

Abstract
Projections from thalamic intralaminar nuclei convey sensory signals to striatal cholinergic interneurons. These neurons respond with a pause in their pacemaking activity, enabling synaptic integration with cortical inputs to medium spiny neurons (MSNs), thus playing a crucial role in motor function. In mice with the DYT1 dystonia mutation, stimulation of thalamostriatal axons, mimicking a response to salient events, evoked a shortened pause and triggered an abnormal spiking activity in interneurons. This altered pattern caused a significant rearrangement of the temporal sequence of synaptic activity mediated by M1 and M2 muscarinic receptors in MSNs, consisting of an increase in postsynaptic currents and a decrease of presynaptic inhibition, respectively. Consistent with a major role of acetylcholine, either lowering cholinergic tone or antagonizing postsynaptic M1 muscarinic receptors normalized synaptic activity. Our data demonstrate an abnormal time window for synaptic integration between thalamostriatal and corticostriatal inputs, which might alter the action selection process, thereby predisposing DYT1 gene mutation carriers to develop dystonic movements.
Introduction
DYT1 dystonia is a common inherited form of primary dystonia associated with the DYT1 mutation in the gene encoding the protein torsinA (Ozelius et al., 1997), but it is still unclear how this leads to the symptoms of the disease.
Abnormal sensorimotor integration is an important pathophysiological mechanism of dystonia. Neurophysiological studies have demonstrated dysfunction of sensory processing in patients, assigning a crucial role to the basal ganglia in linking sensory information to appropriate motor responses (Hallett, 1995; Edwards et al., 2003; Tinazzi et al., 2009). Additionally, neuroimaging studies revealed that both clinically affected patients and DYT1 carriers show overexcitability of the sensorimotor system (Carbon et al., 2010). However, the reasons why the integrative process between sensory inputs and motor function is impaired in DYT1 dystonia remain unknown. One interesting indication derives from the clinical observation that dystonic symptoms are exacerbated by unexpected, salient stimuli that interrupt and redirect the current focus of attention and motor activity, such as anxiety and emotions, whereas dystonic symptoms often disappear during sleep (Marsden and Harrison, 1974; Fish et al., 1991).
The striatum, where thalamostriatal and corticostriatal projections merge, has been proposed as the locus of these attentional mechanisms, providing an output signal that permits a shift in attention and reallocates the resources necessary for action selection (Kimura et al., 2004; Zink et al., 2006; Thorn and Graybiel, 2010). The cortical and thalamic pathways have dissimilar synaptic properties and connections, and code temporally distinct inputs to medium spiny neurons (MSNs), thereby influencing striatal activity and motor function (Smith et al., 2009; Doig et al., 2010). Signals regarding salience of stimuli arise in the caudal intralaminar thalamic nuclei (Matsumoto et al., 2001; Van der Werf et al., 2002), which provide the striatum with a robust glutamatergic innervation. Both MSNs and cholinergic interneurons (ChIs) are targets of this projection (Smith et al., 2004). Recent data suggest that ChIs play a primary role in the integrative process between thalamostriatal and corticostriatal inputs (Ding et al., 2010). Accordingly, lesions of intralaminar nuclei produce attention deficits and sensory neglect (Matsumoto et al., 2001).
Few electrophysiological studies have explored the differential properties of corticostriatal and thalamostriatal synapses (Smeal et al., 2007; Ding et al., 2008), and none have examined these circuits in animal models of dystonia. In previous studies in mice overexpressing human mutant torsinA (hMT1 mice) (Sharma et al., 2005) we found a significant alteration in cholinergic neurotransmission, which was responsible for impairment of striatal synaptic plasticity (Martella et al., 2009). Here, electrophysiological recordings were obtained from slices preserving the integrity of corticostriatal and thalamostriatal axons. We also examined vesicular glutamate transporters (VGluT), 1 and 2, as markers of cortical and thalamic inputs, respectively (Fremeau et al., 2001) and NMDA glutamate receptor subunit composition and function. Our results identify a fundamental alteration in striatal circuit following activation of thalamostriatal axons, which disrupts normal corticostriatal synaptic activity of MSNs and might alter action selection, leading to motor impairment.
Materials and Methods
Mouse models and slice preparation.
Treatment and handling of mice were performed in accordance with the EC and Italian guidelines (86/609/EEC; D.Lvo 116/1992) and approved by the University of Rome Tor Vergata (n. 153/2001A). Breeding colonies of both hWT and hMT1 mice were established at our animal house (Sharma et al., 2005). Male mice were used for the experiments. For each strain, nontransgenic littermates were used as controls and are defined as nontransgenic (NT) mice (C57BL/6). Thus, the genetic background was identical between the control and experimental animals, with the only difference being the presence of the transgene (Smeal et al., 2007). DNA extracted from mouse tail using the Extract-N-Amp Tissue PCR Kit (Catalog #XNAT2; Sigma-Aldrich) was used for genotyping as described previously (Martella et al., 2009). Male knock-in mice heterozygous for ΔE-torsinA (Tor1a ΔGAG+/− mice) and their wild-type littermates (Tor1a ΔGAG/+ mice) (Goodchild et al., 2005) were purchased from The Jackson Laboratory. Mice genotyping was performed on DNA extracted from mouse tail using the Extract-NAmp Tissue PCR Kit (XNAT2 kit; Sigma-Aldrich), as indicated by The Jackson Laboratory (http://jaxmice.jax.org/strain/006251.html), with minor modifications.
Mice were killed by cervical dislocation under ether anesthesia and the brain immediately removed from the skull. Parahorizontal slices (300 μm) were cut with a vibratome in Krebs' solution containing (in 16 mm): 126 NaCl, 2.5 KCl, 1.3 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 10 glucose, and 18 NaHCO3), bubbled with 95% O2 and 5% CO2 as described previously (Smeal et al., 2007; Ding et al., 2008, 2010; Martella et al., 2009). For chronic in vivo treatment, trihexyphenidyl (20 mg/kg, i.p) was injected daily for 3 d (last injection 100 min before killing).
Electrophysiology.
After 30–60 min recovery at room temperature (RT), individual slices were transferred to a recording chamber (0.5–1 ml volume) and continuously superfused with oxygenated Krebs' medium (2.5–3 ml/min, 32–35°C). Recordings were performed on individual MSNs or ChIs visualized by means of IR-DIC videomicroscopy with an Olympus BX-51WIF camera/controller system (Olympus). For all experiments, 50 μm picrotoxin was added to the superfusion medium to block GABAA receptor-mediated synaptic responses. For voltage-clamp experiments from MSNs, pipettes (3–5 MΩ) were filled with Cs+ internal solution containing (in mm): 120 CsMeSO3, 15 CsCl, 8 NaCl, 10 TEA-Cl, 10 HEPES, 2–5 QX-314, 0.2 EGTA, 2 Mg-ATP, and 0.3 Na-GTP, pH 7.3 adjusted with CsOH. For voltage-clamp recordings from interneurons the internal solution consisted of (in mm): 125 K+-gluconate, 10 NaCl, 1 CaCl2, 2 MgCl2, 10 HEPES, 0.3 Na-GTP, and 2 Mg-ATP, pH adjusted to 7.3 with KOH. Holding potential was −60 mV.
Current-clamp recordings were made in the perforated-patch configuration. Gramicidin was used as the pore-forming agent and was added to the pipette solution at an approximate concentration of 20 μg/ml. The perforation process was considered complete when the amplitude of the action potentials was steady and >60 mV, and electrode resistance was steady and <50 MΩ. Data were recorded with Multiclamp 700A (Molecular Devices). Membrane currents were continuously monitored and access resistance measured in voltage-clamp was in the range of 5–30 MΩ before electronic compensation (60–80% routinely used). Steel concentric electrodes (FHC) were used for stimulation (50–400 μs). Electrodes were placed in layer V of the cortex for activation of the cortical projection and close to the border of thalamic reticular nucleus for stimulation of the thalamic fibers (see Fig. 1). To calculate NMDA/AMPA receptor ratios, MSNs were held at +40 mV and cortical or thalamic stimulation was delivered. At least 10 EPSCs at +40 mV were recorded before and after bath-application of MK-801 (30 μm). The average response in the presence of MK-801 (AMPAR-EPSC only) was subtracted from the trace recorded in its absence to calculate an average NMDAR-EPSC. NMDA/AMPA receptor ratio was calculated by dividing the peak of the NMDA-EPSC by the peak of the AMPA-EPSC. Latency time of MSN EPSCs was calculated as described previously (Smeal et al., 2007). For NMDA/NR2B ratio, ifenprodil (3 μm) or TAT2B (1 nm) were applied before MK-801 to block the NR2B-mediated current fraction. The peak of the NR2B-EPSC (calculated by subtraction) was divided by the peak of the NMDAR-EPSC and an average NMDA/NR2B ratio was estimated. AMPA-mediated quantal events were recorded 50 ms after each stimulus (5 pulse, 25 Hz, delivered at 0.01 Hz for both cortical and thalamic stimulation) for a maximum period of a 300 ms (Ding et al., 2008). External Ca2+ was replaced with Sr2+ (2 mm) and MK-801 (30 μm) was added to block NMDA currents. All events were analyzed (Minianalysis; Synaptosoft) as described previously (Sciamanna et al., 2009).
Figure 1.
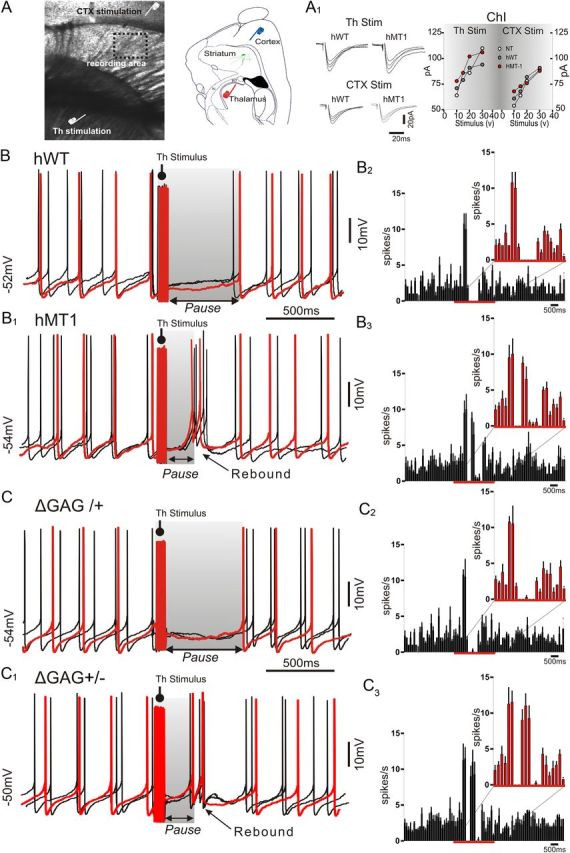
Abnormal pause response evoked by thalamic stimulation in ChIs from hMT1 and knock-in mice. A, Infrared image of a parahorizontal slice used for electrophysiology. Schematic drawing showing the position of stimulating electrodes in the cortex (blue) and thalamus (red). Rt, reticular nucleus; ic, internal capsula. A1, Superimposed traces of EPSCs evoked by thalamic (Th) and cortical (CTX) stimulation in ChIs from hWT and hMT1 mice. Graphs summarize EPSC amplitude changes, following progressive increases in stimulation intensity (10–40 V). B, Sample traces of three consecutive responses in perforated patch-clamp from a cholinergic interneuron after thalamic train stimulation (50 Hz, 10 pulses). In hWT mice, this protocol induced an evident pause in rhythmic firing activity of ChIs lasting ∼500 ms, whereas it resulted in a shorter pause in hMT1 interneurons (B1), followed by a rebound in firing activity. B2, B3, Peristimulus time histograms (bin width, 50 ms) of interneuron firing show the time course of spike discharge following thalamic stimulation in hWT (B2) and hMT1 mice (B3). Note that spiking cessation in hWT mice is more prominent than in hMT1 littermates. C, Superimposed traces of three responses in perforated patch-clamp from a cholinergic interneuron after thalamic stimulation obtained from heterozygous mutant DYT1 knock-in mice and their littermates (ΔGAG+/−, ΔGAG/+). Thalamic activation caused a pause response in the spiking activity in ΔGAG+/− mice that was significantly shorter than in ΔGAG/+ and was followed by a bursting activity (C1). Histograms show the time course of spiking activity after thalamic stimulation in ΔGAG/+ (C2) and ΔGAG+/− mice (C3). In each panel, sample traces are highlighted in red.
To calculate the decay times of averaged currents, curves were fitted with a double exponential equation of the form I(t) =If exp(−t/τf) +Is exp(−t/τs), where If and Is are the amplitudes of the fast and slow decay components, and τf and τs are their respective decay time constants. To compare distinct decay times we used a weighted mean decay time constant τw = [If /(If +Is)]/tf + [Is/(If +Is)]/ts (Stocca and Vicini, 1998). NMDA-mediated membrane currents were induced by focal pressure-ejection of 500 μm NMDA, in tetrodotoxin (1 μm), via a puffer pipette controlled by a Picospritzer (200 μs; puff intensity at 3–5 psi). Coefficient of variation (CV) was calculated as the fraction between SD of interspike intervals and the distribution mean value. For the analysis of firing pattern (see Fig. 2), the onset of suppression in tonic firing was defined as the first of three consecutive bins (bin width, 50 ms) that deviated significantly from the baseline firing frequency before the stimulus, whereas the offset was defined as the first of three consecutive bins that returned to the baseline level (Aosaki et al., 1994a; Ding et al., 2010).
Figure 2.

Long-lasting thalamic activation alters activity pattern of interneurons recorded from hMT1 mice. A, Representative trace from hMT1 mice showing that, without synaptic stimulation, ChIs exhibit a rhythmic pacemaking activity. B, After a repetitive stimulation (0.1 Hz, 10 min), the excitability of interneurons shifts toward a more irregular activity pattern, characterized by brief bursts and pauses of different duration (black arrows). Graphs summarize the changes of CV of firing activity measured in the three genotypes.
Chemicals.
Drugs were from Tocris Cookson, except for BAPTA, trihexyphenidyl, and hemicholinium-3 (Sigma).
Lentiviral vector production.
We used the p277.pCCLsin.PPT.hPGK.GFP.WPRE transfer vector, which carries the green fluorescent protein (GFP) sequence under control of the PGFK promoter. The VSV-G-pseudotyped lentiviral vectors were generated by calcium phosphate transfection of HEK293T cells with a mixture of four plasmids that are essential to produce third-generation lentiviruses (kindly provided by Professor L. Naldini). Cells were cultured in DMEM supplemented with 10% fetal bovine serum, 100 U/ml penicillin G, and 100 μg/ml streptomycin at 37°C in 5% CO2. Cells were plated at 2.7–3 × 106 cells in a 10 cm dish 24 h before transfection. Fifteen hours after incubation with the transfection mix at 37°C, the cells were washed with HBSS, and complete DMEM was added. Virus-containing medium was harvested 40 h after transfection, filtered through a 0.45 μm Durapore Stericup unit, and concentrated by two ultracentrifugation steps. The viral pellet was finally resuspended in PBS with 1% bovine serum albumin (BSA) and stored at −80°C until use. Viral content was measured by p24 antigen enzyme-linked immunosorbent assay (RETROtek; ZeptoMetrix).
Western blot.
To determine VGluT1 and VGluT2 expression levels, protein extracts were obtained by homogenizing striata (from at least five mice) in a buffer containing 50 mm Tris, pH 7.4, 150 mm NaCl, 1% 220 Triton X-100, 0.25% Na deoxycholate, 5 mm MgCl2, 0.1% SDS, 1 mm 221 EDTA, and 1% protease inhibitor mixture (Sigma). Crude lysates were sonicated and incubated on ice for 1 h. Samples were centrifuged at 13,000 rpm for 15 min at 4°C and the supernatants were collected. Five micrograms (VGlut1) and 25 μg (VGluT2) of striatal extract were denaturated in sample buffer without boiling and loaded onto an SDS polyacrylamide gel (8%). Gels were blotted onto a polyvinylidene difluoride (PVDF) membrane. Immunodetection was performed by rabbit polyclonal anti-VGluT1 1:15,000 overnight (ON) at 4°C and anti-VGluT2 1:3000 ON at 4°C (Synaptic Systems; Catalog #135302 and 35403) and mouse monoclonal anti-actin, 1:5000 for 30 min at RT (Sigma; Catalog #T4026) as loading control. Horseradish peroxidase (HRP)-conjugated secondary antibodies (Bio-Rad), ECL-Plus reagent (GE Healthcare Europe), and the storm 840 acquisition system were used for detection. Quantification of the bands intensity on scanned filters was achieved by National Institutes of Health (NIH) ImageJ software (Abramoff et al., 2004).
Immunohistochemistry.
Four weeks after viral injection, mice were deeply anesthetized (avertin), perfused through the aorta with ice-cold 4% paraformaldehyde, and equilibrated with 30% sucrose overnight. Then 30-μm-thick coronal brain sections were cut with a freezing microtome. GFP-positive slices were pre-incubated with 0.5% PBS Triton X-100 for 1 h at RT. After 1 h RT of incubation with a blocking solution containing 5% normal donkey serum and 1% BSA in 0.05% Tween PBS (TwPBS), the sections were incubated with 1% BSA in TwPBS 0.05% and the following antibodies for three overnights: rabbit polyclonal anti-NR2B antibody 1:200 (A6474; Invitrogen), guinea pig polyclonal anti-VGluT1 1:1000 (135–304; Synaptic System), and guinea pig polyclonal anti-VGluT2 1:1300 (AB5907; Millipore). After being washed three times with 0.05% TwPBS, the sections were incubated with Cy-3-conjugated donkey anti-rabbit or anti-guinea pig secondary antibodies (Jackson ImmunoResearch) 1:200 for 2 h at RT and rinsed in TwPBS. Sections were mounted on poly-lysine-coated slides, air-dried, and coverslipped. All the experimental groups (three mice for each group) were scarified and processed for immunofluorescence experiments at the same time to reduce variability due to the experimental procedures. All images for quantitative analysis were acquired with a Zeiss LSM 700 confocal laser scanning microscope and processed using NIH ImageJ software. Images were acquired in the dorsal striatum in the GFP-positive area where isolated dendritic segments of MSNs could be easily identified. To clearly resolve the dendritic branches of MSNs and the relative synaptic afferents, we used a 100×/1.3 oil objective with a digital zoom factor of 2×. Section images (1024 × 1024) in the z-dimension (z-spacing, 0.3 μm) were collected, ensuring that an entire segment of a dendrite (diameter of ∼0.4–0.7 μm), spanning multiple confocal planes, was fully captured from the surface of the section. The confocal pinhole was kept at 1, the gain and offset setting were set to prevent saturation of the brightest signals. The expression of each marker (VGluT1, VGluT2, and NR2B) was analyzed on brain slice images acquired with the same settings, including laser power, photomultiplicator gain, and offset. By means of the ImageJ software, the specific signal in transduced MSN dendrites was isolated and identified within each optical section for its colocalization with GFP. The merged image of the isolated colocalization signal and the GFP channel was then projected on the z-axis and quantified. For each dendritic tract, we calculated the mean percentage of GFP area that overlapped with the specific marker.
Synaptosome preparation.
Brain samples were processed according to the synaptosomal preparation previously described (Musumeci et al., 2009). Briefly, the two striata of each mice were homogenized in 2 ml of buffer [0.32 m sucrose, 1 mm EDTA, 1 mg/ml BSA, 5 mm HEPES (pH 7.4, reached using 1 m NaOH), 10 mm NaF, 25 mm Na3VO4, and 1% protease inhibitor mixture] by use of a Dounce homogenizer (10 strokes and 520 rpm). The crude homogenate was centrifuged for 10 min at 3000 × g. The supernatants were subsequently centrifuged for 12 min at 14,000 × g and resultant pellets were resuspended in 220 μl of Krebs–Ringer buffer (140 mm NaCl, 5 mm KCl, 10 mm HEPES, 1 mm EDTA, 5 mm glucose, pH 7.4, 10 mm NaF, 1 mm Na3VO4, and 1% protease inhibitor mixture) and 180 μl of Percoll (Sigma-Aldrich, P7828). Samples were then centrifuged for 3 min at 14,000 ×g. The synaptosome fraction of the gradient was removed, washed twice with 500 μl of Krebs–Ringer buffer, and centrifuged for 1 min at 14,000 × g. The final pellets were resuspended in 40 μl of Krebs–Ringer buffer. All steps of the procedure were performed at 4°C using ice-cold buffers. Each sample was sonicated in 1% SDS and boiled for 5 min. The protein content was quantified and aliquots were frozen at −80°C until use. Equal amounts of total protein (40 μg) from each sample were loaded onto 10% polyacrylamide gel and transferred onto PVDF membranes. The blots were probed using rabbit polyclonal anti-NR2B 3 ON 4°C (A6474; Invitrogen, 1:250), mouse monoclonal anti PSD-95 1:20,000 30 min RT, and mouse monoclonal anti-actin 30 min RT (Sigma, Catalog #T4026, 1:20,000). HRP-conjugated secondary antibodies (Bio-Rad), ECL-Plus reagent (GE Healthcare Europe GmbH), and the Storm 840 acquisition system were used for detection. Quantification of the band's intensity on scanned filters was achieved by NIH ImageJ software (Abramoff et al., 2004).
Statistical analysis.
Numerical data are presented as means ± SEM. Nonmatched samples were analyzed with the nonparametric Mann–Whitney rank sum test. Matched samples were analyzed with Wilcoxon signed rank test. ANOVA test with a post hoc Tukey's test or Holm–Sidak test (only for morphology experiments) was performed among groups (p < 0.05; α = 0.01). Data were elaborated by means of Origin 8.0 (Microcal) and Prism 5.0 (GraphPad). The significance level was set at p < 0.05. Unless otherwise specified, a single p value >0.05 is meant to indicate the nonsignificance among the three genotypes.
Results
Abnormal pause response in ChIs elicited by thalamostriatal stimulation
Electrophysiology was performed by using mice overexpressing either normal human torsinA (hWT) or its mutant form (hMT1), and comparing the results to NT littermates (Sharma et al., 2005). In another set of recordings, the heterozygous mutant DYT1 knock-in model (ΔGAG+/−) was used (Goodchild et al., 2005). ChIs were recorded from parahorizontal brain slices, in which axonal projections arising both from cortex and intralaminar thalamic nuclei are preserved, allowing selective activation of the two distinct pathways (Fig. 1A) (Smeal et al., 2007; Ding et al., 2008). In such experimental conditions, ChIs exhibit a tonic firing activity, which did not differ among the different strains of mice (NT: 2.3 ± 0.7 Hz, n = 40; hWT: 2.0 ± 0.7 Hz, n = 33; hMT1: 2.4 ± 0.8 Hz, n = 36; p > 0.05) (Sciamanna et al., 2011). Synaptic stimulation from either cortex or thalamus induced an EPSC, comparable among genotypes (Fig. 1A1).
In vivo recordings have shown that during sensorimotor learning, a conditioned stimulus interrupts the pacemaking activity of a class of striatal neurons termed tonically active neurons (TANs) that are believed to correspond to ChIs (Aosaki et al., 1994a). This response consists of an initial depolarizing phase followed by a pause in spiking and ensuing rebound excitation, and requires the integrity of thalamic inputs and activation of dopamine D2 receptors (D2Rs) (Aosaki et al., 1994b; Matsumoto et al., 2001; Goldberg and Reynolds, 2011).
A train of stimuli (five stimuli, 50 Hz) was delivered to thalamostriatal axons to mimic the activity pattern observed in vivo (Aosaki et al., 1994a). In NT and hWT mice, this protocol induced a pause response (NT: 516.8 ± 68 ms; hWT: 533.6 ± 58 ms; n = 8 per group; Fig. 1B) followed by a recovery to tonic firing activity. In ChIs from hMT1 mice the same stimulation elicited a pause, which was significantly shorter in duration (348.6 ± 54 ms; n = 8 p < 0.005), and was followed by a burst of spikes (hMT1: from 0.8 ± 0.2 Hz to 3.5 ± 0.64 Hz; n = 8; p < 0.005; Fig. 1B1–B3). Peristimulus histograms show the significant change in frequency pattern following thalamic stimulation (Fig. 1B2,B3). A similar protocol applied to the cortex failed to induce any pause (data not shown), regardless of the stimulation intensity, in accordance with recent observations (Ding et al., 2010), and with the anatomical evidence that ChIs receive a prominent innervation from caudal intralaminar thalamic nuclei, as compared with the fewer synaptic cortical inputs (Smith et al., 2004).
Then, in an attempt to rule out that the changes in the pause response were determined by the overexpression of mutant torsinA, we replicated these experiments in a different DYT1 model, the heterozygous mutant DYT1 knock-in mouse, where the ΔGAG mutation is inserted in the endogenous gene and regulated by its own promoter (Goodchild et al., 2005). As observed in hWT animals, in control mice (ΔGAG/+), repetitive thalamic activation caused a pause response followed by recovery to rhythmic firing activity (573.8 ± 61 ms; n = 6; Fig. 1C). Conversely, in mutant mice (ΔGAG+/−), the same stimulation protocol induced a much shorter pause in firing activity (290.7 ± 44 ms; n = 8, p < 0.05; Fig. 1C1), followed by a rebound burst of spikes.
The inability of cortical stimulation to induce any pause led us to investigate further the abnormal responsiveness of ChIs to thalamic activation. Because at rest the rhythmic pacemaking activity was normal in hMT1 mice (Fig. 2A), a repetitive stimulation (0.1 Hz, 10 min) was delivered. This protocol modified the activity pattern of interneurons recorded from hMT1 mice, causing a significant reduction in the regularity of autonomous firing pattern (Fig. 2B), as measured by the CV, compared in the three genotypes (hMT1: from 1.06 ± 0.13 to 2.55 ± 0.34; n = 8; p < 0.005; Fig. 2). No change in activity pattern was recorded after cortical stimulation (hMT1: from 1.34.± 0.17 to 1.26 ± 0.14; n = 5; p > 0.05; data not shown).
Mechanisms of the pause response
We have previously reported that ChIs from hMT1 mice exhibit a fundamental alteration in their response to D2R activation, consisting of a paradoxical excitation rather than the expected inhibition (Pisani et al., 2006; Sciamanna et al., 2011). D2R activation is one of the factors contributing to generation of the pause response (Goldberg and Reynolds, 2011). Accordingly, in the presence of sulpiride (3 μm, 15 min), a selective D2R antagonist, thalamic stimulation failed to evoke any pause response both in hWT and hMT1 mice (Fig. 3).
Figure 3.

Mechanisms underlying the pause response evoked by thalamic activation. A, B, Samples traces of perforated patch-clamp recordings of two consecutive responses from ChIs before (control) and after treatment with the D2R antagonist sulpiride in hWT (A) and hMT1 (B) mice. In the presence of sulpiride (3 μm, 10 min) thalamic stimulation (50 Hz, 10 pulses) was unable to elicit the pause response in ChIs. C, In voltage-clamp recordings from hMT1 mice, thalamic stimulation (Th Stim) elicited an inward current in ChIs (HP −60 mV). Superimposed traces show the inward current before and during application of sulpiride (3 μm, 10 min), ω-conotoxin GVIA (ω-conotoxin, 1 μm, 10 min), and apamin (3 μm, 10 min) that suppressed the inward current evoked by thalamic activation. Similarly, intrapipette BAPTA (10 mm) was able to block the thalamically induced current. Results are summarized in the plot (C1).
In another group of experiments, we analyzed more in detail the mechanisms underlying the generation of the pause response. To this aim, ChIs from hMT1 mice were recorded in the voltage-clamp mode in picrotoxin (50 μm) and held at −60 mV. In this condition, repetitive thalamic activation caused an inward current (19.5 ± 2.1 pA, n = 12, Fig. 3C), consistent with the abnormal depolarizing response recorded in the current-clamp mode. We confirmed the D2R dependence of the pause response, as sulpiride abolished the inward current. Because D2Rs are coupled to N-type (Cav2.2) HVA calcium channels, we then explored the capability of ω-conotoxin GVIA, a selective blocker of this class of channels to affect the pause. In the presence of ω-conotoxin GVIA (1 μm) the thalamically induced inward current was significantly reduced (Fig. 3C). The Cav2.2 current activates the small conductance (SK) Ca2+-activated K+ current (Goldberg and Wilson, 2005) that gives rise to the medium-afterhyperpolarization. Accordingly, apamin (3 μm) was able to block the current generated by thalamic activation. Additionally, to demonstrate the calcium dependence of this pause response, we performed a set of experiments by using intrapipette BAPTA, a calcium chelator. With BAPTA in the recording pipette, we were unable to evoke the pause response (Fig. 3C).
Functional and anatomical connectivity of corticostriatal and thalamostriatal afferents
To verify whether the changes in excitability observed in ChIs affected synaptic properties of MSNs, glutamate receptor-mediated EPSCs were evoked in MSNs, in the presence of the GABAA receptor blocker picrotoxin (10 μm), by stimulating either corticostriatal or thalamostriatal afferents (Fig. 1A). EPSCs were fully abolished by antagonists for AMPA and NMDA receptors (6-cyano-7-nitroquinoxaline-2,3-dione [CNQX] 10 μm, MK-801 30 μm, respectively) (Fig. 4A). All the averaged latencies were <1 ms (corticostriatal synapses 0.44 ± 0.07 ms, thalamostriatal synapses 0.50 ± 0.08 ms; n = 13 for each group; p > 0.05; Fig. 4A) consistent with a monosynaptic connection (Smeal et al., 2007). No significant difference was found among MSNs recorded from NT, hWT, and hMT1 mice (n = 15 per genotype, p > 0.05). As previously reported (Ding et al., 2008), measurement of paired-pulse ratio (PPR; interstimulus interval 10–1000 ms) resulted in a synaptic facilitation, whereas the same paradigm elicited a depression at thalamostriatal synapses (Fig. 4B). PPR values were not statistically different between hWT and hMT1 mice, compared with NT controls (interstimulus interval: 50 ms; cortical stimulus: NT: 1.49 ± 0.37, hWT: 1.91 ± 0.33, hMT1: 1.59 ± 0.16; n = 8; p > 0.05; thalamic stimulus: NT: 0.89 ± 0.11, hWT: 0.84 ± 0.18, hMT1: 0.78 ± 0.14; n = 8; p > 0.05; Fig. 4B).
Figure 4.

Synaptic and anatomical connectivity is unaltered in transgenic mice. A, Sample traces represent the average of 10 consecutive EPSCs evoked by cortical (CTX) and thalamic (Th) stimulation in MSNs of hMT1 mice. EPSCs were fully inhibited by a combination of NMDA and AMPA receptor antagonists (MK-801 30 μm, and CNQX 10 μm, respectively, gray traces). Bar plots show the latency values for both cortical (CTX) and thalamic (Th) stimulation. Short latency times (<1 ms) are consistent with a monosynaptic transmission. B, Summary graphs of PPR recorded from hWT and hMT1 MSNs plotted as a function of interstimulus interval for CTX (blue circles) and Th stimulation (red circles). C, D, Confocal images of striatal coronal sections from mice injected with a GFP-lentiviral vector. The infection area is represented in green, while the red immunostaining shows the VGluT2- and VGluT1-positive synaptic terminals. Projection of a series of optical sections showing isolated GFP-positive dendrites and spines (high magnification of the white boxes) contacted by VGluT2 and VGluT1-positive synaptic terminals. C1, D1, The histograms summarize the percentage of GFP area that colocalizes with VGluT2 and VGluT1 signals (normalized to the NT control values).
To determine whether the anatomical connectivity of corticostriatal and thalamostriatal afferents to striatum was modified in hMT1 mice, we performed a Western blot analysis of VGluTs (1 and 2), which are selective markers of cortical and thalamic afferents, respectively (Fremeau et al., 2001). We observed comparable expression of VGluT1 and VGluT2 proteins in the striatum of the three genotypes (NT, VGluT1, n = 7; VGluT2, n = 6; hWT, VGluT1, n = 7; VGluT2, n = 5; hMT1, VGluT1, n = 7; VGluT2, n = 6; p = 0.868 for VGluT2; p = 0.895 for VGluT1; data not shown). Then, we performed a quantitative confocal microscopic analysis on striatal tissue, from mice expressing GFP-positive MSNs, which were immunolabeled to reveal VGluT1 or VGluT2. To this end, we injected highly concentrated lentiviral particles that carried GFP construct under the PGK promoter. GFP-positive MSN dendrites were selected to perform the morphological analysis (Fig. 4C,D). To analyze the VGluT-synaptic density on the GFP-positive MSNs dendrites, we measured the percentage of GFP area in contact with VGluT2 (Fig. 4C, top) or VGluT1 (Fig. 4D, bottom) signal. The mean percentage of synapses formed by terminals derived from both the cortex and thalamus were similar in the three groups. Indeed, hMT1 and hWT mice showed a mean percentage normalized to NT mice (n = 17 and n = 15) of 83% (±0.3, n = 13) and 73.5% (±19.9, n = 13) for VGluT2 and of 82.7% (±9, n = 15) and 84.8% (±8.1, n = 13) for VGluT1, respectively (p = 0.406 for VGluT2 and p = 0.238 for VGluT1) (Fig. 4C,D). Altogether these data indicate that the pattern of glutamatergic innervation of MSNs is similar in the three genotypes.
Altered muscarinic receptor responses in MSNs
Endogenous acetylcholine (ACh), released from ChIs upon thalamic stimulation, triggers a transient presynaptic inhibition of corticostriatal input to MSNs mediated by M2-like muscarinic ACh receptors (mAChRs), followed by an enhancement of MSN postsynaptic responsiveness, which relies on activation of M1 class of mAChRs (Ding et al., 2010). This sequence of synaptic events has been shown to be exclusively elicited by thalamostriatal stimulation. To address the possible consequences of the abnormal activity pattern of ChIs on MSNs, paired recordings were performed from a neighboring MSN and a ChI (Fig. 5A). When cortical stimulation followed thalamic activation within a time range calculated to comprise the pause response elicited in ChIs (50–500 ms), a synaptic depression was recorded in MSNs of NT and hWT mice (measured at 100 ms; NT: 69.2 ± 2.26% of control; hWT: 69 ± 2.9% of control; n = 5 each group, p < 0.05; Fig. 5B,C), an effect that was prevented by preincubation with the M2 mAChR antagonist methoctramine (300 nm; data not shown; n = 6, p > 0.05). Surprisingly, in hMT1 mice, the M2-dependent inhibitory effect (measured at 100 ms: 76.8 ± 1.24%, Fig. 5B,C) rapidly waned, and EPSC amplitude returned nearly to control values within 500 ms (hMT1: 96.4 ± 4.1% of control; n = 12, p > 0.05; Fig. 5B,C), an effect that was significantly different from the other two groups (Fig. 5C, p < 0.05).
Figure 5.
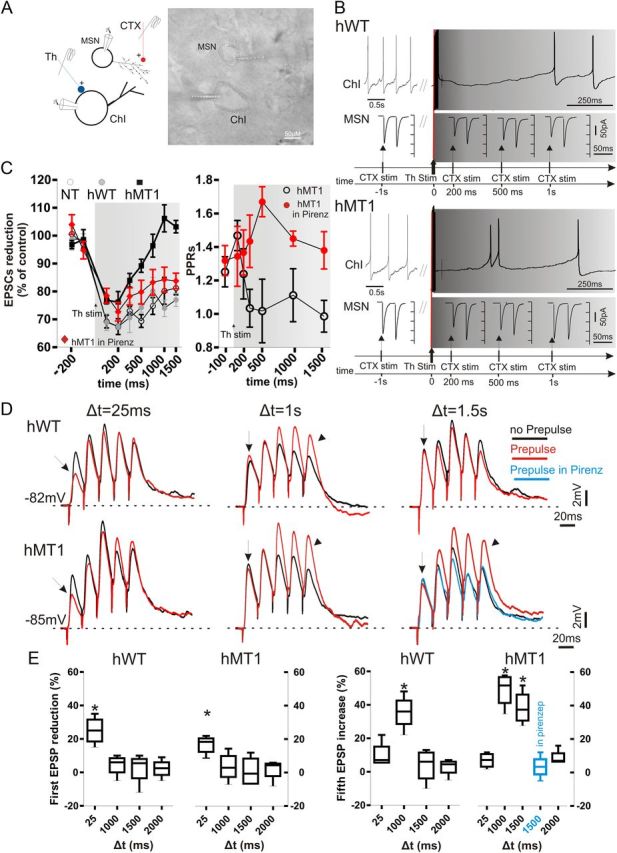
Thalamic stimulation unbalances muscarinic receptor-mediated modulation at corticostriatal synapses. A, Paired recordings were obtained from a ChI and a neighboring MSN during both cortical (CTX, red) and thalamic (Th, blue) stimulation. IR image of the slice preparation (dotted lines mark recording pipettes). B, In hWT mice, the ongoing firing activity of a ChI was interrupted when CTX stimulation was preceded by thalamic stimulation (50 Hz, 10 pulses; gray traces, top right). Such protocol resulted in a decrease of the EPSC amplitude (bottom traces), persisting for up to 1 s after thalamic stimulation (bottom traces). In hMT1 mice (bottom), a similar protocol induced a brief pause in ChI, and a rebound spiking activity. The reduction in EPSC amplitude was smaller than in hWT mice, and declined within 1 s after thalamic stimulation (bottom traces). C, Left, Summary graph of the time dependence of EPSC reduction in the three genotypes, and in hMT1 mice in pirenzepine (red diamonds). C, Right, PPR measurement in hMT1 mice suggests a presynaptic effect at 100 ms, but not at later measurements (500 ms–1.5 s). The presynaptic M2-dependent effect was restored in pirenzepine (red circles). D, A thalamic prepulse (50 Hz, 10 pulses) was given at three times (Δt = 25 ms; Δt = 1 s; Δt = 1.5 s) before the corticostriatal stimulus (5 consecutive pulses, 20 ms interval). Only at short delay (25 ms) a decrease in EPSC amplitude was recorded, suggestive of a presynaptic inhibition in both genotypes (arrows). However, at longer delays (Δt = 1.5) a significant enhancement of EPSP amplitudes was observed only in hMT1 mice (arrowheads). Note that the increase in EPSP amplitude was prevented in the presence of pirenzepine (blue trace). E, Plots summarize the changes (%) in amplitude of the first and fifth EPSP, respectively, at different prepulse delay times.
This rapid decrement of the M2-dependent inhibitory effect in hMT1 mice led us to consider the possibility that a prevailing postsynaptic M1 mAChR action could mask the M2-mediated presynaptic inhibition. This assumption was suggested by the unconventional change in PPR analyzed in hMT1 mice (Fig. 5C), which supported a rapid shift from presynaptic to postsynaptic effect within a short time interval after thalamic stimulation. The M1 mAChR-dependent facilitation normally lasts ∼1 s, whereas in hMT1 mice we observed a long-lasting enhancement up to 1.5 s (Fig. 5C). Thus, we performed paired recordings in the presence of the postsynaptic M1 mAChR antagonist pirenzepine (100 nm, 5 min). In this condition, thalamic stimulation induced a normal presynaptic inhibition in hMT1 mice, not statistically different from that measured in NT and hWT slices (p > 0.05; Fig. 5C), an observation confirmed by the PPR analysis (Fig. 5C). This altered presynaptic effect led us to perform experiments with the M2 agonist oxotremorine. Exogenously applied oxotremorine (300 nm, 5 min) caused a normal presynaptic inhibition of EPSC amplitude, as confirmed by PPR values, similar in the three strains of mice (p < 0.05; n = 8 each group; data not shown), ruling out a loss of receptor sensitivity. In a second set of experiments, we examined the postsynaptic M1-mAChR-dependent effects using a K+-based internal solution to better preserve the integration between somatic and dendritic currents (Ding et al., 2010) in the current-clamp mode. For thalamic stimulation, a thalamic conditioning train (50 Hz, 10 pulses) was given at three different delay times (Δt = 25 ms, Δt = 1 s, and Δt = 1.5 s) before the corticostriatal test stimulus (50 Hz, 5 consecutive pulses). In this condition, only at short delay (25 ms), we observed a decrease in the first EPSP amplitude, suggestive of a presynaptic inhibition, not significantly different among genotypes (NT: 24 ± 3.8%; hWT: 25 ± 4.2%; hMT1: 20 ± 3.2%; p > 0.05; n = 6 per genotype; Fig. 5D,E). The reduction in the first EPSP amplitude was still present when the delay was extended up to 250 ms, but disappeared when the delay was extended to 1 and 1.5 s (Fig. 5D,E), confirming that the mechanisms underlying presynaptic inhibition are preserved. Conversely, with longer delays (1–1.5 s), we recorded a significant enhancement of the fifth EPSP in hMT1 mice, as compared with hWT and NT mice (at 1.5 s: NT: 6 ± 2%; hWT: 3.7 ± 5.1%; hMT1: 38.7 ± 5.2%; p < 0.05; n = 6 per genotype; Fig. 5D,E). Such a significant and persistent enhancement demonstrates an abnormal broadening of the time window for synaptic integration. To prove the involvement of M1 mAChR, pirenzepine (100 nm, 10 min) was able to prevent the increase in EPSP summation in hMT1 mice (compared with predrug, 3.5 ± 3.4%, p < 0.05; n = 6; Fig. 5E).
Enhanced NR2B-dependent EPSC fraction evoked by thalamostriatal stimulation
M1 mAChR activation potentiates NMDA receptor responses in MSNs (Calabresi et al., 1998). Thus, we calculated the effective contribution of AMPA and NMDA receptors to glutamatergic EPSCs evoked in MSNs either by cortical or thalamic stimulation in the hMT1 and control strains. We found a significantly higher NMDA-AMPA/kainate ratio at thalamostriatal synapses in MSNs of hMT1 mice as compared with values collected from NT and hWT mice (NT: 2.2 ± 0.22; hWT: 2.47 ± 0.2; hMT1: 3.65 ± 0.39, n = 8 p = 0.007; Fig. 6A). Conversely, the NMDA-AMPA/kainate ratio measured at corticostriatal synapses was comparable among the three groups of mice (NT: 2.95 ± 0.3; hWT: 2.78 ± 0.23; hMT1: 2.7 ± 0.2, n = 8 p > 0.05; Fig. 6A). This finding led us to examine the component of total NMDA-EPSC carried by NR2B-containing NMDA receptors. Thus, we measured the NMDA/NR2B ratio by using a specific blocker of the NR2B component, ifenprodil, and the selective subunit inhibitor TAT-2B. At corticostriatal synapses measurement of the NMDA/NR2B ratio did not show significant differences (NT: 1.43 ± 0.1; hWT: 1.65 ± 9 0.15; hMT1: 1.57 ± 0.16; n = 5; p = 0.25; Fig. 6B). Conversely, thalamostriatal EPSCs recorded from hMT1 mice showed a robust increment of the NR2B-mediated current, as calculated by current subtraction in pharmacological isolation (NT: 2.13 ± 0.23; hWT: 1.9 ± 0.24; hMT1: 1.19 ± 0.91; n = 5; p = 0.014; Fig. 6B). A different composition of NR2A and NR2B subunits is known to affect not only the pharmacological but also the kinetic properties of central synapses (Stocca and Vicini, 1998). Our analysis revealed a slower decay of NR1/NR2B-mediated EPSC as demonstrated by the increment in τ-weighted constant in hMT1 mice (NT: 62.4 ± 3.3; hWT: 72.1 ± 4.4; hMT1: 113.6 ± 4.3, n = 8 p < 0.005; Fig. 6C), further suggesting that thalamostriatal EPSCs exhibit an increased NR2B current fraction. Next, we performed immunofluorescence experiments and quantitative confocal analysis on striata from mice injected with the GFP-lentiviral particles. We found that the percentage of GFP area expressing the NR2B signal in the isolated GFP-positive MSN dendrites from hMT1 mice was significantly increased compared with the NT group (142.8% ± 6, normalized to NT mice, n = 15; 100% ± 13.2, n = 13; Fig. 6D), although it did not reach statistical significance compared with hWT mice (117% ± 7.1, n = 13; p = 0.017; post hoc Holm–Sidak test, p < 0.05 hMT1 vs NT; p > 0.05 hMT1 vs hWT and hWT vs NT).
Figure 6.
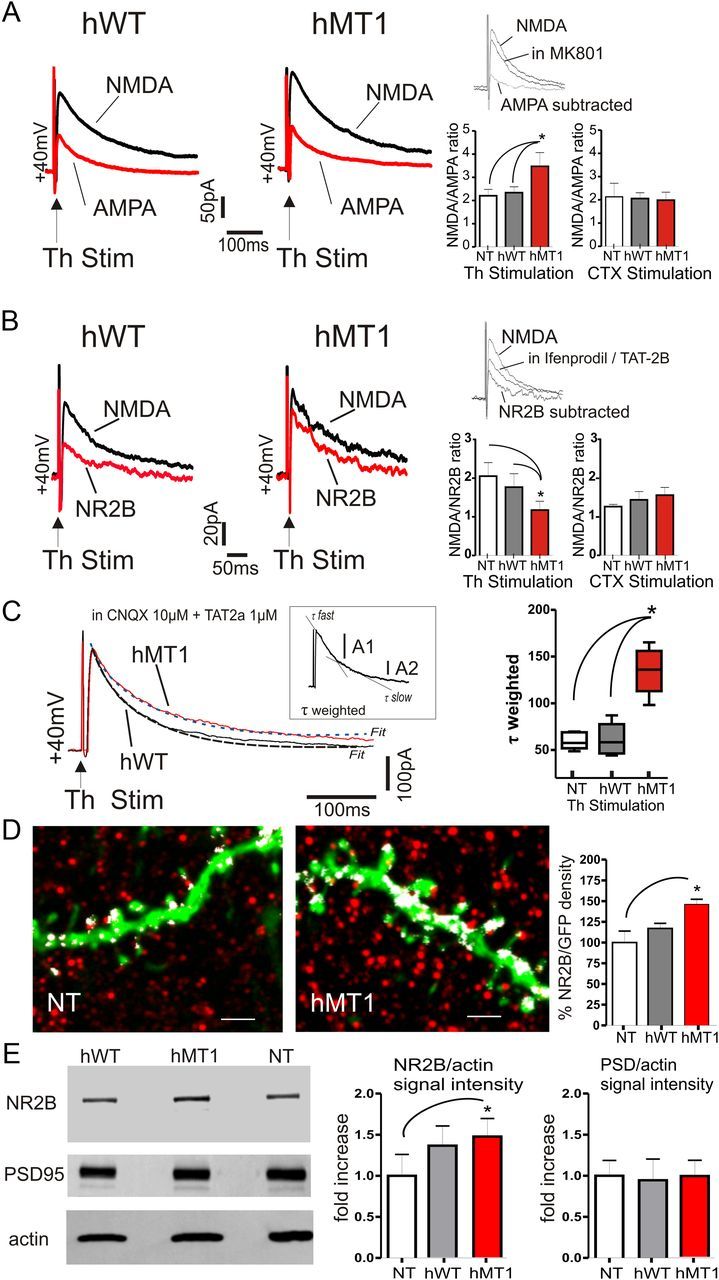
Selective increment of NR2B-mediated current in hMT1 mice. A, Representative traces of EPSCs elicited by thalamic stimulation in MSNs. AMPA- or NMDA-mediated EPSCs were obtained by subtracting the average responses in pharmacological isolation. Summary plots of the differences in NMDA/AMPA ratio. B, Sample traces of NR2B- and NMDA-mediated currents after thalamic stimulation. Plots demonstrate a reduced NMDA/NR2B ratio in hMT1 mice. Insets (in A and B), Single traces pharmacologically isolated. C, Average of 20 consecutive EPSCs evoked by thalamic stimulation in MSN of hWT (gray) and hMT1 (black) mice. A double exponential curve was used to fit the decay phase (dotted lines). Note the slower decay of the NR2B component in hMT1 mice. Consistently, τ-weighted time constant (Tw) was higher in hMT1 mice (plot, right). Inset, Parameters to calculate Tw. D, Projections of optical sections showing the immunolabeling (red) of the NR2B subunit on GFP-positive MSN dendrites of NT and hMT1 mice. White spots represent the colocalization signal between the GFP and NR2B staining. The histogram summarizes the percentage of GFP area that colocalizes with NR2B. Scale bar, 2 μm. E, Western blot analysis of NR2B and PSD-95 expression in striatal synaptosomal fractions from hMT1, hWT, and NT mice. Immunoblots showing synaptosomal expression of NR2B, PSD-95, and actin. NR2B expression is increased in hMT1 mice while PSD-95 is similar in the three groups. Summary of the changes in protein levels of synaptosomal NR2B and PSD-95, respectively, normalized to actin and plotted as fold increase relative of NT mice. *p < 0.05.
To support the immunohistochemical data, we isolated synaptosomes from the striata of each experimental group (n = 4 each group) and measured the level of NR2B by Western blot (Fig. 6E). Interestingly, the protein level of the NR2B subunit in the synaptosomal fraction of hMT1 mice was increased compared with the NT group, while no significant change was observed between hWT and hMT1 mice (p < 0.05 hMT1 vs NT; p > 0.05 hWT vs NT and hWT vs hMT1). Measurement of the levels of PSD-95, a scaffold protein enriched in postsynaptic density of the synaptosomal preparations and an intracellular interactor of NR2B (Kornau et al., 1995), revealed no difference in the three strains of mice (Fig. 6E). Altogether, our findings show a selective increase of the NR2B-mediated component at thalamostriatal synapses, indicating a functional rearrangement of the NR2B subunit in mice with mutant torsinA.
Finally, to exclude an involvement of postsynaptic AMPA receptors, we analyzed miniature AMPA-mediated EPSCs (mEPSCs) (Fig. 7). Because our experimental setting does not allow us to dissect from the synapses the mEPSCs were generated, making it impossible to discriminate between corticostriatal and thalamostriatal quantal release, we performed experiments replacing extracellular Ca2+ with Sr2+ and applying a brief, simultaneous stimulation pulse both at corticostriatal and thalamostriatal fibers (Ding et al., 2008). This experimental protocol causes the asynchronous release of vesicles only from activated synapses. In the presence of Sr2+ (2.5 mm), the frequency of asynchronous quantal EPSCs (qEPSCs) strongly increased after stimulation of both corticostriatal and thalamostriatal synapses, as compared with the changes observed with control extracellular Ca2+ concentrations (CTX stim in NT mice: Ca2+ = 9.10 ± 0.8 Hz; Sr2+ = 24.6 ± 2.0 Hz; in hWT mice: Ca2+ = 10.3 ± 1.2 Hz; Sr2+ = 26.5 ± 2.5 Hz; in hMT1 mice: Ca2+ = 11.8 ± 1.4 Hz; Sr2+ = 25.8 ± 2.2, n = 6 p < 0.05; Fig. 7A,A1 and Th stim: in NT mice: Ca2+ = 10.5 ± 0.7 Hz; Sr2+ = 23.5 ± 3.2 Hz; in hWT mice: Ca2+ = 9.8 ± 1.0 Hz; Sr2+ = 24.6 ± 2.0 Hz; in hMT1 mice: Ca2+ = 11.1 ± 1.4 Hz; Sr2+ = 28.8 ± 2.2, n = 6, p < 0.05; Fig. 7B,B1). The magnitude of the effect mediated by Sr2+ was not significantly different in MSNs recorded from hMT1 mice when compared with NT and hWT mice, demonstrating that AMPA-mediated qEPSCs remain unchanged both at corticostriatal and thalamostriatal synapses (n = 8 p > 0.05). There were no significant differences in amplitude or kinetics of qEPSCs evoked by either cortical or thalamic stimulation (data not shown, n = 8, p > 0.05) suggesting a normal AMPA receptor function.
Figure 7.

Quantal events at corticostriatal and thalamostriatal synapses. Sample traces of mEPSCs recorded after cortical (A, CTX) and thalamic (B, Th) stimulation. Traces were recorded in the voltage-clamp configuration, in the presence of 2 mm Ca2+ or 2 mm Sr2+/0 Ca2+ in the external bathing solution, from hWT and hMT1 mice. Holding potential: −60 mV. A1, B1, The plots show no significant change in asynchronous release frequency in the three genotypes following cortical (A1) and thalamic (B1) stimulation.
Enhancement of NMDA currents by muscarinic M1 receptor agonists
NMDA-dependent currents were recorded in MSNs before and after application of an M1 mAChR agonist. Focal application of 500 μm NMDA generated transient inward currents in MSNs from the three groups of mice (n = 6 per group; Fig. 8A). In the presence of the M1 mAChR agonist MCN-343 (1 μm), NMDA evoked an inward current in hMT1 mice that was significantly larger in amplitude, as compared with hWT and NT mice (p < 0.05; Fig. 8A,A1), supporting the hypothesis of an increased responsiveness of M1 mAChRs. This effect was indeed prevented by pretreatment with the antagonist pirenzepine (data not shown, n = 6).
Figure 8.
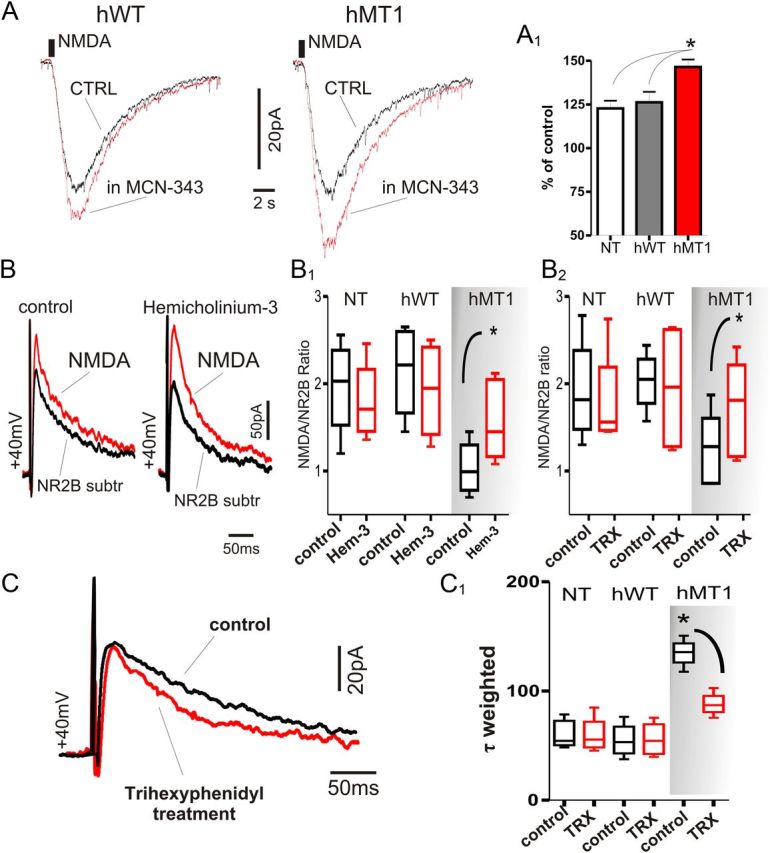
Lowering cholinergic tone or blockade of M1 mAChRs restores normal NMDA receptor-mediated current fraction. A, Focal application of NMDA (500 μm) by pressure ejection caused an inward current in voltage-clamped MSNs of hWT and hMT1 mice. In the presence of MCN-343 (1 μm) a significant increase in the current amplitude was recorded, as summarized in the plot (A1). B, Representative samples of NMDA- and NR2B-subtracted currents elicited by thalamic stimulation in MSN of hMT1 mice. Traces were recorded in control solution (CTRL, left) or hemicholinium-3 (10 μm, right). B1, B2, Treatment with trihexyphenidyl (TRX) and hemicholinium-3 reverted the abnormal NR2B/NMDA ratio in hMT1 mice. C, Average traces of 10 consecutive EPSCs evoked by thalamic stimulation in MSN of hMT1 mice. Chronic treatment with trihexyphenidyl (red line) results in a faster decay phase of NR2B-mediated currents. C1, Summary plot of the reduction of τ-weighted time constants in hMT1 mice treated with trihexyphenidyl.
Anticholinergic drugs restore normal thalamostriatal EPSC composition
These data prompted us to test whether trihexyphenidyl or pirenzepine, preferred M1 antagonists, or hemicholinium-3, which depletes endogenous ACh, could restore a physiological EPSC composition. EPSCs evoked by thalamostriatal stimulation were recorded either in slices pretreated with hemicholinium-3 (10 μm, 2 h), or obtained from mice chronically treated with trihexyphenidyl (20 mg/kg, 3 d, i.p.). In both these conditions, the NR2B-mediated current fraction measured at thalamostriatal synapses returned to levels comparable to those of NT and hWT mice (NR2B/NMDA ratio, n = 6 with trihexyphenidyl and pirenzepine, p = 0.03; Fig. 8B,C), a finding corroborated by the normalization of EPSC kinetic properties (τ-W: control = 110.31 ± 5.52; control = 115.01 ± 2.06, in trihexyphenidyl = 81.04 ± 9.44; n = 5 p = 0.007; Fig. 8C). Similar results were obtained with the ACh-depleting agent, hemicholinium-3, with a nearly complete rescue of NR2B/NMDA ratio (n = 8 with hemicholinium-3, p = 0.02; τ-W: in hemicholinium-3 = 83.5 ± 3.9, n = 5, p = 0.01; Fig. 8B,C).
Discussion
Our results show that, in a model of DYT1 dystonia, repetitive activation of thalamostriatal inputs, mimicking the activity pattern observed in vivo, elicits an abnormal pause response in ChIs. This altered pattern profoundly affects corticostriatal synaptic activity of MSNs both at presynaptic and postsynaptic levels, through the involvement of M2 and M1 mAChRs, respectively (Fig. 9). These data provide the first mechanistic evidence for the close link between mutant torsinA and impairment of the integrative role of the striatal network. Modifications in the temporal window for synaptic integration between thalamostriatal and corticostriatal circuits might provide a plausible explanation of why unpredicted salient stimuli precipitate or worsen dystonic symptoms.
Figure 9.

Schematic representation of the sequence of events and mechanisms following thalamic stimulation in mice with the DYT1 mutation. Normally, glutamatergic thalamic inputs drive ChIs and induce a “pause” in the firing pattern of interneurons. Recruitment of D2Rs is required to generate the pause response. ACh, by acting at M2 receptors (M2R) located on cortical terminals, transiently reduces glutamate release, whereas by activating postsynaptic M1 receptors (M1R) located on MSN dendrites, enhances the responsiveness of MSNs. The precise timing of these events, dictated by the pause response, ensures a correct balance between presynaptic inhibition, mediated by M2Rs and postsynaptic facilitation, dependent upon M1Rs. In the presence of mutant torsinA (hMT1), thalamic activation fails to induce a normal pause response, because of a D2R dysfunction. The loss of the pause response distorts the balance between activation of muscarinic receptor subtypes, favoring M1R activation and its coupling to NR2B-containing NMDA receptors, thereby disrupting synaptic integration between thalamostriatal and corticostriatal inputs.
Altered pause response in ChIs following thalamic activation
The corticostriatal and thalamostriatal pathways form similar numbers of synapses on MSNs, regardless of the neurochemical nature of MSNs (Doig et al., 2010). Conversely, thalamic inputs from caudal intralaminar nuclei provide a robust innervation on ChIs, which receive only relatively weak cortical inputs (Smith et al., 2004). These observations are consistent with electrophysiological in vivo and in vitro studies (Wilson et al., 1990; Ding et al., 2010), and corroborated here by the inability to evoke the stereotyped activity pattern of ChIs by cortical stimulation. ChIs are believed to correspond to TANs that have been characterized in vivo in nonhuman primates. TANs exhibit an ongoing pacemaking activity, interrupted by a burst–pause pattern that is temporally related to behaviorally salient stimuli, predictive of an attentional shift required for the preparation of goal-directed movements (Aosaki et al., 1994a,b; Graybiel et al., 1994; Morris et al., 2004).
Using a slice preparation, which preserves the axons of the thalamostriatal pathway, we observed tonic pacemaking activity in ChIs, which was interrupted by a train of stimuli delivered through the thalamostriatal projections. These features model the responsiveness of TANs to salient stimuli, with the signal of salience deriving from the thalamostriatal pathway (Graybiel et al., 1994; Matsumoto et al., 2001; Kimura et al., 2004; Ding et al., 2010; Thorn and Graybiel, 2010). Since the timing of the pause elicited by thalamic stimulation coincides with a sequence of synaptic events, which is essential for cortically driven action selection (Ding et al., 2010), the alteration of the pause response in ChIs, which we observed both in the hMT1 and in ΔGAG+/− knock-in mice, represents a fundamental alteration in processing thalamostriatal inputs.
A critical issue is the mechanism of the altered responsiveness of ChIs to thalamic stimulation. One possible determinant could reside in an aberrant innervation of MSNs and ChIs by thalamic and cortical axons. However, our results show no significant differences in the expression levels of VGluT1 and VGluT2 between normal and transgenics mice. The pause response is a complex phenomenon requiring not only the integrity of the thalamostriatal projection, but also dopamine D2R activation (Aosaki et al., 1994b, Goldberg and Reynolds, 2011; Ding et al., 2010). Accordingly, we demonstrate that the thalamically driven pause is a D2R-dependent phenomenon, as sulpiride prevented its generation, suggesting that an altered D2R signaling sustains the abnormal ChIs excitability. These results are consistent with the evidence of a lower expression of striatal D2R protein and reduced ability of D2Rs to activate their cognate Go/i proteins in hMT1 mice (Napolitano et al., 2010).
ChIs from hMT1 mice exhibit a substantial alteration in their response to D2R activation, consisting in a paradoxical excitation, rather than the expected inhibition, caused by an enhanced inhibitory coupling of D2Rs to Cav2.2 Ca2+ channels that regulate the opening of Ca2+-dependent K+ channels (Pisani et al., 2006; Sciamanna et al., 2011). Our voltage-clamp recordings provide insights into the mechanistic determinants underlying the abnormal pause in mice with the DYT1 mutation, showing that the inward current generated by thalamic stimulation in ChIs was prevented by intracellular chelation of Ca2+ and blocked by selective inhibitors of Cav2.2 Ca2+ channels. Cav2.2 current activates the small conductance (SK) Ca2+-activated K+ current (Goldberg and Wilson, 2005). Accordingly, blocking these currents with apamin prevented the current generated by thalamic activation.
Consequences of the altered pause response on synaptic integration
Striatal ACh plays a relevant role in motor function and dysfunction (Pisani et al., 2007). The thalamically evoked pattern of interneuron activity has a notable impact on cortical regulation of MSNs, creating a temporal window for synaptic integration between corticostriatal and thalamostriatal inputs (Ding et al., 2010), through a temporally defined sequence of synaptic events consisting in a fast, transient presynaptic inhibition mediated by M2 mAChRs, followed by a slower potentiation, dependent on postsynaptic M1 mAChRs, virtually matching the duration of the pause response. Surprisingly, in hMT1 mice the aberrant firing recorded in ChIs after thalamic activation led to a persistent facilitation of the M1-dependent postsynaptic component, which exceeded the expected duration of the pause response, broadening the window for cortical inputs to modulate striatal network activity. A broader time window could expose striatal network to nonspecific cortical inputs, rendering DYT1 gene mutation carriers more susceptible to generate aberrant motor patterns.
Paired recordings from hMT1 mice demonstrated that the M2-dependent presynaptic inhibition rapidly waned. Several mechanisms could account for the decline of the M2-dependent presynaptic effect. The inability to recruit M2 mAChRs by an inappropriate stimulation was ruled out by the efficacy of such a protocol also in NT and hWT mice. Moreover, the observation that M2 mAChR agonists were effective excluded an altered receptor sensitivity. A further potential clue came from measurement of PPR values, an index of presynaptic inhibition. The transient inhibition was indeed accompanied by a change in PPR suggestive of a presynaptic effect, but then (>200 ms) the PPR values returned to levels indicating that the presynaptic nature of the effect was lost. Thus, we considered the possibility of an abnormal role of M1 receptors, which might alter the M1/M2 balance, disrupting the timing of this process. Accordingly, pirenzepine, an M1 antagonist, rescued the presynaptic effect, suggesting that a prevailing M1-dependent activity overwhelmed the inhibition by M2 mAChRs. This assumption fits also with the timing of different mAChR subtype activation, as presynaptic effects occur rapidly, whereas the M1-dependent facilitation is slower (Pakhotin and Bracci, 2007; Ding et al., 2010).
While the M2-dependent presynaptic inhibition of cortical input occurs similarly in both classes of principal neurons, striatonigral and striatopallidal MSNs, the postsynaptic M1-mediated facilitation has been described only in striatopallidal cells (Ding et al., 2010). In our recordings, we did not detect differences in subgroups of MSNs. One possible explanation could reside in the abnormal cholinergic modulation of MSN excitability. ACh enhances MSN excitability by promoting the closure of different K+ channel subtypes (Shen et al., 2005, 2007). Most of these effects have been shown to occur through the activation of M1 mAChRs, which are robustly expressed in both types of MSNs (Hersch et al., 1994). In hMT1 mice, the sustained activation of M1 mAChRs might determine the loss of specificity between the two cell types, promoting indistinct closure of K+ channels, enhancing cell excitability in both MSN subgroups.
Postsynaptic M1 mAChRs increase MSN excitability also through the potentiation of NMDA-receptor-mediated currents (Calabresi et al., 1998). In hMT1 mice, we found an increase of the NMDA/NR2B current fraction, but only following thalamic stimulation. This change in synaptic currents was corroborated by the evidence that NMDA currents were robustly potentiated by M1 mAChR agonists. Accordingly, treatment with M1 antagonists partially restored normal EPSC composition and kinetics.
These electrophysiological results match with an increase of the NR2B subunit expression observed on the spiny dendrites of MSNs. While the Western blot shows increased NR2B in both striatal synaptosomal preparations, the immunohistochemical data on MSNs show increased colocalization of NR2B with spines only in hMT1 mice. A plausible explanation for this apparent discrepancy is that although the increase in protein is present on both, synaptic localization of NR2B increases only in hMT1 MSNs.
The dynamic nature of NMDA receptor trafficking does not allow, at present, the establishment of whether such an increase was indeed determined by an activity-dependent recruitment of extrasynaptic receptors or by torsinA overexpression. This latter hypothesis could be suggested by the observation that both hWT and hMT1 mice exhibited similar increases in NR2B protein levels. On the other hand, NMDA currents were potentiated only in hMT1 mice, suggesting that mutant torsinA may drive synaptic localization of NR2B subunits. Our data argue for an increased responsiveness of striatal M1 mAChR, which might foster the recruitment of extrasynaptic NMDA receptors, enhancing NMDA function (Lan et al., 2001).
Salient stimuli conveyed by thalamostriatal fibers alters action selection in dystonia
Unexpected changes of behavioral significance interrupt ongoing motor activity and convey attention toward the salient stimulus. This multimodal information arising from intralaminar thalamic nuclei is primarily directed to the striatum, which plays an integrative role in cognition and motor function (Graybiel et al., 1994, Kimura et al., 2004). Functional MRI imaging studies support this notion, showing that activity in the human striatum reflects the level of saliency associated with a stimulus, providing a signal to reallocate resources for action selection (Zink et al., 2006). Likewise, the intralaminar thalamic nuclei are selectively activated when human subjects switch from a relaxed state to an active state demanding attention (Kinomura et al., 1996). Thus, to optimize selection of neural signals, it is essential that a correct sensorimotor integration between thalamic sensory inputs and cortically driven motor patterns occurs within the striatum, where these signals merge.
Dystonic symptoms worsen with anxiety and disappear during sleep (Marsden and Harrison, 1974; Fish et al., 1991). Our data show that an abnormal cholinergic signaling perturbs the integration between corticostriatal and sensory thalamostriatal inputs, suggesting that in gene mutation carriers disruption of the filtering activity might result in an altered motor output, favoring appearance of dystonic symptoms.
Footnotes
This study was supported by the Bachmann-Strauss Dystonia-Parkinson Foundation, Dystonia Medical Research Foundation, Istituto Superiore di Sanitá, and National Institutes of Health P50NS037409. We thank M. Tolu and V. Batocchi for their excellent technical support. We thank Professor L. Naldini for providing the plasmids for lentiviral production, and Dr. W. Dauer for making the DYT1 knock-in mice available for these studies.
References
- Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- Aosaki T, Tsubokawa H, Ishida A, Watanabe K, Graybiel AM, Kimura M. Responses of tonically active neurons in the primate's striatum undergo systematic changes during behavioral sensorimotor conditioning. J Neurosci. 1994a;14:3969–3984. doi: 10.1523/JNEUROSCI.14-06-03969.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aosaki T, Graybiel AM, Kimura M. Effects of the nigrostriatal system on acquired neural responses in the striatum of behaving monkeys. Science. 1994b;265:412–415. doi: 10.1126/science.8023166. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Gubellini P, Pisani A, Bernardi G. Endogenous Ach enhances NMDA-responses via M1-like muscarinic receptors and PKC activation. Eur J Neurosci. 1998;10:2887–2895. doi: 10.1111/j.1460-9568.1998.00294.x. [DOI] [PubMed] [Google Scholar]
- Carbon M, Argyelan M, Habeck C, Ghilardi MF, Fitzpatrick T, Dhawan V, Pourfar M, Bressman SB, Eidelberg D. Increased sensorimotor network activity in DYT1 dystonia: a functional imaging study. Brain. 2010;133:690–700. doi: 10.1093/brain/awq017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Peterson JD, Surmeier DJ. Corticostriatal and thalamostriatal synapses have distinctive properties. J Neurosci. 2008;28:6483–6492. doi: 10.1523/JNEUROSCI.0435-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding JB, Guzman JN, Peterson JD, Goldberg JA, Surmeier DJ. Thalamic gating of corticostriatal signaling by cholinergic interneurons. Neuron. 2010;67:294–307. doi: 10.1016/j.neuron.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doig NM, Moss J, Bolam JP. Cortical and thalamic innervation of direct and indirect pathway medium-sized spiny neurons in mouse striatum. J Neurosci. 2010;30:14610–14618. doi: 10.1523/JNEUROSCI.1623-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards MJ, Huang YZ, Wood NW, Rothwell JC, Bhatia KP. Different patterns of electrophysiological deficits in manifesting and non-manifesting carriers of the DYT1 gene mutation. Brain. 2003;126:2074–2080. doi: 10.1093/brain/awg209. [DOI] [PubMed] [Google Scholar]
- Fish DR, Sawyers D, Smith SJ, Allen PJ, Murray NM, Marsden CD. Motor inhibition from the brainstem is normal in torsion dystonia during REM sleep. J Neurol Neurosurg Psychiatry. 1991;54:140–144. doi: 10.1136/jnnp.54.2.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ, Bellocchio EE, Fortin D, Storm-Mathisen J, Edwards RH. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron. 2001;31:247–260. doi: 10.1016/s0896-6273(01)00344-0. [DOI] [PubMed] [Google Scholar]
- Goldberg JA, Reynolds JN. Spontaneous firing and evoked pauses in the tonically active cholinergic interneurons of the striatum. Neuroscience. 2011;198:27–43. doi: 10.1016/j.neuroscience.2011.08.067. [DOI] [PubMed] [Google Scholar]
- Goldberg JA, Wilson CJ. Control of spontaneous firing patterns by the selective coupling of calcium currents to calcium-activated potassium currents in striatal cholinergic interneurons. J Neurosci. 2005;25:10230–10238. doi: 10.1523/JNEUROSCI.2734-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodchild RE, Kim CE, Dauer WT. Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron. 2005;48:923–932. doi: 10.1016/j.neuron.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Graybiel AM, Aosaki T, Flaherty AW, Kimura M. The basal ganglia and adaptive motor control. Science. 1994;265:1826–1831. doi: 10.1126/science.8091209. [DOI] [PubMed] [Google Scholar]
- Hallett M. Is dystonia a sensory disorder? Ann Neurol. 1995;38:139–140. doi: 10.1002/ana.410380203. [DOI] [PubMed] [Google Scholar]
- Hersch SM, Gutekunst CA, Rees HD, Heilman CJ, Levey AI. Distribution of m1–m4 muscarinic receptor proteins in the rat striatum: light and electron microscopic immunocytochemistry using subtype-specific antibodies. J Neurosci. 1994;14:3351–3363. doi: 10.1523/JNEUROSCI.14-05-03351.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, Minamimoto T, Matsumoto N, Hori Y. Monitoring and switching of cortico-basal ganglia loop functions by the thalamo-striatal system. Neurosci Res. 2004;48:355–360. doi: 10.1016/j.neures.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Kinomura S, Larsson J, Gulyás B, Roland PE. Activation by attention of the human reticular formation and thalamic intralaminar nuclei. Science. 1996;271:512–515. doi: 10.1126/science.271.5248.512. [DOI] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- Lan JY, Skeberdis VA, Jover T, Grooms SY, Lin Y, Araneda RC, Zheng X, Bennett MV, Zukin RS. Protein kinase C modulates receptor trafficking and gating. Nat Neurosci. 2001;4:382–390. doi: 10.1038/86028. [DOI] [PubMed] [Google Scholar]
- Marsden CD, Harrison MJ. Idiopathic torsion dystonia (dystonia musculorum deformans). A review of forty-two patients. Brain. 1974;97:793–810. doi: 10.1093/brain/97.1.793. [DOI] [PubMed] [Google Scholar]
- Martella G, Tassone A, Sciamanna G, Platania P, Cuomo D, Viscomi MT, Bonsi P, Cacci E, Biagioni S, Usiello A, Bernardi G, Sharma N, Standaert DG, Pisani A. Impairment of bidirectional synaptic plasticity in the striatum of a mouse model of DYT1 dystonia: role of endogenous acetylcholine. Brain. 2009;132:2336–2349. doi: 10.1093/brain/awp194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto N, Minamimoto T, Graybiel AM, Kimura M. Neurons in the thalamic CM-Pf complex supply striatal neurons with information about behaviorally significant sensory events. J Neurophysiol. 2001;85:960–976. doi: 10.1152/jn.2001.85.2.960. [DOI] [PubMed] [Google Scholar]
- Morris G, Arkadir D, Nevet A, Vaadia E, Bergman H. Coincident but distinct messages of midbrain dopamine and striatal tonically active neurons. Neuron. 2004;43:133–143. doi: 10.1016/j.neuron.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Musumeci G, Sciarretta C, Rodríguez-Moreno A, Al Banchaabouchi M, Negrete-Díaz V, Costanzi M, Berno V, Egorov AV, von Bohlen Und Halbach O, Cestari V, Delgado-García JM, Minichiello L. TrkB modulates fear learning and amygdala synaptic plasticity by specific docking sites. J Neurosci. 2009;29:10131–10143. doi: 10.1523/JNEUROSCI.1707-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano F, Pasqualetti M, Usiello A, Santini E, Pacini G, Sciamanna G, Errico F, Tassone A, Di Dato V, Martella G, Cuomo D, Fisone G, Bernardi G, Mandolesi G, Mercuri NB, Standaert DG, Pisani A. Dopamine D2 receptor dysfunction is rescued by adenosine A2A receptor antagonism in a model of DYT1 dystonia. Neurobiol Dis. 2010;38:434–445. doi: 10.1016/j.nbd.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, de Leon D, Brin MF, Raymond D, Corey DP, Fahn S, Risch NJ, Buckler AJ, Gusella JF, Breakefield XO. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet. 1997;17:40–48. doi: 10.1038/ng0997-40. [DOI] [PubMed] [Google Scholar]
- Pakhotin P, Bracci E. Cholinergic interneurons control the excitatory input to the striatum. J Neurosci. 2007;27:391–400. doi: 10.1523/JNEUROSCI.3709-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani A, Martella G, Tscherter A, Bonsi P, Sharma N, Bernardi G, Standaert DG. Altered responses to dopaminergic D2 receptor activation and N-type calcium currents in striatal cholinergic interneurons in a mouse model of DYT1 dystonia. Neurobiol Dis. 2006;24:318–325. doi: 10.1016/j.nbd.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Pisani A, Bernardi G, Ding J, Surmeier DJ. Re-emergence of striatal cholinergic interneurons in movement disorders. Trends Neurosci. 2007;30:545–553. doi: 10.1016/j.tins.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Sciamanna G, Bonsi P, Tassone A, Cuomo D, Tscherter A, Viscomi MT, Martella G, Sharma N, Bernardi G, Standaert DG, Pisani A. Impaired striatal D2 receptor function leads to enhanced GABA transmission in a mouse model of DYT1 dystonia. Neurobiol Dis. 2009;34:133–145. doi: 10.1016/j.nbd.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciamanna G, Tassone A, Martella G, Mandolesi G, Puglisi F, Cuomo D, Madeo G, Ponterio G, Standaert DG, Bonsi P, Pisani A. Developmental profile of the aberrant dopamine D2 receptor response in striatal cholinergic interneurons in DYT1 dystonia. PLoS One. 2011;6:e24261. doi: 10.1371/journal.pone.0024261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N, Baxter MG, Petravicz J, Bragg DC, Schienda A, Standaert DG, Breakefield XO. Impaired motor learning in mice expressing torsinA with the DYT1 dystonia mutation. J Neurosci. 2005;25:5351–5355. doi: 10.1523/JNEUROSCI.0855-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Hamilton SE, Nathanson NM, Surmeier DJ. Cholinergic suppression of KCNQ channel currents enhances excitability of striatal medium spiny neurons. J Neurosci. 2005;25:7449–7458. doi: 10.1523/JNEUROSCI.1381-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Tian X, Day M, Ulrich S, Tkatch T, Nathanson NM, Surmeier DJ. Cholinergic modulation of Kir2 channels selectively enhances dendritic excitability in striatopallidal neurons. Nat Neurosci. 2007;10:1458–1466. doi: 10.1038/nn1972. [DOI] [PubMed] [Google Scholar]
- Smeal RM, Gaspar RC, Keefe KA, Wilcox KS. A rat brain slice preparation for characterizing both thalamostriatal and corticostriatal afferents. J Neurosci Methods. 2007;159:224–235. doi: 10.1016/j.jneumeth.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Smith Y, Raju DV, Pare JF, Sidibe M. The thalamostriatal system: a highly specific network of the basal ganglia circuitry. Trends Neurosci. 2004;27:520–527. doi: 10.1016/j.tins.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Smith Y, Raju D, Nanda B, Pare JF, Galvan A, Wichmann T. The thalamostriatal systems: anatomical and functional organization in normal and parkinsonian states. Brain Res Bull. 2009;78:60–68. doi: 10.1016/j.brainresbull.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocca G, Vicini S. Increased contribution of NR2A subunit to synaptic NMDA receptors in developing rat cortical neurons. J Physiol. 1998;507:13–24. doi: 10.1111/j.1469-7793.1998.013bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorn CA, Graybiel AM. Pausing to regroup: thalamic gating of cortico-basal ganglia networks. Neuron. 2010;67:175–178. doi: 10.1016/j.neuron.2010.07.010. [DOI] [PubMed] [Google Scholar]
- Tinazzi M, Fiorio M, Fiaschi A, Rothwell JC, Bhatia KP. Sensory functions in dystonia: insights from behavioral studies. Mov Disord. 2009;24:1427–1436. doi: 10.1002/mds.22490. [DOI] [PubMed] [Google Scholar]
- Van der Werf YD, Witter MP, Groenewegen HJ. The intralaminar and midline nuclei of the thalamus. Anatomical and functional evidence for participation in processes of arousal and awareness. Brain Res Brain Res Rev. 2002;39:107–140. doi: 10.1016/s0165-0173(02)00181-9. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Chang HT, Kitai ST. Firing patterns and synaptic potentials of identified giant aspiny interneurons in the rat neostriatum. J Neurosci. 1990;10:508–519. doi: 10.1523/JNEUROSCI.10-02-00508.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink CF, Pagnoni G, Chappelow J, Martin-Skurski M, Berns GS. Human striatal activation reflects degree of stimulus salience. Neuroimage. 2006;29:977–983. doi: 10.1016/j.neuroimage.2005.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]