

Abstract
Alcoholism is frequently co-morbid with posttraumatic stress disorder (PTSD) but it is unclear how alcohol impacts neural circuits mediating recovery from trauma. We found that chronic intermittent ethanol (CIE) impaired fear extinction and remodeled the dendritic arbor of medial prefrontal cortical (mPFC) neurons in mice. CIE impaired extinction encoding by infralimbic (IL) mPFC neurons in vivo, and functionally downregulated burst-mediating NMDA GluN1 receptors. These findings suggest alcohol may increase risk for trauma-related anxiety disorders by disrupting mPFC-mediated extinction of fear.
Alcoholism and anxiety disorders are some of the most commonly occurring neuropsychiatric disorders, and often co-occur in the same individuals. Current theories emphasize the occurrence of anxiety symptoms arising as a result of a history of heavy drinking. By comparison, little attention has been paid to the effects of alcohol abuse on the subsequent risk for anxiety disorders such as PTSD even though excessive drinking increases exposure to traumas such as car accidents and domestic violence1.
Fear extinction, the learned inhibition of a trauma-related fear response, provides a valuable assay for translational studies of PTSD. Studies in rodents have found evidence of impaired extinction following acute2,3 or repeated4,5 alcohol exposure, but have either been unable to clearly parse effects of alcohol on extinction from fear per se, or have not established the mechanisms involved. In this context, fear extinction heavily recruits analogous regions of the mPFC across species6. In rodents, IL- inactivation7, stress-induced IL neuronal atrophy8, or blockade of mPFC N-methyl-D-aspartate receptors (NMDARs)9 and downstream signaling pathways10 impairs fear extinction, whereas activation of the prelimbic cortex (PL) correlates with poor extinction11,12 and PL inactivation reduces fear expression7. Although extinction deficits have not yet been reported in alcoholism, alcoholics exhibit deficits in PFC-mediated cognitive and executive functions13 that are coupled with abnormalities in PFC volume, grey matter and activation14. Together, these prior observations lead to the hypothesis that chronic alcohol impairs extinction by disrupting mPFC function.
To test this idea, we subjected mice to a regimen of chronic intermittent ethanol exposure and withdrawal (CIE) (Supplementary Information) that is designed to mimic repeated cycles of heavy abuse typifying alcoholism and drives neural adaptations in circuits regulating higher-order behaviors. Mice received continuous vaporized alcohol (yoked controls received vaporized air) for 16 hr (to achieve 175±25 mg/dL in blood, Fig. S1), followed by 8-hr withdrawal, every day for 4 consecutive days. After a longer, 80-hr withdrawal, the 4-day cycle was repeated - for a total of 4 cycles. Mice were trained to fear an auditory conditioned stimulus (CS) repeatedly paired with footshock 2 days after CIE to avoid non-specific behavioral disturbances during acute withdrawal. Extinction acquisition was tested the day after (3 days post-CIE) via 50xCS-alone presentations. To evaluate the strength of the extinction memory, a retrieval test was conducted 1 day later, followed by a context-renewal test. CS-related freezing was measured as an index of fear.
CIE and air groups had equivalent freezing during conditioning and the first trial-block of extinction showing that, unlike acute alcohol2, CIE did not disrupt the fear memory formation or expression. During extinction training, both groups showed clear decreases in freezing, but CIE mice had higher freezing during the second trial-block, indicating a modest retardation of extinction learning (CIE × trial-block ANOVA interaction: F9,225=2.43, p<.01, post hoc p<.05) (Fig. 1a). Most strikingly, however, CIE significantly impaired extinction retention, as evidenced by higher fear relative to air controls during extinction retrieval (t-test: t=2.07, df=25, p<.05) (Fig. 1a). Both groups exhibited fear renewal although this trended (t-test: t=1.51, df=25, p=0.15) higher in the CIE group. In a separate cohort of mice, CIE did not alter unconditioned anxiety-like behavior in the light-dark test (2 days post-CIE) (t-test: t=0.84, df=24, p=0.41) (Fig. 1b), and did not produce tolerance to the rotarod-ataxic (t-test: t=1.40, df=24, p=0.18) or loss of righting reflex (t-test: t=0.78, df=24, p=0.44) effects of acute alcohol challenge (3-4 days post-CIE) (Fig. 1b). These results demonstrate CIE impaired extinction, and did so in the absence of alterations in fear, anxiety or tolerance, at least on the tests employed.
Fig. 1. CIE impairs fear extinction.
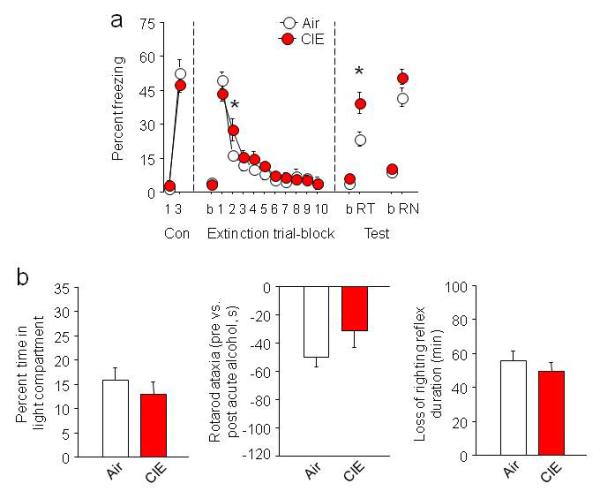
(a) CIE transiently retarded extinction acquisition (higher fear on trial-block 2) and significantly impaired extinction retrieval, relative to air controls (air n=10, CIE n=12). (b) CIE did not affect anxiety-like behavior in the light-dark exploration test (transitions: air=19.8±10.1, CIE=13.8±2.2, p>.05), or produce tolerance to the effects of acute alcohol challenge, as measured by rotarod ataxia (=pre versus post acute alcohol) (Fig. S2: rotarod training) and the loss of righting reflex (air n=10, CIE n=12). b=baseline, Con=conditioning, RT=retrieval, RN=renewal. Data are Means ±SEM. *P<.05 CIE versus air
Because impaired extinction is associated with altered dendritic morphology in mPFC neurons15, we sacrificed behaviorally-naïve mice 3 days after CIE for visualization of IL and PL dendritic arbors from Golgi-Cox impregnated tissue. In IL pyramidal neurons, dendritic arborization was not different between groups in the apical (ANOVA distance from soma effect: F34,476=3009.58, p<.01, CIE effect: F1,14=0.38, p=.55) (Fig. 2d-f) or basilar (Fig. S3c,d) tree. By contrast, PL neurons of CIE mice had significantly longer dendrites than air controls in the apical (Fig. 2a,b), not basilar (Fig. S3a,b) tree. Furthermore, this increase was limited to non-terminal branches (t-test: t(14)=3.83, p<.01) and to branches relatively distal to the soma, with a corresponding degree of dendritic retraction near the soma (ANOVA CIE × distance interaction: F34,476=3.81, p<.01, post hocs p<.05) (Fig. 2c). Thus, CIE produced significant dendritic remodeling in mPFC neurons, characterized by PL hypertrophy. Underscoring the specificity of remodeling, we observed normal dendritic arborization after CIE in other cortical regions (orbitofrontal cortex, Fig. S4) and nodes in the extinction circuit (basolateral amygdala, Fig. S5).
Fig. 2. CIE remodels mPFC neurons.

(a) Coronal cartoon of PL. Examples of reconstructed neurons. (b) CIE increased overall length of PL apical dendrites at non-terminal branches (air n=7, CIE n=9, n=6-8 neurons/mouse for b,c). (c) Sholl analysis revealed that PL apical dendritic length was lesser close to the soma and greater farther from the soma after CIE. (d) Coronal cartoon of IL. Examples of reconstructed neurons (scale bar=50 μm). (e) CIE did not alter overall IL apical length (air n=14, CIE n=12, n=6-8 neurons/mouse for e,f). (f) Sholl analysis found no alcohol-induced change in IL dendritic length as a function of distance from the soma. Data are Means ±SEM. *P<.05 CIE versus air
Dendritic dysmorphology represents a structural correlate of the CIE-induced extinction deficit, but does not reveal changes in vmPFC neuronal function. To more directly examine function, we performed in vivo neuronal recordings from multichannel electrode arrays. In rat mPFC, an extinction-encoding neuronal signal is characterized by a rapid and transient increase in firing16. Replicating this pattern in mouse, we observed a 100-msec increase in CS-related firing during early extinction in IL (ANOVA time effect: F29,4466=7.81, p<.01) (Fig. 3a) and PL (ANOVA time effect: F29,7192=13.47, p<.01) (Fig. S6a). This was equivalent between air and CIE groups. CS-related firing was also evident during late extinction and, more strongly still, extinction retrieval, but was significantly attenuated after CIE during both late extinction (ANOVA IL=CIE × time interaction: F29,4408=1.42, p=.066, PL=CIE × time interaction: F29,7192=1.89, p<.01, post hocs p<.05) and retrieval (ANOVA IL=CIE × time interaction: F29,4118=1.61, p<.05, PL=CIE × time interaction: F29,6177=2.61, p<.01, post hocs p<.05) (Fig. 3a, S6a). These data demonstrate blunting of extinction-related neuronal encoding in mPFC neurons.
Fig. 3. CIE disrupts mPFC extinction encoding and decreases mPFC NMDAR-mediated currents.
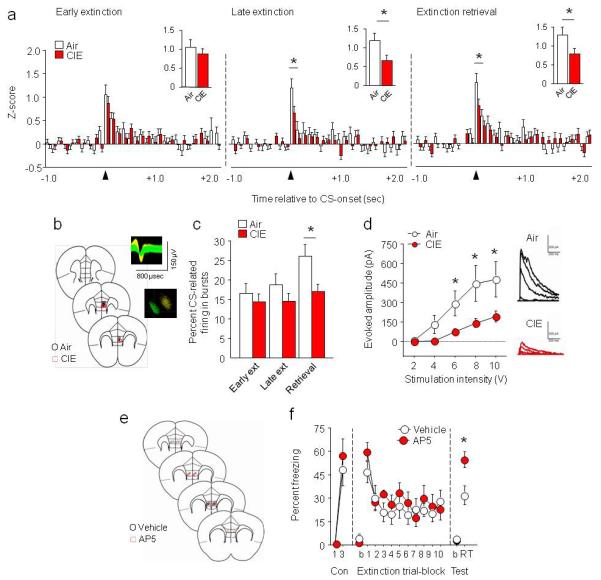
(a) In vivo freely moving recordings of IL neurons demonstrated CIE decreased neuronal activity during the first 100 milliseconds (redrawn in insets) of CS onset during late extinction (trial-block 10) and extinction retrieval (air n=7, CIE n=7, n=69-85 neurons, for a-c). (b) Coronal cartoon showing electrode placements in IL. Example extracellular waveforms and principal components separation of simultaneously recorded neurons. (c) CIE decreased CS-related IL neuronal bursting during extinction retrieval. (d) CIE decreased stimulation-evoked synaptic NMDAR-mediated currents in IL neurons, with example traces (air n=7, CIE n=8). (e) Coronal cartoon showing cannula placements in IL. (f) NMDAR antagonist AP5 infused into IL immediately after extinction training impaired extinction retrieval in alcohol-naive mice (air n=8, CIE n=7). Data are Means ±SEM. *P<.05 CIE versus air
CIE mice also showed significantly less CS-related burst activity during extinction retrieval. Notably, this was specific to IL (t-test: t(143)=2.65, p<.01) (Fig. 3c), with no loss of bursting in the PL after CIE (Fig. S6c). Although we did not see IL versus PL differences in extinction-related neuronal firing per se, as has been seen in rats7, the subregion dissociation in bursting fits well with the prior observation that low bursting in IL, but not PL, predicts poor extinction retrieval9.
Post-extinction training antagonism of NMDARs in IL impairs extinction retrieval in tandem with reduced IL neuronal buirsting9, while chronic NMDAR blockade produces PL apical dendritic hypertrophy17 similar to that produced by CIE. This suggests that CIE may have caused loss of mPFC NMDAR. Consistent with this, we found significantly reduced expression of the functionally obligatory GluN1 NMDAR subunit in mPFC tissue (IL+PL) 3 days after CIE (air=100±8%, CIE=61±10% normalized to air, t-test: t(14)=2.96, p<.0, Fig. S7). PSD-95 and other NMDAR subunits showed normal expression, indicating no general loss of mPFC synapses (Fig. S7). Furthermore, ex vivo slice physiology analysis indicated diminished stimulation-evoked excitatory NMDAR-mediated currents in IL and PL neurons at the same time point post-CIE (ANOVA IL=CIE × stimulation interaction: F4,52=3.94, p<.01, post hocs p<.05, Fig. 3d; PL=CIE × trial-block interaction: F4,48=3.77, p<.01, Fig. S6d). Finally, in alcohol naive mice, local bilateral micro-infusion of the NMDAR antagonist, AP5 (1 μg, 0.1 μl/side), immediately after extinction training mimicked the extinction retrieval deficit produced by CIE (t(13)=2.90, p<.05) (Fig. 3e-f).
Collectively, these data propose a model whereby CIE drives NMDAR downregulation in mPFC, with attendant abnormalities in dendritic morphology and neuronal encoding of extinction. A threshold of chronicity appears necessary to produce these effects, as subchronic CIE (2 cycles) was insufficient to downregulate mPFC NMDAR expression, alter IL/PL dendritic morphology or impair extinction (Fig. S8). The finding that mPFC-mediated extinction is vulnerable to CIE implies that a chronic history of alcohol abuse may increase risk of persistent fear after psychological trauma by degrading PFC-mediated capacity for extinction.
Online Methods
(See supplemental file).
Supplementary Material
Acknowledgements
We thank Yavin Shaham and Cara Wellman for valuable comments, and Benita Hurd, Aaron Plitt, Daniel Fisher, and Rachel Daut for assistance with the CIE procedure. Support for this work was from the Department of Defense in the Center for Neuroscience and Regenerative Medicine (AH, SF, OGC), NIH Grants AA019454, AA017668, AA020911 (TK, CL, CM) AA021043 (KP), DoD Grant PT090344 (TK, KP), NARSAD (TK), Bowles Center for Alcohol Studies (TK), and the NIAAA Intramural Research Program (AH, PJF, KM, LD, PF, SM, GC, OGC, MC).
Footnotes
Author contributions: AH, PJF, TLK, MC designed the experiments. PJF, KPM, LD, GC, SMF, SM, KEP, CL, CAM, TLK, OGC, and MC collected the data. AH, PJF, KPM, LD, GC, SMF, SM, KEP, CL, CAM, TLK, OGC, and MC analyzed the data. AH wrote the manuscript.
References
- 1.WHO. World Health Organization 2006 [Google Scholar]
- 2.Lattal KM. Behav Neurosci. 2007;121:1280–92. doi: 10.1037/0735-7044.121.6.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baum M. J Comp Physiol Psychol. 1969;69:238–40. doi: 10.1037/h0028188. [DOI] [PubMed] [Google Scholar]
- 4.Ripley TL, O’ Shea M, Stephens DN. Eur J Neurosci. 2003;18:441–8. doi: 10.1046/j.1460-9568.2003.02759.x. [DOI] [PubMed] [Google Scholar]
- 5.Bertotto ME, Bustos SG, Molina VA, Martijena ID. Neuroscience. 2006;142:979–90. doi: 10.1016/j.neuroscience.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 6.Milad MR, et al. Biol Psychiatry. 2009;66:1075–82. doi: 10.1016/j.biopsych.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sierra-Mercado D, Padilla-Coreano N, Quirk GJ. Neuropsychopharmacology. 2011;36:529–38. doi: 10.1038/npp.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Izquierdo A, Wellman CL, Holmes A. J Neurosci. 2006;26:5733–8. doi: 10.1523/JNEUROSCI.0474-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burgos-Robles A, Vidal-Gonzalez I, Santini E, Quirk GJ. Neuron. 2007;53:871–80. doi: 10.1016/j.neuron.2007.02.021. [DOI] [PubMed] [Google Scholar]
- 10.Herry C, Trifilieff P, Micheau J, Luthi A, Mons N. Eur J Neurosci. 2006;24:261–9. doi: 10.1111/j.1460-9568.2006.04893.x. [DOI] [PubMed] [Google Scholar]
- 11.Burgos-Robles A, Vidal-Gonzalez I, Quirk GJ. J Neurosci. 2009;29:8474–82. doi: 10.1523/JNEUROSCI.0378-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whittle N, Hauschild M, Lubec G, Holmes A, Singewald N. J Neurosci. 2010;30:13586–96. doi: 10.1523/JNEUROSCI.0849-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldstein RZ, et al. Neuropsychologia. 2004;42:1447–58. doi: 10.1016/j.neuropsychologia.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 14.Pfefferbaum A, Sullivan EV, Rosenbloom MJ, Mathalon DH, Lim KO. Arch Gen Psychiatry. 1998;55:905–12. doi: 10.1001/archpsyc.55.10.905. [DOI] [PubMed] [Google Scholar]
- 15.Holmes A, Wellman CL. Neurosci Biobehav Rev. 2009;33:773–83. doi: 10.1016/j.neubiorev.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Milad MR, Quirk GJ. Nature. 2002;420:70–4. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- 17.Martin KP, Wellman CL. Cereb Cortex. 2011;21:2366–73. doi: 10.1093/cercor/bhr021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.