ν is the 14N nuclear Zeeman frequency. For 14N nitrogen, as opposed to protons, the hyperfine term, 14A/2, rather than the nuclear Zeeman term 14ν, is the dominant term measured by ENDOR. As with 17O, for CW rapid passage Q-band ENDOR, the 14ν+ENDOR branch is frequently the branch observed.
Protons/Deuteron Frequencies and Hyperfine Analysis
The frequencies of proton or deuteron ENDOR features, 1νENDOR or 2νENDOR, center, to first order, at the respective free proton or free deuteron nuclear Zeeman frequency, 1ν or 2ν, and in the proton or deuteron ENDOR spectra we show the spectra centered at 1ν or 2ν. For example, taking 1A as the proton hyperfine coupling, one finds that the frequencies, 1ν±ENDOR, are split away from the nuclear Zeeman frequency by ± 1/21A for protons coupled to the electron spin ½ doublet. Proton ENDOR frequencies, occurring as “+” or as “−” Zeeman branches, are:53
| (4) |
With the neglect of small quadrupolar terms, a similar expression holds for deuterons:
| (5) |
First-order expressions hold here because 1ν > |1A/2| and 2ν > |2A/2|.
The strong, largely dipolar coupling to terminal water protons localized on the Mn(III) of the (2-OH-3,5-Cl2-SALPN)2 Mn(III)-Mn(IV) complex has previously been resolved by ENDOR-ESEEM measurements.40, 54 The theory to explain the terminal water dipolar coupling, called extended dipole theory, incorporates the quantum mechanical projection factors for spin localized on the Mn(III) (S = 2) and Mn(IV) (S = 3/2) where these two ions are an antiferromagnetically coupled S = ½ pair. (The projection factor expresses the spin on an individual metal ion in terms of the total spin of the dimer. See, for example, the discussion and formula A3 on p. 4923, Khangulov et al.40) The theory provides the diagonalized hyperfine tensor54 where both metal ions contribute in a non-colinear fashion to the dipolar Hamiltonian. A more complete discussion of the extended dipole method applied to 17O–μ-oxo and to the terminal water protons in MnCat is provided in the Supporting Information.
Results
X-band EPR 17O Line Broadening of MnCat
The first derivative CW X-band spectra were examined in the 16O- and 17O-exchanged states. The extent of broadening from the 17O ligand is clear in Figure 2, which compares the overlaid line shapes of the 16O-exchanged and 17O-exchanged. The broader line width implies 17O exchanged into the Mn(III)-Mn(IV) coordination sphere.
Figure 2.

CW first derivative X-band EPR spectra are presented for MnCat exchanged in H216O (black) and 90% H217O (red). The inset shows an enlarged view of several hyperfine lines to demonstrate the broadening. The spectra from the MnCat in H216O and 90% H217O have been normalized to the integrated spin count. Samples were incubated for two hours after addition of labeled or non-labeled water before freezing.
CW Q-band 17O ENDOR of MnCat
CW rapid-passage Q-band ENDOR was obtained for water-derived ligands that exchanged into the active site of MnCat. (Q-band rapid-passage EPR of the 16-line spectra of MnCat are shown in Figure 2S of the Supporting Information.) Figure 3A provides evidence for the appearance after a two hour exchange of 17O-water of a broad signal labeled 17OA, similar in line shape and ENDOR frequency to the 17O from the μ-oxo oxygens in the di-μ-oxo Mn(III)-Mn(IV) bipyridine model complex.22 The frequency of 17OA is 13.7 ± 0.5 MHz, and this ENDOR frequency translates via Eq. 2a into a 17O hyperfine coupling of 13.0 ± 1.0 MHz. To give insight into the approximate time course and origin for appearance of peaks, the MnCat sample was exchanged with 17O-water for ~ 1 min, and as shown in Figure 3B, the higher frequency 17OA feature near 14 MHz was then not present but features in the 7–11 MHz region were. When 16O-water was used, a peak in the 6–7 MHz region remained, as shown in Figure 3C. This remaining peak had an ENDOR frequency of 6.5 ± 0.3 MHz, and its 14N coupling per Eq. 3 for 14ν+ is 14A = 5.4 ± 0.6 MHz. This coupling is consistent with the coupling of 14A = 5.75 MHz recently reported by multi-frequency ESEEM and assigned to the histidine nitrogen ligated to Mn(III) of L. plantarum MnCat.39 Although there is evidence from Figure 3C for a broad underlying baseline in the 10–25 MHz region, neither of the peaks, 17OA and 17OB, was at all evident in this baseline.
Figure 3.

This figure provides a comparison of the ENDOR signals in the 1–30 MHz range from MnCat prepared as follows: A) in 66 %17O-water with a two hour incubation before freezing; B) in 66% 17O water with a 1 min incubation before freezing; C) 16O-water. These spectra were obtained at a magnetic field of 1.243 T. Conditions: microwave power = 0.22 μW, modulation amplitude = 5 G p.t.p., time constant = 20 ms, RF power ≈ 10 W, sweep rate = 6 MHz/s. Spectrum A was the result of 3300 5 s sweeps, and B and C were the result of 1000 5 s sweeps. Spectra A, B, and C were normalized to the number of sweeps and to their respective underlying EPR intensities, and the vertical intensity unit scale is the same for A, B, and C. Inset: The inset was obtained from MnCat exchanged in 17O-water for 1 minute, but to obtain better resolution of weakly coupled features, a field modulation of 2.5 Gauss p.t.p., a slower sweep of 3 MHz/s over a 1–16 MHz range, and a higher RF power by a factor of 2. The inset spectrum was the result of 1100 5 s sweeps.
Figure 3S in the Supporting Information provides a comparison of spectra from the same sample as that of Figure 3A at two different magnetic fields, H = 1.243 T near the center of the EPR pattern and H = 1.267 T at its high field end. Other than a difference in intensity, the ENDOR spectra at the two fields in Figure 3S are essentially identical. A more highly resolved spectrum for this two-hour 17O-exchanged sample is shown in the Supporting Information, Figure 4S, where there is still the 17OA peak near 14 MHz, but where there is evidence for lower frequency features in the 7–11 MHz region, notably those labeled 17OB.
By use of lower field modulation in the CW ENDOR technique, higher resolution of the 17OB signal than that in Figure 3B is provided in the inset adjacent to Figure 3B using the sample prepared with 1 min 17O exchange. The 17O hyperfine coupling of the 17OB peak labeled with an arrow in the inset near 9 MHz was 3.8 ± 0.6 MHz. Above 10 MHz there were two shoulders noted in the inset to 3B with splittings of about 1.3 MHz, while overlap with the 14N signal prevented additional resolution of any 17O details below 9 MHz. In the Supporting Information, Figure 5S, a comparison is made of the spectrum of the inset to Figure 3B showing the 17OB signals and the MnCat sample prepared with H216O which shows the 14N signal but not 17OB signals.
CW Q-band – Proton and Deuteron ENDOR of MnCat
A comparison of CW proton ENDOR from a sample prepared in protonated buffer to a sample prepared in deuterated buffer provided evidence for exchangeable protons at the active site. Detailed features, especially the starred outlying ones, were best resolved in the first derivative presentation of Figure 4. (The standard absorption-like ENDOR spectra before a derivative was numerically taken are shown as Figure 6S in the Supporting Information.) These outlying features, per Eq. 4, had hyperfine couplings of 17.2 ± 0.4 and 12.2 ± 0.4 MHz, larger than any previously reported from MnCat by frequency-modulated X-band ENDOR of the 1990’s,40 but comparable with the largest component of the anisotropic proton hyperfine tensor of terminal water ligated to Mn(III) in the (2-OH-3,5-Cl2-SALPN)2 Mn(III)-Mn(IV) dimer.54 There may be broad underlying exchangeable features with proton hyperfine couplings (A3) less than 10 MHz, but such features are obscured by non-exchangeable protons.
Figure 4.

This figure presents the first derivative 1H-ENDOR spectra, centered at the free proton NMR frequency, 1ν, to compare exchangeable features from MnCat prepared in protonated solution with MnCat prepared in deuterated solution. Conditions: 1.243 T (1ν = 52.92 MHz), microwave power = 0.22 μW, field modulation = 5 G p.t.p., time constant = 20 ms, sweep rate = 3 MHz/s, 750 scans for the protonated sample and 250 for the deuterated sample. Inset: 2H-ENDOR of D2O-exchanged MnCat following subtraction of the underlying spectrum from a protonated MnCat sample. Deuteron ENDOR conditions: (2ν = 8.12 MHz), sweep rate = 1 MHz/s, field modulation = 0.5 G p.t.p., scans = 1000. The inset spectrum is centered at the free deuteron NMR frequency, and the scale above it is the proton frequency scale (obtained with a multiplier of 6.52) corresponding to the deuteron scale. Exchange of D2O was accomplished in about a minute and the sample frozen; the sample was then unfrozen, deuterated glycerol added, and refrozen in about 5 minutes.
Evidence for exchangeable protons implied that there are corresponding exchangeable deuterons, and so ENDOR from exchangeable deuterons was sought near the deuteron Larmor frequency, 2ν = 8.12 MHz, as shown in the inset to Figure 4. The CW deuteron ENDOR spectrum does not have the resolution or signal to noise of the proton spectrum, but it complements the proton ENDOR information and shows evidence of exchangeable deuterons. Protons with hyperfine couplings less than ~10 MHz are obscured in Figure 4 by non-exchangeable protons; whereas, the associated deuterons with couplings less than about 1.5 MHz (i.e., with a splitting away from 2ν of ~±0.75 MHz) are obvious even at low resolution and occur only from exchanged deuterons.
CW Q-band 17O and 14N ENDOR of the S2 State of PSII
The 17O information from MnCat provided motivation to obtain corresponding information from the S2 state of PSII. Twenty four hours of exchange with H217O produced a definite, light-induced 17O hyperfine interaction from the S2 state of PSII. A comparison (Figure 5) of S2 ENDOR signals from PSII samples, respectively exchanged with H216O and H217O, indicated a 17O ENDOR signal at 12.8 ± 0.5 MHz, similar to that of MnCat here and to that of the previously reported 17O-bridged MnIII-MnIV bipyridine dimer.22 The 17O hyperfine coupling, derived via Eq. 2a, was |17A| = 11 ± 1 MHz. An additional signal occurred at 7.3 ± 0.2 MHz from all S2 samples, and its hyperfine coupling, assigned to 14N and interpreted by Eq. 3, is |14A| = 7.0 ± 0.4 MHz. This coupling is similar to the 7.2 MHz coupling reported from ESEEM studies of the OEC and assigned to the D1-H332 histidine nitrogen 55, 56 that is ligated to Mn No. 2 (Numbering system of Sproviero et al.57)
Figure 5.

ENDOR spectra of white light-illuminated PSII in H217O (red) and H216O (black) and a dark-adapted sample in H217O (green) at field positions: (A) 1.24 T and (B) & (C) 1.27 T. A light-induced feature (17ν+ENDOR) from the 17O sample occurs at 12.8 ± 1.0 MHz. Acquisition conditions: field modulation 2.5 G, time constant 82 msec, mw power = 0.28 μW, RF sweep rate 3 MHz/sec, number of scans (A) 1200, (B) 600, and (C) 200. Spectral amplitudes were normalized to the peak labeled 14N. Quoted ENDOR and hyperfine frequencies in text are the average of upward and downward frequency sweeps shown in the Supporting Information, Figure 7S.
Discussion
μ-oxo 17O Hyperfine Interaction
There are now three structurally related systems into which 17O has been exchanged from 17O-water. The first is the di-μ-oxo Mn(III)-Mn(IV) bipyridine complex, known by mass spectroscopic quantitation to exchange both of its μ-oxo oxygens with isotopically enriched 18O-water.21, 24 It has no other oxygen ligands to exchange and, therefore, its 17O ENDOR signal has to be μ-oxo. Both ENDOR and EPR line broadening of the 17O-di-μ-oxo Mn(III)-Mn(IV) bipyridine complex showed a 17O hyperfine coupling of 12.8 MHz.22 For the di-μ-oxo Mn(III)-Mn(IV) bipyridine complex, the time for induction of the 17O signal by exchange from 17O-water was of order twenty minutes. The present study on MnCat showed an even longer time for induction of its 17O signal (17OA in Figure 3A), a signal having a very similar hyperfine coupling to that of the di-μ-oxo Mn(III)-Mn(IV) bipyridine complex of 13.0 MHz. MnCat is known to have a di-μ-oxo center,27, 37 and based on the slow 17O kinetic induction from 17O-water and the similar 17O hyperfine to that in the di-μ-oxo model, we assign the 17O ENDOR signal (17OA in Figure 3A) of MnCat to a μ-oxo oxygen in its Mn(III)-Mn(IV) center.
The S2 state of the OEC has resonance Raman evidence for slow oxygen isotope exchange into a μ-oxo group,23 and the implication is that there is at least one μ-oxo oxygen in the OEC that can exchange very slowly with solvent. The OEC showed a similar 17O signal to the slowly exchanging 17O signal of the 17O-μ-oxo Mn(III)-Mn(IV) bipyridine and also to the 17OA signal in MnCat. The hyperfine coupling from the 17O signal in the OEC was 11 MHz, which is ~ 15 % less than the hyperfine coupling of the 17O-μ-oxo in the di-μ-oxo Mn(III)-Mn(IV) bipyridyl complex. A simple explanation for the smaller coupling in the OEC could be that the spin in the tetranuclear Mn center of the OEC is delocalized beyond one di-μ-oxo bridged Mn(III)-Mn(IV) pair.58, 59 The tetranuclear center of the OEC is less well defined at present than the bi-nuclear center of Mn(III)-Mn(IV) bipyridine, and so we can not be as confident in our assignment of our 17O ENDOR signal from the OEC. However, it is extremely tempting to suggest that the 17O ENDOR signal of the OEC may be from a μ-oxo oxygen within the OEC, in particular, to the μ-oxo oxygen between the distal Mn(III) (Mn No. 4) and its adjacent Mn(IV) partner (Mn No. 2),9, 57 which is the oxygen postulated to originate from a “slow exchangeable water” (See Fig. 13 of Kulik et al.58).
Beyond the obvious presence of high-valent Mn in all of these systems, their common aspect is μ-oxo oxygen. μ-oxo oxygen mediates via covalent superexchange the antiferromagnetic coupling between paramagnetic metals,60 and the detailed electronic nature of antiferromagnetic superexchange is critical to redox/catalytic properties.61 Broken symmetry DFT has been used to predict spin density at μ-oxo oxygens and pathways for superexchange. Hyperfine couplings for 55Mn, 14N, and 1H, have been calculated, but not for 17O.62–65 Given the central importance of μ-oxo oxygens to all these high-valent multi-manganese complexes, especially the OEC, prediction of the μ-oxo 17O hyperfine coupling would be a sensitive test of the capabilities of broken symmetry DFT.
The antiferromagnetically coupled Fe(III)-Fe(IV) center of ribonucleotide reductase (RNR) has been the subject of considerable previous 17O ENDOR.46, 66 The μ-oxo 17O hyperfine tensor in RNR was found to be highly anisotropic (17Axx, 17Ayy, 17Azz = 0, 23.5, 23.5 MHz). The μ-oxo 17O isotropic hyperfine coupling of RNR is 15–16 MHz,44 comparable to the isotropic coupling we estimate from our μ-oxo 17O findings. There was additional hyperfine coupling to a terminal 17O-water of the Fe(III) with an isotropic component about double that of the μ-oxo, and the μ-oxo 17O of RNR was distinguished kinetically from the terminal 17O-water through rapid freeze quench methods to follow rapid incorporation from 17O2. The RNR system has advantages over the multi-Mn system that lead to better ENDOR signal to noise and resolution. Due to absence large 55Mn nuclear hyperfine couplings, the RNR EPR signal at Q-band is only ~0.1 T wide67 versus 1.2–1.8 T wide for di-Mn(III)-Mn(IV), accounting for greater EPR-ENDOR sensitivity for RNR. The RNR system has sufficient g-anisotropy for good angle selection of 17O ENDOR hyperfine anisotropy; this is not the case for di-Mn(III)-Mn(IV). The μ-oxo couplings of RNR should be larger than those in di-Mn(III)-Mn(IV) systems because Fe tends to be more covalent than Mn and because the S = 5/2 ferric ion has a spin-containing d(x2-y2) orbital directed for σ bonding toward the oxygen 2s and 2p σ-bonding orbital.22
In analyzing the 17O ENDOR information, we have simply provided the ENDOR frequency and hyperfine coupling (Eq. 2a) from the peak of the ENDOR feature assigned to 17O in Figures 3 and 5. Without additional information, we would take this peak frequency as approximately from an isotropic coupling of ~11–13 MHz magnitude. There should, however, be anisotropy in the μ-oxo 17O hyperfine coupling. A starting point for theoretically estimating hyperfine anisotropy is to calculate the dipolar coupling of the μ-oxo oxygen with electron spin on its Mn(III) and Mn(IV) partners. As explained in the Supporting Information, we applied to μ-oxo 17O the extended point-dipole model originally developed for predicting dipolar hyperfine interactions of protons near the Mn(III)-Mn(IV) with both metal spins,40, 54, 62 and Figure 8S in the Supporting Information provides a simulation68 of the μ-oxo 17O ENDOR feature broadened by hyperfine anisotropy. A goal of future advanced EPR studies of 17O in the OEC and models for it should be to resolve this hyperfine anisotropy.
17O ENDOR Assigned to the Terminal Water Ligand on Mn(III) of MnCat
The more weakly coupled 17OB inset to Figure 3B with hyperfine coupling (per Eq. 2a) of about 3.8 MHz became obvious from the MnCat sample that had been exchanged only one minute with 17O-water before freezing. Previous ESEEM18,69 has provided a hyperfine coupling of about 2 MHz for a 17O-water sharing π–bonding electrons with low-spin ferric heme in an S = ½ system. Because of the +2 projection factor for Mn(III), the hyperfine coupling for π–bonded 17O-water on Mn(III) of the Mn(III)-Mn(IV) pair would be about 4 MHz, comparable to that which is obtained here. The quadrupole tensor of 17O for water ligands of aqua-Mn(II)70 and aqua-vanadyl complexes,71 has two large components (−17Pxx) = 17Pyy = ~0.33 MHz and a third, 17Pzz ~ 0. The expected 17O-water quadrupole splittings in the ENDOR expressions of Eq. 2 will be |3 17Pxx| ~ |3 17Pyy| ≈ 1 MHz, comparable with the poorly resolved 1.3 MHz splitting of the shoulders marked in the inset to Figure 3B. We assign the peak feature labeled 17OB to terminal 17O-water on the Mn(III) of the Mn(III)-Mn(IV) pair because there is ENDOR evidence for 17O with hyperfine coupling and possibly quadrupole couplings consistent with water, because there is a terminal water on Mn in the crystal structure of MnCat, and because there are also proton and deuteron ENDOR data consistent with the protons of that very same water. Further, as discussed below, the terminal water, similar in size to the superoxide substrate of MnCat, would be expected to diffuse into the coordination sphere of the metal center in subsecond times, not hours, and the 17OB feature assigned to terminal water does rapidly appear. We point out that a water σ-bonded along a Jahn-Teller distorted axis of Mn(III) might in principle be expected to have larger hyperfine coupling than water π-bonded to Mn(III) or Mn(IV), but we do not observe a large coupling to a rapidly incorporated 17O.
Exchangeable Proton and Deuteron ENDOR Assigned to the Terminal Water Ligand on Mn(III) of MnCat
The largest proton coupling of 17.2 MHz (singly starred in Figure 4) is comparable to the largest coupling of 17.6 MHz observed from terminal water on Mn(III) from the (2-OH-3,5-Cl2-SALPN)2 Mn(III)-Mn(IV) complex.54 In the crystal structure of the di-Mn active site in MnCat (Figure 1) there is a carboxylate oxygen of Glu178 within a 2.5 Å hydrogen-bonding distance of the terminal water oxygen with a Mn-O-H-O bond angle of 120°. We have positioned a hydrogen-bonded proton along the water-oxygen-to-carboxylate-oxygen direction at 1.0 Å from the water oxygen. The geometric parameters for that water proton are provided in the Supporting Infomration (See Scheme 1S and pages S11 and S12 in the supporting Information.) The dipolar coupling to this proton is 1Axxdip, 1Ayydip, 1Azzdip = −8.6, −6.6, +15.2 MHz. In addition to the dipolar coupling of 1Azzdip = 15.2 MHz, the total hyperfine coupling of 1Azz = 17.2 MHz of the starred features in Figure 4 would require a small +2.0 MHz isotropic component, comparable to that suggested by Randall et al.54 We assign the proton that gives rise to the observed coupling of 17.2 MHz in Figure 4 to the water that also gives the 17O coupling of 3.8 MHz for 17OB. The predicted smaller perpendicular dipolar components, 1Axxdip, 1Ayydip, which are 6–9 MHz in magnitude, would be largely buried here in the proton ENDOR spectra of Figure 4 under non-exchangeable proton ENDOR signals, but the deuteron ENDOR shown in the inset to Figure 4 does have intensity in the 0.9–1.4 MHz region that may correspond to the 6–9 MHz proton region. Water ligated to Mn(III) may have a second proton located further from the Mn(III) than the one with the coupling of 17.2 MHz. Dipolar calculations performed on a second water proton a few tenths of an Å further from the Mn(III) predicted a smaller maximal proton dipolar coupling in the 12–14 MHz range, consistent with the outlying doubly starred proton hyperfine signals having a splitting of 12.2 MHz in Figure 4.
Exchange of Water into the Manganese Active Sites
Q-band ENDOR and X-band EPR showed that the solvent-derived 17O ligands appeared in the di-μ-oxo core of Mn(III)-Mn(IV) bipyridyl22 within tens of minutes and in the di-μ-oxo core of MnCat within several hours. Q-band 17O ENDOR and Resonance-Raman-detectable 18O at a μ-oxo site23 within PSII occurred on a similar time scale. If the μ-oxo 17O observed by ENDOR of PSII is the same μ-oxo that Resonance Raman has resolved after slow exchange,23 then this oxygen would not be catalytically relevant in the sense that it rapidly appears in the O2 product, although it is relevant to the structure of the catalytic center. (It is likely that the bridge opening to allow oxygen exchange requires a proton from a nearby base,24 such as a nearby arginine in the OEC.)
MnCat functions with a time for superoxide turnover of 5 μs, close to the diffusion limit.30 Superoxide, and very likely the similar-sized water, can diffuse into the active site very easily, so that diffusion of water into the interior of the MnCat hexamer,27 as opposed to exchange into the μ-oxo Mn(III)-Mn(IV) core, is unlikely to be an impediment to oxygen isotope exchange for MnCat. The evidence for terminal water ligation from H217O samples frozen within a minute of the initiation of water exchange, is consistent with such a rapid exchange, which indeed may even occur in a much shorter time than a minute.
The large ENDOR-resolved proton coupling in MnCat (1Azz > 15 MHz) indicating ligation of a terminal water to Mn(III) of the di-μ-oxo Mn(III)-Mn(IV) pair in MnCat has not yet been observed in PSII. We would interpret the hyperfine couplings of so far unresolved exchangeable proton/deuterons in the OEC derived from ENDOR and ESEEM,72, 73 as being too small to be those of water protons on a Mn(III) of a Mn(III)-Mn(IV) antiferromagnetically coupled pair. We cannot at this point exclude hydrogen bonding to a bridging μ-oxo between Mn and Ca as a candidate for ESEEM-observable hydrogen bonds in PSII because to date there is not a Mn-Ca model to test this hypothesis.
Conclusion
Although there was previous ENDOR evidence for exchange of 17O from 17O-water into the μ-oxo ligands of the Mn(III)-Mn(IV) bipyridine model complex,21, 22, 24 the spectroscopic signatures had not been explored for 17O exchanged from 17O-water into a protein active site having the potential to exchange both μ-oxo and terminal water oxygens. MnCat, di-μ-oxo Mn(III)-Mn(IV) bipyridine, and the S2 signal of the OEC have now all provided a similar 17O ENDOR signal that arose after slow exchange with 17O-water. This signal is assigned to a μ-oxo oxygen that joins a Mn(III)-Mn(IV) antiferromagnetically coupled pair. This μ-oxo oxygen, an important structural aspect of multi-Mn centers, exchanged on the scale of tens of minutes into the Mn(III)-Mn(IV) bipyridine complex and on the time scale of hours into the μ-oxo Mn(III)-Mn(IV) centers of MnCat and the OEC. Thus, the rapid water binding sites for the OEC are most likely to be terminal, or even more distant, water binding sites. As judged by the small magnitude of exchangeable 1H and 2H hyperfine couplings for the OEC72,73 when compared to the > 15 MHz exchangeable proton coupling from the assigned terminal water ligand of Mn(III) in Mn(III)-Mn(IV) MnCat, these sites in the OEC are not of a terminal water to the Mn(III) of a Mn(III)-Mn(IV) pair. Hydrogen bonding to a μ-oxo ligand between Mn and Ca cannot be excluded.
For MnCat, ENDOR evidence was obtained for terminal water exchange with H217O on a minute (or less) time scale. 17O hyperfine and possible 17O quadrupole ENDOR signals, which differed significantly from 17O-μ-oxo ENDOR signals, indicated terminal water. In addition there were large exchangeable proton couplings consistent with the dipole interaction of a water ligand proton with a near Mn(III) and a more distant Mn(IV); exchangeable deuteron signals, although less well resolved, were also consistent with the deuterium on the same terminal water.
In Scheme 1 we succinctly present the outcomes reported in this article. Scheme 1 summarizes the time span for the appearance of 17O-ENDOR peak positions from MnCat and the OEC and for our assignment of the physical location of these 17O within the manganese centers of MnCat and the OEC. The assignment of exchangeable proton/deuteron ENDOR to the exchangeable water on the Mn(III) of MnCat is also noted.
Scheme 1.

shows the following information: a) The time course for H217O binding and 17O-migration within the limits of the present experiments. b) The location of the 17O peaks as the time course of the experiment progresses., including the labels 17OA for μ-oxo and 17OB from a 17O-water ligand. c) The location of the exchangeable H on terminal water ligand of Mn(III) in MnCat. is shown. d) The labels of Mn(4) and Mn(3) are indicated per Sproviero et al. 55
Supplementary Material
Acknowledgments
This research was supported by NIH GM32715 (GWB) and EB00326929 & GM066253-01A1 (CPS).
Footnotes
Supporting Information Available
Protocols A and B, including Figure 1S for preparation of concentrated PSII highly enriched in exchangeable H217O; Figure 2S - Comparison of Q-band rapid passage EPR signals from the Mn-Cat prepared with 17O-water and with 16O-water; Figure 3S - Comparison of ENDOR spectra obtained at 1.243 T and 1.267 T; Figure 4S – 17O ENDOR spectra of MnCat at high RF power and higher signal to noise. Figure 5S – Comparison of ENDOR from MnCat with 1 min H217O exchange to ENDOR from MnCat exchanged with H216O. Figure 6S - Rapid passage absorption-like ENDOR signals of MnCat in protonated and deuterated solvent; Figure 7S - A comparison of the ENDOR spectra of OEC – S2 with frequency sweeps in the upward and the downward directions; Figure 8S - The simulated 17O ENDOR dipolar-broadened spectrum of μ-oxo oxygen compared to the experimental ENDOR spectrum; Theory for extended dipole interaction of a nuclear spin with both Mn(III) and Mn(IV);. The material in the supporting information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.McConnell I, Li G, Brudvig GW. Chemistry and Biology. 2010;17:434–447. doi: 10.1016/j.chembiol.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore GF, Brudvig GW. Annu Rev Condensed Matter Phys. 2011;2:303–327. [Google Scholar]
- 3.Lewis NS, Nocera DG. Proc Natl Acad Sci U S A. 2006;103:15729–15735. doi: 10.1073/pnas.0603395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lewis NS. MRS Bull. 2007;32:808–820. [Google Scholar]
- 5.Lewis NS. Science. 2007;315:798–801. doi: 10.1126/science.1137014. [DOI] [PubMed] [Google Scholar]
- 6.McEvoy JP, Brudvig GW. Chem Rev. 2006;106:4455–4483. doi: 10.1021/cr0204294. [DOI] [PubMed] [Google Scholar]
- 7.Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- 8.Guskov A, Kern J, Gabdulkhakov A, Broser M, Zouni A, Saenger W. Nat Struct Mol Biol. 2009;16:334–342. doi: 10.1038/nsmb.1559. [DOI] [PubMed] [Google Scholar]
- 9.Umena Y, Kawakami K, Shen J-R, Kamiya N. Nature. 2011;472:55–60. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- 10.Messinger J, Badger M, Wydrzynski T. Proc Natl Acad Sci U S A. 1995;92:3209–3213. doi: 10.1073/pnas.92.8.3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hillier W, Messinger J, Wydrzynski T. Biochemistry. 1998;37:16908–16914. doi: 10.1021/bi980756z. [DOI] [PubMed] [Google Scholar]
- 12.Hillier W, Wydrzynski T. Biochemistry. 2000;39:4399–4405. doi: 10.1021/bi992318d. [DOI] [PubMed] [Google Scholar]
- 13.Hillier W, Wydrzynski T. Physical Chemistry Chemical Physics. 2004;6:4882–4889. [Google Scholar]
- 14.Hillier W, Wydrzynski T. Coordination Chemistry Reviews. 2008;252:306–317. [Google Scholar]
- 15.Dismukes GC, Siderer Y. FEBS lett. 1980;121:78–80. [Google Scholar]
- 16.Hansson Ö, Andréasson L-E, Vänngård T. FEBS lett. 1986;195:151–154. [Google Scholar]
- 17.Evans MC, Nugent JH, Ball RJ, Muhiuddin I, Pace RJ. Biochemistry. 2004;43:989–994. doi: 10.1021/bi035489y. [DOI] [PubMed] [Google Scholar]
- 18.Goldfarb D, Bernardo M, Thomann H, Kroneck PMH, Ullrich V. J Am Chem Soc. 1996;118:2686–2693. [Google Scholar]
- 19.Su JH, Lubitz W, Messinger J. Am Chem Soc. 2008;130:786–787. doi: 10.1021/ja076620i. [DOI] [PubMed] [Google Scholar]
- 20.Su JH, Lubitz W, Messinger J. J Am Chem Soc. 2011;133:12317. [Google Scholar]
- 21.Tagore R, Chen HY, Crabtree RH, Brudvig GW. J Am Chem Soc. 2006;128:9457–9465. doi: 10.1021/ja061348i. [DOI] [PubMed] [Google Scholar]
- 22.Usov OM, Grigoryants VM, Tagore R, Brudvig GW, Scholes CP. Journal of the American Chemical Society. 2007;129:11886–11887. doi: 10.1021/ja073179n. [DOI] [PubMed] [Google Scholar]
- 23.Chu HA, Sackett H, Babcock GT. Biochemistry. 2000;39:14371–14376. doi: 10.1021/bi001751g. [DOI] [PubMed] [Google Scholar]
- 24.Tagore R, Crabtree RH, Brudvig GW. Inorganic Chemistry. 2007;46:2193–2203. doi: 10.1021/ic061968k. [DOI] [PubMed] [Google Scholar]
- 25.Kono Y, Fridovich I. J Biol Chem. 1983;258:6015–6019. [PubMed] [Google Scholar]
- 26.Barynin VV, Vagin AA, Melik-Adamyan VR, Grebenko AI, Khangulov SV, Popov AN, Andrianova ME, Vainshtein BK. Sov Phys-Dokl (Engl Transl) 1986;31:457–459. [Google Scholar]
- 27.Barynin VV, Whittaker MM, Antonyuk SV, Lamzin VS, Harrison PM, Artymiuk PJ, Whittaker JW. Structure. 2001;9:725–738. doi: 10.1016/s0969-2126(01)00628-1. [DOI] [PubMed] [Google Scholar]
- 28.Waldo GS, Yu S, Penner-Hahn JE. J Am Chem Soc. 1992;114:5869–5870. [Google Scholar]
- 29.Waldo GS, Penner-Hahn JE. Biochemistry. 1995;34:1507–1512. doi: 10.1021/bi00005a006. [DOI] [PubMed] [Google Scholar]
- 30.Wu AJ, Penner-Hahn JE, Pecoraro VL. Chem Rev. 2004;104:903–938. doi: 10.1021/cr020627v. [DOI] [PubMed] [Google Scholar]
- 31.Waldo GS, Fronko RM, Penner-Hahn JE. Biochemistry. 1991;30:10486–10490. doi: 10.1021/bi00107a017. [DOI] [PubMed] [Google Scholar]
- 32.Khangulov SV, Barynin VV, Antonyuk-Barynina SV. Biochim Biophys Acta. 1990;1020:25–33. [Google Scholar]
- 33.Khangulov SV, Barynin VV, Voevodskaya NV, Grebenko AI. Biochim Biophys Acta. 1990;1020:305–310. [Google Scholar]
- 34.Whittaker MM, Barynin VV, Antonyuk SV, Whittaker JW. Biochemistry. 1999;38:9126–9136. doi: 10.1021/bi990499d. [DOI] [PubMed] [Google Scholar]
- 35.Meier AE, Whittaker MM, Whittaker JW. Biochemistry. 1996;35:348–360. doi: 10.1021/bi952126s. [DOI] [PubMed] [Google Scholar]
- 36.Schäfer K-O, Bittl R, Lendzian F, Barynin V, Weyhermller T, Wieghardt K, Lubitz W. J Phys Chem B. 2003;107:1242–1250. [Google Scholar]
- 37.Stemmler TL, Sturgeon BE, Randall DW, Britt RD, Penner-Hahn JE. J Am Chem Soc. 1997;119:9215–9225. [Google Scholar]
- 38.Larson E, Lah MS, Li X, Bonadies JA, Pecoraro VL. Inorg Chem. 1992;31:373–378. [Google Scholar]
- 39.Stich TA, Whittaker JW, Britt RD. J Phys Chem B. 2010;114:14178–14188. doi: 10.1021/jp908064y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khangulov S, Sivaraja M, Barynin VV, Dismukes GC. Biochemistry. 1993;32:4912–4924. doi: 10.1021/bi00069a028. [DOI] [PubMed] [Google Scholar]
- 41.Fronko RM, Penner-Hahn JE, Bender CJ. J Am Chem Soc. 1988;110:7554–7555. [Google Scholar]
- 42.Berthold DA, Babcock GT, Yocum CF. FEBS lett. 1981;134:231–234. [Google Scholar]
- 43.Sienkiewicz A, Smith BG, Veselov A, Scholes CP. Rev of Sci Instrum. 1996;67:2134–2138. [Google Scholar]
- 44.Lee B, Usov OM, Grigoryants VM, Myers WK, Shapleigh JP, Scholes CP. Biochemistry. 2009;48:8985–8993. doi: 10.1021/bi900833f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao Y, Lukoyanov DA, Toropov YV, Wu K, Shapleigh JP, Scholes CP. Biochemistry. 2002;41:7464–7474. doi: 10.1021/bi0256274. [DOI] [PubMed] [Google Scholar]
- 46.Burdi D, Willems J-P, Riggs-Gelasco P, Antholine WE, Stubbe J, Hoffman BM. J Am Chem Soc. 1998;120:12910–12919. [Google Scholar]
- 47.Willems J-P, Lee H-I, Burdi D, Doan PE, Stubbe J, Hoffman BM. J Am Chem Soc. 1997;119:9816–9824. [Google Scholar]
- 48.Usov OM, Choi PS, Shapleigh JP, Scholes CP. J Am Chem Soc. 2006;128:5021–5032. doi: 10.1021/ja055323f. [DOI] [PubMed] [Google Scholar]
- 49.Myers WK, Scholes CP, Tierney DL. J Am Chem Soc. 2009;131:10421–10429. doi: 10.1021/ja900866y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Werst MM, Kennedy MC, Beinert H, Hoffman BM. Biochemistry. 1990;29:10526–10532. doi: 10.1021/bi00498a015. [DOI] [PubMed] [Google Scholar]
- 51.Veselov A, Sun H, Sienkiewicz A, Taylor H, Burger RM, Scholes CP. J Am Chem Soc. 1995;117:7508–7512. [Google Scholar]
- 52.Veselov AV, Osborne JP, Gennis RB, Scholes CP. J Am Chem Soc. 2000;122:8712–8716. [Google Scholar]
- 53.Hoffman BM, DeRose VJ, Doan PE, Gurbiel RJ, Houseman ALP, Telser J. In: Biological Magnetic Resonance, Vol. 13: EMR of Paramagnetic Molecules. Berliner LJ, Reuben J, editors. Vol. 13 Plenum; New York: 1993. [Google Scholar]
- 54.Randall DW, Gelasco A, Caudle MT, Pecoraro VL, Britt RD. J Am Chem Soc. 1997;119:4481–4491. [Google Scholar]
- 55.Yeagle GJ, Gilchrist ML, Jr, Walker LM, Debus RJ, Britt RD. Philos Trans R Soc Lond B Biol Sci. 2008;363:1157–1166. doi: 10.1098/rstb.2007.2211. discussion 1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yeagle GJ, Gilchrist ML, McCarrick RM, Britt RD. Inorg Chem. 2008;47:1803–1814. doi: 10.1021/ic701680c. [DOI] [PubMed] [Google Scholar]
- 57.Sproviero EM, Gascon JA, McEvoy JP, Brudvig GW, Batista VS. J Am Chem Soc. 2008;130:3428–3442. doi: 10.1021/ja076130q. [DOI] [PubMed] [Google Scholar]
- 58.Kulik LV, Epel B, Lubitz W, Messinger J. J Am Chem Soc. 2007;129:13421–13435. doi: 10.1021/ja071487f. [DOI] [PubMed] [Google Scholar]
- 59.Peloquin JM, Campbell KA, Randall DW, Evanchik MA, Pecoraro VL, Armstrong WH, Britt RD. J Am Chem Soc. 2000;122:10926–10942. [Google Scholar]
- 60.Anderson PW. Exchange in Insulators: Superexchange, Direct Exchange, and Double Exchange. In: Rado GT, Suhl H, editors. Magnetism. I. Academic Press; New York: 1963. pp. 28–83. [Google Scholar]
- 61.Noodleman L, Peng CY, Case DA, Mouesca JM. Coord Chem Rev. 1995;144:199–244. [Google Scholar]
- 62.Sinnecker S, Neese F, Noodleman L, Lubitz W. J Am Chem Soc. 2004;126:2613–2622. doi: 10.1021/ja0390202. [DOI] [PubMed] [Google Scholar]
- 63.Sinnecker S, Neese F, Lubitz W. J Biol Inorg Chem. 2005;10:231–238. doi: 10.1007/s00775-005-0633-9. [DOI] [PubMed] [Google Scholar]
- 64.Schinzel S, Kaupp M. Can J Chem. 2009;87:1521–1539. [Google Scholar]
- 65.Pantazis DA, Orio M, Petrenko T, Zein S, Lubitz W, Messinger J, Neese F. Phys Chem Chem Phys. 2009;11:6788–6798. doi: 10.1039/b907038a. [DOI] [PubMed] [Google Scholar]
- 66.Burdi D, Sturgeon BE, Tong WH, Stubbe J, Hoffman BM. J Am Chem Soc. 1996;118:281–282. [Google Scholar]
- 67.Sturgeon BE, Burdi D, Chen S, Huynh B-H, Edmondson DE, Stubbe J, Hoffman BM. J Am Chem Soc. 1996;118:7551–7557. [Google Scholar]
- 68.Stoll S, Schweiger A. J Magn Reson. 2006;178:42–55. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 69.Thomann H, Bernardo M, Goldfarb D, Kroneck PMH, Ullrich V. J Am Chem Soc. 1995;117:8243–8251. [Google Scholar]
- 70.Tan X, Bernardo M, Thomann H, Scholes CP. J Chem Phys. 1995;102:2675–2690. [Google Scholar]
- 71.Baute D, Goldfarb D. J Phys Chem A. 2005;109:7865–7871. doi: 10.1021/jp052132q. [DOI] [PubMed] [Google Scholar]
- 72.Britt RD, Campbell KA, Peloquin JM, Gilchrist ML, Aznar CP, Dicus MM, Robblee J, Messinger J. Biochim Biophys Acta. 2004;1655:158–171. doi: 10.1016/j.bbabio.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 73.Su JH, Messinger J. Appl Magn Reson. 2010;37:123–136. doi: 10.1007/s00723-009-0051-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.