

Background: Integrin α1β1 binding to collagen IV down-regulates collagen synthesis, but how modulating integrin α1β1 affinity alters collagen homeostasis is unknown.
Results: Cells carrying mutations of the α1 subunit with enhanced collagen binding show increased ERK activation and increased collagen down-regulation.
Conclusion: Enhancing the affinity of the α1 subunit to collagen potentiates integrin α1β1-mediated outside-in signaling.
Significance: These findings have major implications for pathological conditions such as fibrotic diseases.
Keywords: Collagen, Fibrosis, Integrins, MAP Kinases (MAPKs), NMR, I Domains
Abstract
Integrin α1β1 binding to collagen IV, which is mediated by the α1-inserted (I) domain, down-regulates collagen synthesis. When unligated, a salt bridge between Arg287 and Glu317 is thought to keep this domain in a low affinity conformation. Ligand binding opens the salt bridge leading to a high-affinity conformation. How modulating integrin α1β1 affinity alters collagen homeostasis is unknown. To address this question, we utilized a thermolysin-derived product of the α1α2α1 network of collagen IV (α1α2α1(IV) truncated protomer) that selectively binds integrin α1β1. We show that an E317A substitution enhanced binding to the truncated protomer, consistent with a previous finding that this substitution eliminates the salt bridge. Surprisingly, we show that an R287A substitution did not alter binding, whereas R287E/E317R substitutions enhanced binding to the truncated protomer. NMR spectroscopy and molecular modeling suggested that eliminating the Glu317 negative charge is sufficient to induce a conformational change toward the open state. Thus, the role played by Glu317 is largely independent of the salt bridge. We further show that cells expressing E317A or R287E/E317R substitutions have enhanced down-regulation of collagen IV synthesis, which is mediated by the ERK/MAPK pathway. In conclusion, we have demonstrated that modulating the affinity of the extracellular α1 I domain to collagen IV enhances outside-in signaling by potentiating ERK activation and enhancing the down-regulation of collagen synthesis.
Introduction
Fibrosis, the process by which tissues are replaced by extracellular matrix, is an irreversible event that leads to loss of tissue and organ functions. Interactions between cells with the surrounding extracellular matrix have emerged as a key factor involved in the initiation and progression to fibrosis (1). Integrins α1β1 and α2β1 are the key extracellular matrix receptors that regulate collagen turnover in many organs, including skin (2, 3) and kidneys (4, 5). These integrins bind to collagens via their extracellular domains, resulting in the activation of intracellular signaling pathways via their cytoplasmic tails. Integrin α1β1 activation results in the down-regulation of endogenous collagen synthesis, whereas integrin α2β1 activation promotes collagen synthesis (2, 6). Integrin α1β1 is the best studied collagen receptor in the context of fibrosis. We demonstrated previously that integrin α1-null mice develop more severe renal fibrosis following injury, characterized by excessive deposition of collagen IV and generation of reactive oxygen species (4, 7). The protective role of integrin α1β1 resides in its ability to down-regulate the activation of the pro-fibrotic EGF receptor (8) as well as regulate the level and phosphorylation state of caveolin-1, a scaffolding protein that negatively regulates EGF receptor activation (9, 10). Thus, integrin α1β1 is a negative regulator of collagen production, and its engagement is beneficial in the setting of fibrosis.
One of the unique features of the integrin α1 and α2 subunits is that their extracellular domains contain an insertion of ∼200 amino acids called the inserted (I)3 domain. X-ray crystallography of the α2 I domain revealed that this domain consists of a central hydrophobic β-sheet surrounded by seven amphipathic α-helices with a metal ion-dependent adhesion site motif at the top of the β-sheet (11), which forms the major site for ligand binding (12). The I domain confers ligand specificity; studies on isolated I domains show that the α1 I domain preferentially binds to collagen IV, whereas the α2 I domain preferentiality binds to collagen I (13–15). In addition, the integrin α1 I domain binds, although with lower affinity, to collagen I and laminin-111, -211, and -511 (16–18). The use of chimeric α1/α2 I domains demonstrates a role for the α3, α5, and a portion of the α6 helices within the α1 I domain in regulating collagen IV binding (16). Point mutation studies of the α1 I domain have suggested that Glu317 on helix α7 forms a salt bridge with Arg287 on helix αC, thus keeping the α1 I domain in a closed/low affinity state. Mutating Glu317 into an alanine results in a gain of function with increased binding affinity to both collagens I and IV (17). Moreover, when Arg218 in the α1 I domain (in the loop between helices α3 and α4) is mutated to alanine, it leads to decreased affinity to collagen IV, suggesting that Arg218 plays an important role in determining binding specificity to collagen IV (17). Although these data indicate that more than one amino acid is necessary for determining the specificity of the α1 I domain to its natural ligand, the contribution of Arg287 in keeping the α1 I domain in a closed/low affinity conformation, as well as whether the salt bridge between Arg287 and Glu317 is indeed responsible for keeping the α1 I domain in a low affinity conformation has not been analyzed. Most importantly, how modulating the affinity of integrin α1β1 to collagen IV results in integrin α1β1-dependent alterations of collagen IV homeostasis has not been explored.
In this study we performed mutagenesis studies in combination with molecular modeling and nuclear magnetic resonance spectroscopy to delineate the molecular mechanisms that modulate integrin α1β1 affinity and integrin-mediated cell function. We show that an E317A substitution resulted in a gain-of-function mutant with increased affinity to both collagens I and IV, as reported previously (17). However, we show that a R287A substitution increased affinity to collagen I but did not change affinity to collagen IV. Surprisingly, charge reversal of Arg287 and Glu317 (R287E/E317R) resulted in a gain-of-function mutant. Molecular modeling and nuclear magnetic resonance spectroscopy of wild type and E31A α1 I domains indicated that eliminating the Glu317 negative charge was sufficient to favor an activate conformational change. This novel result suggests that key roles played by Glu317 are largely independent of its interaction with Arg287 as a salt bridge. Finally, we demonstrate that cells expressing E317A or R287E/E317R substitutions show not only increased binding to collagen IV but also enhanced down-regulation of endogenous collagen synthesis, which is mediated by the ERK/MAPK signaling pathway. Thus, modulating integrin α1β1 affinity to collagen plays a key role in altering endogenous collagen synthesis. Altogether, these results suggest that increasing the affinity of the α1 I domain to its natural ligand results in enhanced outside-in signaling. Our findings identify a novel mechanism whereby integrin α1β1 signaling is regulated, which has major implications for pathological conditions such as fibrotic diseases.
EXPERIMENTAL PROCEDURES
Antibodies
Anti-human integrin α1 TS2/7 and anti-FAK antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Hamster anti-mouse CD49a (integrin α1 chain), hamster anti-mouse CD49b (integrin α2 chain), and rabbit anti-mouse collagen IV were purchased from BD Biosciences. Mouse anti-human integrin α1 (mAb 1973) antibodies were obtained from Chemicon (Temecula, CA). Anti-ERK and anti-phospho-ERK antibodies were purchased from Cell Signaling (Danvers, MA). Mouse anti-vinculin (V9264) antibodies were purchased from Sigma-Aldrich.
Generation and Purification of the Human Integrin α1 I Domain Mutants
The human integrin α1 I domain (residues 140–336 of the mature integrin α1 subunit (16)) (supplemental Fig. 1A) was subcloned into a pET27 (Novagen) derived from the expression vector pBG101 (Vanderbilt Center for Structural Biology) to produce an N-terminal His6-GST fusion protein. This vector was used as a template for the generation of the integrin α1 I domain mutants using a QuikChange kit (Stratagene). Primers used for mutagenesis were as follows: R218D mutant (forward, 5′-GTCCAGAGAGGTGGCGACCAGACTATGACAGC-3′; reverse, 5′-GCTGTCATAGTCTGGTCGCCACCTCTCTGGAC-3′); T220A mutant (forward, 5′-TGGCCGCCAGGCTATGACAGCTCTTGGAATAG-3′; reverse, 5′-CTATTCCAAGAGCTGTCATAGCCTGGCGGCCAC-3′); R287A mutant (forward, 5′-GCTATTCTTGGCAGCTATAACGCAGGAAATTTAAGCACTG-3′; reverse, 5′-CAGTGCTTAAATTTCCTGCGTTATAGCTGCCAAGAATAGC-3′); E317A mutant (forward, 5′-CTTCAATGTCTCTGATGCATTGGCTCTAGTCACC-3′; reverse, 5′-GGTGACTAGAGCCAATGCATCAGAGACATTGAAG-3′); R287E mutant (forward, 5′-GCTATTCTTGGCAGCTATAACGAAGGAAATTTAAGCACTG-3′; reverse, 5′-CAGTGCTTAAATTTCCTTCGTTATAGCTGCCAAGAATAGC-3′); E317R mutant (forward, 5′-CTTCAATGTCTCTGATAGATTGGCTCTAGTCAC-3′; reverse, 5′-GGTGACTAGAGCCAATCTATCAGAGACATTGAAG-3′). The fusion proteins were overexpressed in Escherichia coli BL-21(DE3) cells (Novagen) in LB medium supplemented with 30 μg/ml kanamycin and 0.5 mm isopropyl-1-thio-β-d-galactopyranoside for 16–24 h at 16 °C. The cells were suspended in 50 mm Tris-HCl (pH 8.0), 500 mm NaCl, 10 mm imidazole, 20% (v/v) glycerol, and 2 mm β-mercaptoethanol and lysed with an Emusilflex C3 homogenizer (Avestin). Proteins were purified using Ni-NTA (Qiagen) affinity chromatography followed by on-column PreScission Protease (GE Healthcare) cleavage overnight at 4 °C (supplemental Fig. 1B). Mutant proteins were further purified over a 320-ml Superdex 200 gel filtration preparative grade column (GE Healthcare) equilibrated in 50 mm Tris-HCl (pH 8.0), 300 mm NaCl, 10% (v/v) glycerol, and 1 mm dithiothreitol. The yield of purified wild type (WT) and mutant α1 I domains was ∼10 mg/liter culture.
Cell Culture
Immortalized mesangial cells were isolated from integrin α1-null mice crossed onto the Immortomouse background as described previously (8) and propagated at 33 °C in the presence of 100 IU/ml γ-interferon. Cells were cultured at 37 °C without γ-interferon for at least 4 days prior to experiments, as this is the optimal time for immortalized mesangial cells to acquire a phenotype similar to that of freshly isolated primary mesangial cells (8).
Generation of Mesangial Cells Expressing the Human Integrin α1 I Domain Mutants
The wild type full-length integrin α1 cDNA was digested with BamHI and EcoRI to generate a 2.7-kb fragment containing the I domain. This fragment was subcloned into pBluescript vector and used as a template to generate the α1 I domain mutants described above. The mutants were then released from pBluescript by BamHI and EcoRI digestion and subcloned into pcDNA3.1 vector. For the generation of integrin α1-null mesangial cells expressing the various integrin α1 I domain mutants, cells were transfected with an empty pcDNA3.1 vector or the various cDNA constructs using Lipofectamine Plus reagent (Invitrogen) followed by Zeocin selection (Sigma-Aldrich) according to the manufacturer's instructions. Stable cell populations of α1-null mesangial cells expressing equal levels of the various mutants were selected by fluorescence-activated cell sorting using antibodies recognizing the extracellular I domain of human integrin α1 subunit (TS2/7). Polyclonal cell populations were used to avoid some of the pitfalls associated with isolation of monoclonal cell lines.
Preparation of the Collagen IV α1α2α1 Truncated Protomer
Peeled bovine lens capsules were suspended in 1 m NaCl plus protease inhibitors and then sonicated and centrifuged to remove connective tissues. The clear lens capsules (∼3 mg) were digested with 100 μg/ml thermolysin (protease type X, Sigma-Aldrich) in TBS buffer with 2 mm CaCl2 at 14 °C. After 24 h, the digested mixture was centrifuged at 14,000 rpm at 4 °C for 15 min, and the supernatant was collected followed by the addition of EDTA (10 mm final) and NaCl (1.8 m final). After a 4-h incubation at 4 °C, the mixture was centrifuged at 14,000 rpm at 4 °C for 15 min, and the pellet was collected and dissolved in TBS buffer containing 0.02 n acetic acid. The α1α2α1(IV) truncated protomer was purified by FPLC gel filtration chromatography using a Superdex S200 column (GE Healthcare) in TBS buffer (pH 7.4). In certain experiments, the α1α2α1(IV) truncated protomer was further digested with pepsin (Sigma) in 0.1 m acetic acid at a 1:100 ratio to remove non-collagenous (NC1) domain hexamers or with bacterial collagenase (CLSPA grade, Worthington) at a 1:100 ratio to remove the collagenous domain of the protomer.
Collagen Biotinylation
The α1α2α1(IV) truncated protomer and rat tail collagen I (BD Biosciences) were biotinylated using EZ-Link NHS-PEG4-biotin (Thermo Scientific) following the manufacturer's protocol after rapid buffer exchange to phosphate-buffered saline (PBS) using centrifugal ultrafiltration filters (Millipore, MWCO 10 kDa). Labeled proteins were purified from an excess of biotinylation reagent using a PD-10 gel filtration column (GE Healthcare), equilibrated, and eluted with TBS buffer. Efficiency of labeling was verified by Western blotting and ELISA using avidin conjugated with alkaline phosphatase (Sigma).
FACS Analysis
To determine the ability of the α1α2α1(IV) truncated protomer to bind cells in suspension, biotinylated protein (2.5 μg/ml) was preincubated with avidin-FITC (5 μg/ml) in the presence of 1 mm MgCl2 for 1 h and then added to mesangial cells in suspension. In some experiments, mesangial cells were preincubated with hamster anti-integrin α1 or hamster anti-integrin α2 antibodies (30 μg/ml, final) before incubation with biotinylated α1α2α1(IV) truncated protomer. Cells incubated with avidin-FITC alone were used as negative control. After 1 h, cells were washed and analyzed with a FACScan (BD Biosciences). To determine the cell surface expression of the various integrin α1 I domain mutants, mesangial cells were incubated with anti-human integrin α1 antibodies (TS2/7) for 45 min, washed, and then incubated with the appropriate phycoerythrin-conjugated secondary antibodies and analyzed with a FACScan (BD Biosciences). Data collected in flow cytometry experiments were analyzed using CellQuest software (BD Biosciences).
Solid Phase Binding
Microtiter 96-well plates (Maxisorb, Nunc) were coated overnight at 4 °C with 10 μg/ml mouse EHS (Engelbreth-Holm-Swarm) collagen IV (BD Biosciences), fibronectin (Sigma-Aldrich), α1α2α1(IV) truncated protomer, pepsin-digested truncated protomer, collagenase-digested truncated protomer, or α1α2α1(IV) NC1 hexamers in TBS. The plates were washed and incubated with 1% BSA in TBS to block nonspecific binding. After 1 h, the plates were washed and incubated for 2 h at 30 °C with purified integrin α1β1 (Chemicon, 2 μg/ml) or integrin αvβ3 (Chemicon, 2 μg/ml) in TBS with 1 mm MgCl2, 0.2 mm MnCl2 or in TBS with 10 mm EDTA. After washing, the plates were incubated with anti-integrin α1 (Santa Cruz Biotechnology, 1:5000)- or αvβ3 (Chemicon, 1:5000)-specific monoclonal antibodies for 1 h followed by anti-mouse secondary antibodies conjugated to alkaline phosphatase (Sigma, 1:2000) in TBS supplemented with 1 mm MgCl2, 0.2 mm MnCl2 or with 10 mm EDTA. After 1 h at 30 °C, the plates were washed and incubated with p-nitrophenyl phosphate and absorbance was measured with a SpectraMax microplate reader (Molecular Devices) at 405 nm. Specific binding was calculated as the difference in absorbance between wells incubated in the presence of divalent cations (1 mm MgCl2, 0.2 mm MnCl2) and those incubated with 10 mm EDTA. Binding experiments were performed in triplicate and repeated three times.
In some experiments, wells were coated as described above with wild type and the various human integrin α1 I domain mutants or the human integrin α2 I domain (10 μg/ml in 0.1 m Na2CO3/NaHCO3 (pH 9.5)) followed by biotinylated α1α2α1(IV) truncated protomer or biotinylated collagen I (0–2 μg/ml in TBS with or without 1 mm MgCl2) followed by avidin plus HRP-conjugated biotin (Bio-Rad, 5 μg/ml). Binding was detected by incubating the wells with HRP substrate and reading the plates at 595 nm with a microtiter plate reader. Collagen bound to 3% BSA-coated wells was used as background, and this optical density was subtracted from that obtained in the experimental wells. Three independent experiments were performed in quadruplicate.
To verify that the various integrin α1 I domains coated the wells equally, wells were coated as indicated above with wild type or the α1 I domain mutants (10 μg/ml) followed by mouse anti-human integrin α1 antibody (mAb 1973, 2 μg/ml) followed by anti-mouse IgG antibodies conjugated to HRP. Three independent experiments were performed in triplicate (supplemental Fig. 1C).
Cell Adhesion
Cell adhesion was performed as described previously (4). Briefly, U-bottom 96-well plates were coated at 4 °C with α1α2α1(IV) truncated protomer or collagen I (10 μg/ml in 0.1 m Na2CO3/NaHCO3 (pH 9.5)). After 12 h, the plates were washed, and nonspecific binding sites were blocked with 1% BSA in PBS. After 1 h at 37 °C, the plates were washed and 100-μl cell suspensions (5 × 104 cells in serum-free medium supplemented with 0.25% BSA) were added to the wells. After a 1-h incubation at 37 °C, nonadherent cells were removed, and the attached cells were fixed with 4% paraformaldehyde, stained with 0.1% crystal violet, and lysed with 10% acetic acid. Cell adhesion was quantified by reading the plates at 595 nm with a microtiter plate reader. The absorbance of the wells coated with 1% BSA only was subtracted from each well. Three experiments were performed in triplicate.
Cell Proliferation
Mesangial cells (5 × 103/well) were plated in low serum (1% fetal calf serum) onto 96-well plates coated with α1α2α1(IV) truncated protomer or collagen I (10 μg/ml, both). After 12 h, the cells were gently washed and incubated with serum-free medium with or without the MEK inhibitor PD98059 (10 μm) in the presence of [3H]thymidine (0.5 μCi/well). After 48 h, the cells were collected, and the amount of incorporated [3H]thymidine was analyzed with a Beckman β-counter. Four independent experiments were performed in quadruplicate.
Cell Spreading
Mesangial cells (3,000/well) were plated in serum-free medium onto chamber slides coated with α1α2α1(IV) truncated protomer or collagen I (10 μg/ml, both). After 4 h, cells were fixed in 4% formaldehyde for 10 min and permeabilized with 0.1% Triton X-100 in PBS for 5 min. After blocking with 3% BSA in PBS, cells were incubated with rhodamine-phalloidin (1:5,000; Molecular Probes) or anti-vinculin antibodies (1:500) followed by anti-mouse antibodies conjugated to FITC (1:1000). Slides were mounted with antifade mounting medium (Vectastain, Vector Laboratories) and examined under an epifluorescence microscope (Nikon).
Western Blot Analysis
To determine the basal levels of collagen IV and ERK activation, 3 × 104 cells were plated into 6-well plates in complete medium. After 8 h, the cells were gently washed and cultured in 0.5% serum with or without the MEK inhibitor PD98059 (10 μm). After 48 h, the cells were gently washed and incubated in serum-free medium with or without the MEK inhibitor for a further 16 h. Cells were then lysed in 50 mm Tris (pH 7.5), 150 mm NaCl, 1% Triton X-100, and 1% sodium deoxycholate. Equal amounts of total proteins (20 μg/lane) were resolved in 8% SDS-PAGE, transferred onto nitrocellulose, and then probed with anti-mouse collagen IV or anti-phospho-ERK. Immunoreactive proteins were visualized with an appropriate HRP-conjugated secondary antibody and an ECL kit. Membranes were stripped and reblotted with anti-FAK or anti-ERK antibodies to verify equal loading. Collagen IV, FAK, phospho-ERK, and ERK bands were quantified by densitometry analysis, and the collagen IV or phospho-ERK signals were expressed as the collagen IV/FAK or phospho-ERK/ERK ratio, respectively. The collagen IV/FAK or phospho-ERK/ERK ratio in control samples (i.e. integrin α1KO mesangial cells) was arbitrarily assigned the value of 1, and all of the other ratios were expressed as -fold changes relative to the assigned control value.
Expression and Purification of Wild Type and E317A α1 I Domain
Wild type and α1 I domain mutants were expressed from a pET-27 derivative vector as a His6-GST fusion protein in E. coli BL-21(DE3) cells (Novagen) grown in M9 minimal medium supplemented with 30 μg/ml kanamycin. Protein expression was induced at A600 nm ∼0.7 by adding 0.5 mm isopropyl-1-thio-β-d-galactopyranoside (IPTG) followed by incubation for 36 h at 16 °C. The cell pellet was suspended in buffer A (50 mm Tris-HCl (pH 8.0), 500 mm NaCl, 10 mm imidazole, 20% (v/v) glycerol, and 2 mm β-mercaptoethanol). Cells were lysed by sonication and the lysate clarified by centrifugation. The wild type α1 I domain fusion protein was purified from the clarified lysate using Ni-NTA (Qiagen) affinity chromatography and dialyzed against buffer B (50 mm Tris (pH 8.0), 300 mm NaCl, 10% glycerol, and 0.5 mm dithiothreitol (DTT)) using MWCO 15,000 Da tubing (SpectraPor 2.1 Biotech). The His-GST tag was removed by treatment with PreScission Protease (GE Healthcare) for 3 h at 4 °C followed by Ni-NTA chromatography in buffer B. Mutant α1 I domain proteins were expressed and purified similar to the wild type but with the following minor changes to increase efficiency. Mutant protein expression was induced with 1 mm IPTG for 24 h at 16 °C. Cells were lysed with an EmusilFlex C3 homogenizer (Avestin). The fusion protein was cleaved overnight on the Ni-NTA column at 4 °C by incubation with PreScission Protease. The eluted proteins were concentrated using an Amicon spin concentrator (Millipore) and further purified over a 320-ml Superdex 200 gel filtration preparative grade column (GE Healthcare) equilibrated in buffer B except with 1 mm DTT. The yield of purified wild type and mutant α1 I domain proteins was ∼10 mg/liter culture.
NMR Spectroscopy
15N-Labeled α1 I-domain samples were expressed and purified as described above, except that expression was in 15N-labeled M9 minimal medium. Purified proteins were buffer-exchanged into 50 mm sodium phosphate (pH 7.0), 150 mm NaCl, 5 mm DTT, 2 mm MgCl2, and 10% D2O using an Amicon Ultra-15 (MWCO 10,000 Da) centrifugal ultrafiltration concentrator. The samples were concentrated to 150 μm, and sodium azide was added to 0.01%. 1H-15N TROSY HSQC experiments were acquired at 298 K with 32 transients/increment and 128 increments on a Bruker 800 MHz spectrometer equipped with a triple resonance cryoprobe. NMR data were processed with NMRPipe software and analyzed in Sparky.
Homology Modeling of the α1 I Domain Open Conformation
SWISS PdbViewer was used to thread the amino acid sequence of α1 I domain (residues 141–325) onto the crystal structure of the α2 I domain bound to a collagen-like peptide containing the GFOGER sequence (Protein Data Bank ID code 1DZI) (12). Only 24 side chains showed steric clashes, and these were removed by energy minimization using the SWISS-MODEL web server (19).
Statistical Analysis
Two-tailed Student's t test was used for comparisons between two groups, and analysis of variance with Sigma-Stat software was used for the determination of statistically significant differences among multiple groups. p < 0.05 was considered statistically significant.
RESULTS
Experimental Strategy
Integrins α1β1 and α2β1 have distinct binding affinities: α1β1 has a higher affinity for collagen IV, whereas α2β1 has a higher affinity for collagen I (13–15). In the present study, we have explored the structural basis for binding affinity of integrin α1β1 for collagen IV and whether modulation of the affinity influences collagen IV homeostasis. For reference purposes, binding to collagen I was compared in most experiments. The known binding site for integrin α1β1 within the α1α2α1 network of collagen IV is located near the N terminus of the long triple helical protomer. This site was characterized for binding activities by excision with cyanogen bromide cleavage, yielding a triple helical fragment designated as CB3 (20, 21). However, this fragment also harbors a binding site for integrin α2β1 (20), a property that can confound studies aimed specifically at integrin α1β1. Herein, we report a novel site within the α1α2α1 network of collagen IV, excised by thermolysin digestion, which binds only integrin α1β1. This thermolysin-derived product was characterized and used as a tool for structure/function studies of integrin α1β1.
Characterization of a Novel Integrin α1β1 Binding Site in the α1α2α1 Network of Collagen IV
Our earlier studies revealed that pseudolysin digestion of the α1α2α1 network of collagen IV of lens capsule basement membrane solubilized a large truncated protomer of about 100 nm in length, representing about one-fourth of the full-length protomer, with retention of the C-terminal noncollagenous (NC1) domain and devoid of the CB3 region (22). Pseudolysin is no longer available, but we found that thermolysin yields the same truncated protomer (supplemental Fig. 2). The absence of the CB3 region in the thermolysin-derived α1α2α1(IV) truncated protomer was demonstrated by the lack of a molecular weight shift under reducing conditions (supplemental Fig. 2), which is expected for CB3-containing polypeptides because of the presence of unique disulfide knots within this region (20). To determine whether the α1α2α1(IV) truncated protomer contains integrin α1β1 binding site(s), we performed solid phase binding assays with purified integrins. As shown in Fig. 1A, integrin α1β1, but not integrin αvβ3 (used as a negative control), bound to the immobilized α1α2α1(IV) truncated protomer. Similar results were obtained when full-length collagen IV from mouse EHS tumor was used as substrate. In contrast, integrin αvβ3, but not integrin α1β1, bound to immobilized fibronectin.
FIGURE 1.
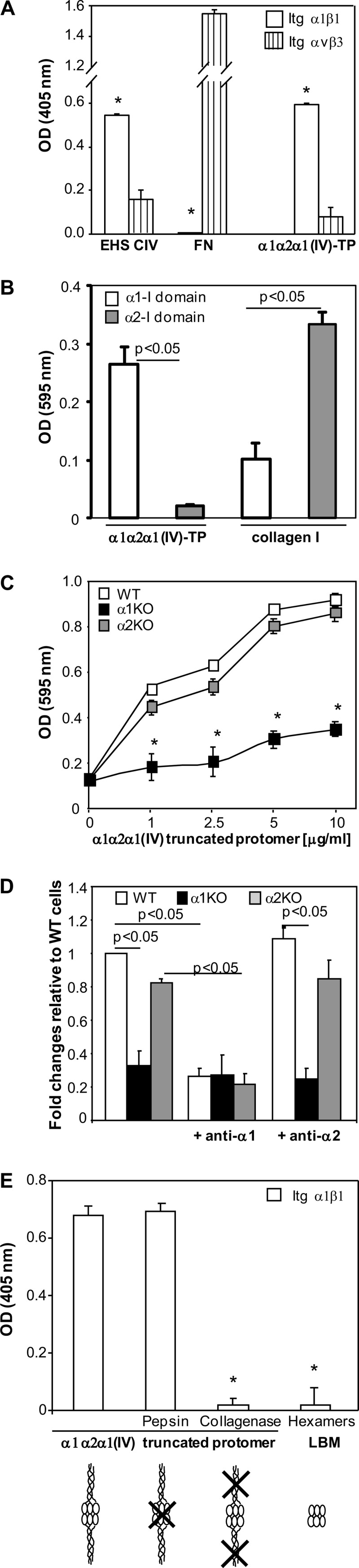
The integrin α1 I domain binds the collagen α1α2α1(IV) truncated protomer. A, binding of integrins α1β1 and αvβ3 to the immobilized matrices indicated was determined in a solid phase binding assay. Note that integrin α1β1 binds to both mouse collagen IV (EHS CIV) and the α1α2α1(IV) truncated protomer but not to fibronectin (FN). The values are expressed as binding in the presence of 1 mm MgCl2 minus the binding in the presence of 10 mm EDTA. Values are the mean ± S.E. of representative experiment performed in triplicates. *, differences between α1β1 versus αvβ3 were significant, with p < 0.05. B, α1 I domain binds the α1α2α1(IV) truncated protomer. Binding of biotinylated α1α2α1(IV) truncated protomer or collagen I to immobilized α1 or α2 I domain was detected in a solid phase binding assay in the presence of 1 mm MgCl2. Note that the α1α2α1(IV) truncated protomer binds to the α1 I but not the α2 I domain. Values are the mean ± S.D. of a representative experiment performed in triplicates. C, cell adhesion to α1α2α1(IV) truncated protomer. An equal number of mesangial cells in serum-free medium was allowed to adhere for 1 h to immobilized α1α2α1(IV) truncated protomer (0–10 μg/ml). Adhesion was evaluated as described under “Experimental Procedures.” Values are the mean ± S.D. of three independent experiments performed in quadruplicate. *, indicates significant differences compared with WT mesangial cells. D, binding of α1α2α1(IV) truncated protomer to cell in suspension. An equal number of mesangial cells was preincubated with or without blocking anti-mouse integrin α1 or anti-mouse integrin α2 antibodies followed by incubation with biotinylated α1α2α1(IV) truncated protomer in serum-free medium. Protomer binding was evaluated by FACS following incubation with FITC-biotin. Note that addition of anti-α1 antibodies prevents the binding of α1α2α1(IV) truncated protomer to WT and integrin α2-null mesangial cells. Values are the mean ± S.D. of three independent experiments and represent -fold changes relative to binding to WT cells (arbitrarily assigned the value of 1). E, integrin α1β1 binds to the collagenous portion of the α1α2α1(IV) truncated protomer. Integrin α1β1 binding to the immobilized substrata was determined in a solid phase binding assay. Note that integrin α1β1 binds to undigested or pepsin-digested α1α2α1(IV) truncated protomer but not to collagenase-digested protomer or NC1 hexamers. The values are expressed as binding in the presence of 1 mm MgCl2 minus the binding in the presence of 10 mm EDTA. Values are the mean ± S.E. of a representative experiment performed in triplicate. *, indicates significant differences (p < 0.05) compared with undigested truncated protomer. Itg, integrin; LBM, lens basement membrane.
As both integrins α1β1and α2β1 bind collagen IV in the CB3 region (20), we next determined whether the α1α2α1(IV) truncated protomer also contains integrin α2β1 binding sites by performing solid phase binding assays with purified integrin α1 and α2 I domains. As shown in Fig. 1B, biotinylated α1α2α1(IV) truncated protomer bound to immobilized integrin α1 I but not integrin α2 I domain. Opposite results were obtained when binding of biotinylated collagen I was analyzed, with strong binding to α2 and less binding to the α1 I domain, demonstrating that the α2 I domain is functionally active under these experimental conditions.
To further confirm that the α1α2α1(IV) truncated protomer contains only integrin α1 but not α2 I domain binding sites, we evaluated the ability of wild type, integrin α1-null, and integrin α2-null mesangial cells to adhere to immobilized α1α2α1(IV) truncated protomer. Whereas both wild type and integrin α2-null mesangial cells adhere equally to α1α2α1(IV) truncated protomer, the adhesion of cells lacking integrin α1β1 was significantly impaired (Fig. 1C). Finally, we determined by FACS the ability of biotinylated α1α2α1(IV) truncated protomer to bind mesangial cells in suspension. Similar to the adhesion data, wild type and integrin α2-null but not integrin α1-null mesangial cells bound α1α2α1(IV) truncated protomer (Fig. 1D). Furthermore, binding of this collagen IV fragment to wild type and integrin α2-null cells was inhibited significantly by preincubation of cells with blocking anti-integrin α1 but not anti-integrin α2 antibodies (Fig. 1D).
As the α1α2α1(IV) truncated protomer contains the NC1 domain and a collagenous triple helical domain, we next determined where the integrin α1β1 binding site(s) resides. To do this, the α1α2α1(IV) truncated protomer was digested with either pepsin (to digest the NC1 domain) or bacterial collagenase (to digest the collagenous domain). Although pepsin treatment did not affect integrin α1β1 binding, collagenase digestion completely abolished it (Fig. 1E), suggesting that the integrin α1β1 binding site resides within the collagenous triple helical domain. Thus, the thermolysin-derived α1α2α1(IV) truncated protomer contains a novel specific integrin α1β1 binding site that resides within its collagenous triple helical part.
Charge Reversal of Arg287 and Glu317 Does Not Keep the Integrin α1 I Domain in a Low Affinity Conformation
Superposition of unligated α1 and α2 I domains show that the two domains are virtually identical in structure (11), yet they show differences in ligand specificity. We used the available α1 and α2 I domain crystal structures (11, 23) together with published mutagenesis studies to model sequence and structural differences between these two domains in order to identify key amino acids responsible for the binding of the α1 I domain to the α1α2α1(IV) truncated protomer. The following integrin α1 I domain mutants were generated: 1) R218D, as this amino acid is an important determinant of the binding specificity to collagen IV (17); 2) T220A, as the corresponding Thr221 in the α2 I domain is important for coordinating the metal ion and binding to collagen I (24); 3) E317A and R287A, as these two amino acids form a salt bridge, thus keeping the α1 I domain in a closed/low affinity conformation (17); and 4) R287E/E317R, based on the assumption that charge reversal should be sufficient to keep the I domain in a closed conformation.
We performed solid phase binding to determine the ability of either the biotinylated α1α2α1(IV) truncated protomer or collagen I to bind to the various integrin α1 I domain mutants. As reported previously with binding to human collagen IV (17), both α1α2α1(IV) truncated protomer and collagen I bound better to the E317A mutant than to the WT α1 I domain (Fig. 2). Binding of the α1α2α1(IV) truncated protomer to the R218D mutant was significantly lower than that detected for the WT α1 I domain (Fig. 2A). However, no differences in binding of collagen I to either the WT or R218D mutants were observed (Fig. 2B). In contrast to the reported T221A mutation in the α2 I domain that leads to decreased adhesion to collagen I (24), binding of the α1α2α1(IV) truncated protomer or collagen I to the T220A mutant was not significantly different from binding to the WT α1 I domain (Fig. 2). Two surprising results were obtained when R287A and R287E/E317R were analyzed. In contrast to the gain-of-function E317A, the R287A substitution significantly enhanced binding of collagen I with no significant changes with respect to binding of the α1α2α1(IV) truncated protomer when compared with the WT α1 I domain (Fig. 2). Finally, the R287E/E317R double mutant significantly increased binding of both the α1α2α1(IV) truncated protomer and collagen I (Fig. 2).
FIGURE 2.

Characterization of integrin α1 I domain mutants by solid phase binding. Binding of biotinylated α1α2α1(IV) truncated protomer (A) or biotinylated collagen I (B) (both at 0–2 μg/ml) to immobilized integrin α1 I domain mutants (10 μg/ml) was determined in a solid phase binding assay. Binding was performed in the presence or absence of 1 mm MgCl2. Values are the mean ± S.D. of three independent experiments performed in quadruplicate. *, indicates significant differences (p < 0.05) compared with WT α1 I domain.
In conclusion, our solid phase binding assays confirm that the R218D and E317A are loss-of-function and gain-of-function mutations, respectively, with respect to α1α2α1(IV) truncated protomer binding. In addition, they reveal the following novel findings: 1) T220A does not significantly modify α1α2α1(IV) truncated protomer or collagen I binding; 2) R287A enhances collagen I binding without significantly affecting α1α2α1(IV) truncated protomer binding; and 3) the R287E/E317R mutant, similar to the E317A mutant, is a gain-of-function mutation.
The Properties of the Various α1 I Domain Mutants Are Retained When the Same Mutations Are Expressed in the Full-length α1 Subunit
We next generated populations of integrin α1-null mesangial cells expressing comparable levels of full-length integrin α1 subunit carrying the mutations described above (Fig. 3A) to determine whether the changes in ligand affinity identified in the isolated α1 I domain mutants are retained in the context of the full-length α1 subunit. We performed FACS analysis to analyze the ability of these cell populations to bind biotinylated truncated protomer in suspension. Truncated protomer was used instead of commercially available collagen IV or collagen I as it carries a specific binding site for integrin α1β1 (Fig. 1). As shown in Fig. 3B, integrin α1-null mesangial cells expressing full-length WT integrin α1 (α1-WT) bound the α1α2α1(IV) truncated protomer in solution better than parental integrin α1-null cells (α1-KO). Similar to the results obtained with solid phase binding assay, no differences in α1α2α1(IV) truncated protomer binding were observed between α1-WT cells and α1-T220A or α1-R287A cells. In contrast, α1-R218D cells bound the α1α2α1(IV) truncated protomer less efficiently than α1-WT cells, whereas α1-E317A and α1-R287E/E317R cells bound the α1α2α1(IV) truncated protomer more efficiently than α1-WT cells.
FIGURE 3.

Characterization of integrin α1-null mesangial cells expressing integrin α1 subunits mutated within the I domain. A, integrin α1-null mesangial cells were transfected with either empty vector (α1KO) or the human integrin α1 carrying the mutations indicated; cell populations with equal levels of expression of the integrin α1 subunits were sorted by FACS using anti-human integrin α1 antibodies. B, binding of biotinylated α1α2α1(IV) truncated protomer to the mesangial cell populations carrying the mutations indicated was analyzed by FACS in serum-free medium as described under “Experimental Procedures.” Values are the mean ± S.D. of three independent experiments and are expressed as -fold changes relative to binding to integrin α1KO mesangial cells (arbitrarily assigned the value of 1). C and D, the cell populations carrying the mutations indicated were plated in serum-free medium on α1α2α1(IV) truncated protomer (C) or collagen I (D) (both at 10 μg/ml) for 1 h and their adhesion determined as described under “Experimental Procedures.” Values are mean ± S.D. of three independent experiments performed in quadruplicate.
Cell adhesion to immobilized α1α2α1(IV) truncated protomer and collagen I was also analyzed. As shown in Fig. 3C, the binding of cells to the truncated protomer paralleled the FACS data, with α1-R218D cells binding to this protomer less than α1-WT cells, whereas α1-E317A and α1-R287E/E317R cells adhered to this protomer more than α1-WT cells. Cell adhesion on immobilized collagen I (Fig. 3D) revealed that α1-WT cells adhere more than α1-KO cells, and α1-R287A, α1-E317A, and α1-R287E/E317R cells bound more efficiently than α1-WT cells.
Engagement of integrins by their natural ligand leads to cell spreading and formation of focal adhesions. On the α1α2α1(IV) truncated protomer, only α1-WT, α1-R287A, α1-E317A, and α1-R287E/E317R cells spread and formed focal adhesions, as measured by vinculin staining (Fig. 4A). On collagen I, in addition to α1-WT and the two gain-of-function mutants, the mutant α1-R287A was able to spread and recruit vinculin to focal adhesions (Fig. 4B). Altogether these data indicate that the differences in binding of the various α1 I domain mutants to either the α1α2α1(IV) truncated protomer or collagen I are retained when the same mutation(s) is expressed in the full-length α1 subunit.
FIGURE 4.

Integrin α1 I domain mutants differentially promote focal adhesion formation in mesangial cells. Populations of integrin α1KO mesangial cells transfected either with empty vector or with the integrin α1 I domain mutants, as indicated, were plated in serum-free medium on 10 μg/ml α1α2α1(IV) truncated protomer (A) or collagen I (B). After 4 h, the cells were fixed, permeabilized, and stained with rhodamine-phalloidin or anti-vinculin antibodies. A representative cell is shown for each cell population. Bar represents 10 μm. *, indicates areas that have been enlarged and shown in the bottom panel.
E317A- and R287E/E317R-expressing Cells Show Increased Proliferation on Both Collagen IV and Collagen I
Activation of integrin α1β1 upon binding to its natural ligand collagen IV promotes cell proliferation (25). To determine whether modulating the affinity of integrin α1β1 to collagen IV alters this cell function, we investigated the ability of the various mutant cells to proliferate on collagenous substrata. α1-WT cells proliferated significantly more than α1-KO cells when plated on the α1α2α1(IV) truncated protomer or collagen I (Fig. 5). Cells expressing the two gain-of-function mutations (α1-E317A and α1-R287E/E317R) proliferated significantly more than α1-WT cells on both α1α2α1(IV) truncated protomer and collagen I, whereas the α1-R287A cells grew more than α1-WT cells only when plated on collagen I (Fig. 5). An intermediate phenotype was observed when α1-R218D and α1-T220A cells were analyzed. Both of these cell lines proliferated significantly more than α1-KO cells when plated on truncated collagen IV protomer or collagen I but significantly less than α1-WT cells (Fig. 5). Thus, mutations that lead either to a gain of function or increased affinity to collagen I alter cell proliferation proportionately to the ability of the cell to adhere to either α1α2α1(IV) truncated collagen IV or collagen I.
FIGURE 5.

Integrin α1 I domain mutants differentially regulate proliferation of mesangial cells. Populations of integrin α1KO mesangial cells transfected either with empty vector or the integrin α1 I domain mutants, as indicated, were plated in 96-well plates coated with 10 μg/ml α1α2α1(IV) truncated protomer (A) or collagen I (B). Eight hours later, the cells were incubated with serum-free medium containing [3H]thymidine (0.5 μCi/well) for a further 48 h, and proliferation was then evaluated as described under “Experimental Procedures.” Values are the mean ± S.D. of three independent experiments performed in quadruplicate and are expressed as -fold change related to the proliferation of α1KO cells (arbitrarily assigned the value of 1).
E317A -and R287E/E317R-expressing Cells Show Decreased Collagen IV Synthesis with a Concomitant Increase in ERK Activation
Binding of integrin α1β1 to its natural ligand leads to down-regulation of endogenous collagen synthesis as well as activation of ERK (2, 25). We therefore determined whether cells expressing gain-of-function mutants down-regulate collagen IV synthesis and activate ERK more efficiently than α1-WT cells. α1-WT cells produce less collagen and have increased base-line ERK activation compared with α1KO cells (Fig. 6). Interestingly, α1-E317A and α1-R287E/E317R cells revealed a more prominent decrease in collagen IV synthesis with a concomitant increase in ERK activation (Fig. 6). Cells expressing α1-R218D, α1-T220A, and α1-R287A behaved similarly to α1-WT cells with respect to their ability to control collagen synthesis and ERK activation. Thus, increasing the ability of cells to adhere to collagen IV results in increased ERK activation and decreased collagen production.
FIGURE 6.

Integrin α1-null mesangial cells expressing E317A and R287E/E317R mutants show reduced basal levels of collagen IV and increased ERK activation. A, total cell lysates (20 μg/lane) of serum-starved populations of integrin α1KO mesangial cells, which were transfected either with empty vector or with the integrin α1 I domain mutants as indicated, were analyzed by Western blot for basal levels of collagen IV and activated ERK. Note that cells expressing E317A and R287E/E317R mutants show reduced levels of CIV with concomitant increased ERK activation. B and C, CIV, FAK, pERK, and ERK bands were quantified by densitometry analysis; the collagen IV and pERK signals are expressed as CIV/FAK (B) and pERK/ERK (C) ratios, respectively. Values are the mean ± S.D. of three experiments and represent -fold change relative to integrin α1KO cells (assigned the arbitrary value of 1). Differences between integrin α1KO and mutant-expressing cells (*) or between cells expressing WT and E317A or R287E/E317R mutants (**) were significant with p < 0.05.
ERK Inhibition Leads to Decreased Cell Proliferation and Increased Collagen Synthesis
To determine whether the significant increase in ERK activation observed in the α1-E317A and α1-R287E/E317R cells regulates both proliferation and collagen synthesis, we analyzed the effect of the MEK inhibitor PD98059 on these cell functions. Treatment with 10 μm PD98059 significantly decreased the proliferation of α1-WT, α1-E317A, and α1-R287E/E317R cells (Fig. 7A) with a concomitant inhibition of ERK phosphorylation (Fig. 7, B and C). As no significant changes in cell proliferation were observed when α1-KO cells were treated with the same dose of the MEK inhibitor (Fig. 7A), we excluded the possibility of cytotoxic effects. In contrast to proliferation, treatment with the MEK inhibitor led to a significant increase in collagen IV synthesis in α1-WT, α1-E317A, and α1-R287E/E317R cells (Fig. 7, B and C). Thus, our data indicate that the ability of gain-of-function mutants to drive proliferation and down-regulate collagen synthesis is dependent on the degree of ERK activation.
FIGURE 7.

ERK inhibition prevents E317A- and R287E/E317R-mediated cell function. A, populations of integrin α1KO mesangial cells were transfected either with empty vector or the integrin α1 I domain mutants, as indicated, and were plated in 96-well plates coated with 10 μg/ml α1α2α1(IV) truncated protomer with or without the MEK inhibitor PD98059. Eight hours later, the cells were incubated with serum-free medium containing [3H]thymidine (0.5 μCi/well) with or without the ERK inhibitor for a further 48 h, and proliferation was then evaluated as described under “Experimental Procedures.” Values are the mean ± S.D. of three independent experiments performed in quadruplicate and are expressed as -fold change related to the proliferation of α1KO cells (arbitrarily assigned the value of 1). Differences between integrin α1KO and mutant-expressing cells (*) or untreated versus PD98059-treated cells (**) were significant, with p < 0.05. B, total cell lysates (20 μg/lane) of the cell populations indicated above were serum-starved for 24 h with or without the MEK inhibitor PD98059 and were analyzed by Western blot for basal levels of collagen IV and activated ERK. Note that cells expressing E317A and R287E/E317R mutants treated with PD98059 show increased levels of CIV with a concomitant decrease in ERK activation. C, CIV, FAK, pERK, and ERK bands were quantified and expressed as described in the legend for Fig. 6. Values are the mean ± S.D. of three experiments and represent -fold change relative to integrin α1KO cells (assigned the arbitrary value of 1). * and **, as described in A.
The E317A Mutation Perturbs the Conformation of the α1 I Domain
Activation of the α1 I domain to the high collagen IV affinity form has been attributed to a shift in the position of helix α7 that releases the Arg287–Glu317 salt bridge linking helices α7 and αC, resulting in the unwinding of αC (17). The E317A substitution in the α1 I domain enhances collagen binding (17), presumably as a result of disruption of the Arg287–Glu317 salt bridge. Our binding data show that alanine and arginine substitutions at position Glu317 result in a gain of affinity for collagen IV, suggesting that the increased binding affinity is a result of a change of the protein to the open conformation. To determine whether the E317A mutation perturbs the conformation of the α1 I domain, we performed 1H-15N TROSY HSQC NMR experiments on the WT and E317A α1 I domains (Fig. 8). In this experiment, the chemical shift positions of the peaks report on the structural environment of the various amide bonds in the protein. Accordingly, these experiments are commonly used to qualitatively evaluate both the structural integrity and the conformational state of proteins. As seen in the spectra in Fig. 8, both the WT and E317A α1 I domains have excellent proton chemical shift dispersion, which indicates that both are well folded. However, there are significant chemical shift perturbations in the spectrum of the E317A mutant relative to the spectrum of the WT α1 I domain. Given that both the E317A mutant and the WT domain are well folded and that the sample conditions are identical (see “Experimental Procedures”), the significant and widespread changes in resonance chemical shift positions must result from differences in the conformational states between the E317A mutant and the WT domain. These results are consistent with, although not definitive of, the hypothesis that the WT α1 I domain resides in a closed/low affinity conformation, whereas the E317A mutant may adopt an open/high affinity state by disrupting the proposed salt bridge with Arg287.
FIGURE 8.

1H-15N TROSY HSQC spectra of wild type and E317A integrin α1 I domains. Experiments were recorded at 800 MHz (1H) on the 150 μm I domain at 298 K in 50 mm sodium phosphate (pH 7.0), 150 mm NaCl, 5 mm DTT, 2 mm MgCl2, and 10% D2O. The wild type spectrum (left panel) is shown in black and the E317A spectrum (center panel) in red. An overlay of the two spectra is shown in the right panel. The E317A mutation induces significant chemical shift perturbations relative to the wild type α1 I domain, suggestive of conformational change.
DISCUSSION
The goals of this study were to define the key amino acids involved in the binding of the integrin α1 I domain to its natural ligand, collagen IV, and to determine whether altering the affinity of integrin α1β1 to collagen IV would affect integrin α1β1-dependent regulation of cell function. We investigated the ability of integrin α1β1 to regulate collagen homeostasis, as increased collagen production is the hallmark of all fibrotic diseases and integrin α1β1 plays a protective role in the course of fibrosis by negatively regulating collagen synthesis.
Nine mammalian integrin α-subunits contain an I domain (26). All α I domains have a typical Rossmann fold in which a central β-sheet is surrounded by seven α-helices forming a divalent metal binding site, referred to as a metal ion-dependent adhesion site (MIDAS) (27). In the α I domains, the natural ligands are in direct contact with Mg2+ coordinated by residues forming the MIDAS (28). These α I domains have a very dynamic structure, especially during ligand-induced integrin activation when the conformation changes from a close to an open state (11). Although the α I domains share highly conserved structural similarity, it has been proposed that the small differences present within the 200 amino acids may provide specific ligand binding characteristics for different integrins (18, 29–31). The integrin α1 I domain preferentially binds collagen IV, although binding to laminin-111, -211, and -511 has been also reported (17). Arg218 in the α1 I domain has been reported as an important determinant of the binding specificity to collagen IV (17). Glu317 in the helix α7 forms a salt bridge with Arg287 in the helix αC, thus keeping the α1 I domain in the closed/low affinity conformation (17). However, the contribution of Arg287, and whether the salt bridge between the helices αC and α7 plays indeed a role in keeping the integrin α1 I domain in the closed conformation, has not been investigated. To address these issues and to identify key amino acids able to bind the thermolysin-derived α1α2α1(IV) truncated protomer we generated various α1 I domain mutants. When we examined the effects of the R218D mutant binding to the α1α2α1(IV) truncated protomer, we found its affinity was decreased, thus confirming results of recent binding studies of the same mutant to human collagen IV (17). We also confirmed that the E317A substitution resulted in a gain-of-function mutant with increased affinity for the α1α2α1(IV) truncated protomer and collagen I, suggesting a key role for Glu317 in keeping the α1 I domain in a close conformation (17).
Studies from the α2 I domain suggest that Thr221 resides in proximity to the MIDAS and is critically important for coordinating the metal ion and binding to collagen I (24). Whether the corresponding Thr220 in the α1 I domain also coordinates the metal ion and binding to collagen IV has not been evaluated. We show that an integrin α1 I domain carrying the T220A substitution binds to the α1α2α1(IV) truncated promoter similar to the WT α1 I domain. Similarly, integrin α1-null cells expressing the T220A mutant do not show any difference in their ability to bind the α1α2α1(IV) truncated promoter when compared with α1-WT cells. This surprising result suggests that, unlike the Thr221 residue in the α2 I domain, Thr220 in the α1 I domain is not important for coordinating the metal ion and binding to collagen IV.
Activation of the α1 I domain to the open form has been attributed to a shift in the position of helix α7 and concomitant release of the Arg287–Glu317 salt bridge linking helices α7 and αC, resulting in the unwinding of αC (Fig. 9A). The expectation, therefore, was that charge reversal of the R287E/E317R mutant should retain this interaction and have no or little effect on collagen binding. Indeed, molecular models we constructed from the existing α1 and α2 I domains indicated that this electrostatic interaction could easily be accommodated in the R287E/E317R double mutant. However, we provided functional and structural evidence that both E317A and R287E/E317R mutations result in gain-of-function conformation mutants, demonstrating that elimination of the negative charge in Glu317 is sufficient for increased activity of the mutant. Our NMR results confirmed that the E317A mutant adopts a different conformation than to the WT α1 I domain, and the extent of chemical shift differences between the WT and E317A spectra shown in Fig. 8 is consistent with a significant shift difference between helix α7 and associated global structural changes toward open conformation (12). It is surprising, therefore, that this helix was unperturbed in a recent crystal structure of the α1 I domain containing a double C139S/E317A substitution (32). This inconsistency may be due to the different experimental conditions used in the NMR and crystallization experiments and the fact that the crystal lattice may impose a constraint that favors a conformation that is likely to be only one of multiple interconverting conformations present under solution.
FIGURE 9.

Molecular model for E317 stabilization of the closed form of the α1 I domain. A, schematic representations of the closed (green) and open (gold) forms of the α1 I domain, with α helices depicted as cylinders and Glu317 and Arg287 side chains shown in ball-and-stick form. The closed form was taken from the crystal structure of the α1 I domain (Protein Data Bank ID code 1QC5) (23). The open form was constructed by homology modeling as described under “Experimental Procedures” using as a template the crystal structure of the α2 I domain bound to a collagen-like peptide containing the GFOGER sequence (Protein Data Bank ID code 1DZI) (12). B and C, schematic of parallel helices α1 and α7 in WT (B) and E317R (C, left) and E317A (C, right). The dipole moments are shown next to the cylinders. Partial charges resulting from the dipoles are shown as δ+ and δ−, and formal charges from the Glu317 side chain are shown as (+) and (−). D, overlay of the α1/α7 helix region of the α1 I open conformation model (gold) and the α2 I crystal structure (magenta). E, side and top views of the α1/α7 helices in the α1 I closed (green) conformation.
The activating conformational change resulting from elimination of the Glu317 negative charge can be rationalized on the basis of the crystal structures of the open and closed forms of the α1 and α2 I domains (11, 12, 23) and the model proposed in Fig. 9. In the closed form, the negatively charged Glu317 side chain is well positioned to undergo a favorable monopole/helical dipole interaction with helix α7 and perhaps also with helix α1, both of which have their N→C dipole moments aligned in a parallel orientation. Thus, disruption of this negative charge in any Glu317 mutant (i.e. E317A or E317R) would abrogate any favorable monopole/dipole interaction, stabilizing the position of helix α7, and may even increase electrostatic repulsion between α7 and α1 dipoles, consequently destabilizing the closed form and shifting the equilibrium toward the higher affinity open form. In addition, substitution of the glutamate side chain will disrupt stabilizing interactions proximal to the MIDAS. Furthermore, electrostatic interactions between the Glu317 side chain and Asp167 on helix α1 may also influence the switch between open and closed conformations (Fig. 9, B–D).
We also provide evidence that the R287A substitution leads to increased affinity toward collagen I and unchanged affinity toward collagen IV. The differential binding of the R287A mutant toward collagens I and IV is likely a result of the fact that in the open form Arg287 is exposed to the collagen binding surface, suggesting that this residue directly contacts collagen in the bound form in a way that it is collagen IV-specific. Taken together, these data suggest that the key roles played by Arg287 and Glu317 seem to be largely independent of their interaction as a salt bridge.
A key question addressed in this study was whether changes in affinity observed in the isolated mutated I domains were retained in the context of the full-length α1 subunit in cells. This is important, as cells expressing the single D219N or D292N substitution in the α2 I domain are still able to bind collagen I, despite the loss of binding in the isolated α2 I domain carrying the same mutations (33). On the other hand, mutation of the Thr221 in the α2 I domain results in loss of adhesion to collagen I in the isolated α2 I domain as well as when it is expressed in the full-length integrin α2 subunit (34). Finally, the E317A substitution in the α1 I domain results in a domain with increased binding to both collagens I and IV, an effect that is maintained when the same mutation is expressed in the whole integrin α1 subunit in cells (32). Thus, it is clear that not all mutations that alter the binding of the α1 I domain to its natural ligand result in the same changes in the context of the whole integrin. We have provided evidence that integrin α1-KO cells expressing the R287A and T220A mutations, as well as the two gain-of-function E317A and R287E/E317R mutations, adhere to either the α1α2α1(IV) truncated protomer or collagen I with the same pattern as observed with the isolated I domains, suggesting that expressing the mutation in the full integrin α1 subunit does not change the ligand specificity.
Finally, another key point we addressed in this study was whether modulating the affinity to ligand affected integrin α1β1-mediated cell function. Upon binding to their natural ligand, integrins activate intracellular pathways controlling different cell functions, including cell proliferation, migration, differentiation, and matrix homeostasis. The best characterized mechanisms of integrin-mediated signaling are those mediated by their cytoplasmic tails. Integrin α1subunit has a short cytoplasmic tail consisting of 15 amino acids, and truncation and point mutants have revealed a role for specific amino acids within the tail in controlling selective cell functions (10, 35–37). In addition, the transmembrane domains of integrins have been shown to play a key role in integrin activation as well as outside-in signaling. In this context, mutations that disrupt the interaction between integrin αIIb and β3 transmembrane domains result in the activation of intracellular signaling pathways (38). Moreover, we have demonstrated that substitution of the transmembrane domain of the integrin α2 subunit with that of α1 results in decreased cell adhesion, migration, and tubulogenesis, clearly suggesting a role for the integrin α2 transmembrane domain in regulating cell functions (37). In this study, we demonstrated that altering the affinity of the integrin α1 I domain to collagen affected α1β1-mediated intracellular signaling. Integrin α1-KO cells carrying mutations within the α1 I domain that enhance binding to both the α1α2α1(IV) truncated protomer and collagen I (i.e. E317A and R287E/E317R) have a significantly increased ERK activation compared with cells carrying WT or loss-of-function mutant α1 I domains. We further showed that α1-KO cells carrying the E317A or R287E/E317R substitutions proliferate significantly more than α1-KO or α1-WT cells, suggesting that enhanced ERK activation in these mutant cells leads to robust cell proliferation. These results are consistent with our previous findings that ligated integrin α1β1 activates the Shc/Grb2/ERK signaling pathway, which promotes cell proliferation, and that cells lacking integrin α1β1 fail to activate this mitogenic pathway and to proliferate when plated on collagen IV (25).
ERK not only promotes proliferation but also plays a role in regulating collagen synthesis. ERK can be pro-fibrotic as seen in the situation of increased mechanical load, which induces ERK activation, a key step in promoting collagen I expression (39). In contrast, it has been shown recently that matrix-induced activation of the MEK/ERK pathway promotes inhibition of collagen I synthesis (40). We previously showed that binding of integrin α1β1 to collagen IV results in the down-regulation of collagen IV and that cells lacking this collagen receptor show increased basal levels of endogenous collagen IV (2, 4, 8). Our current finding that cells that express α1 I domain mutants, which are able to promote sustained ERK activation, show enhanced down-regulation of endogenous collagen IV synthesis, suggesting a key role for ERK activation in mediating the down-regulation of collagen IV synthesis.
In conclusion, we have demonstrated that increasing the affinity of the extracellular domain of the integrin α1 I domain to its natural ligand enhances activation of intracellular kinases and, most importantly, down-regulation of collagen synthesis. This finding has major implications for our understanding of the pathogenesis of fibrotic disease and suggests that targeting collagen receptors may be a therapeutic strategy to halt the progression of this ubiquitous pathological process.
Supplementary Material
These studies were supported, in whole or in part, by National Institutes of Health Grants 2P01DK065123 (to A. P., B. F. E., R. Z., and B. G. H.), DK069221 and DK075594 (to R. Z.), DK083187 (to R. Z. and C. R. S.), and P30DK79341 (the O'Brien) (to A. P. and R. Z.). This work was also supported by a Department of Veterans Affairs Merit Review (to A. P. and R. Z.) and by an American Heart Association (AHA) Established Investigator Award (to R. Z.) and AHA Grant 10POST4210008 (to W. D. V.-H.).

This article contains supplemental Figs. 1 and 2.
- I
- inserted
- Ni-NTA
- nickel-nitrilotriacetic acid
- MWCO
- molecular weight cutoff
- MIDAS
- metal ion-dependent adhesion site
- CIV
- collagen IV
- FAK
- focal adhesion kinase.
REFERENCES
- 1. Pozzi A., Voziyan P. A., Hudson B. G., Zent R. (2009) Regulation of matrix synthesis, remodeling, and accumulation in glomerulosclerosis. Curr. Pharm. Des. 15, 1318–1333 [DOI] [PubMed] [Google Scholar]
- 2. Gardner H., Broberg A., Pozzi A., Laato M., Heino J. (1999) Absence of integrin α1β1 in the mouse causes loss of feedback regulation of collagen synthesis in normal and wounded dermis. J. Cell Sci. 112, 263–272 [DOI] [PubMed] [Google Scholar]
- 3. Zweers M. C., Davidson J. M., Pozzi A., Hallinger R., Janz K., Quondamatteo F., Leutgeb B., Krieg T., Eckes B. (2007) Integrin α2β1 is required for regulation of murine wound angiogenesis but is dispensable for reepithelialization. J. Invest. Dermatol. 127, 467–478 [DOI] [PubMed] [Google Scholar]
- 4. Chen X., Moeckel G., Morrow J. D., Cosgrove D., Harris R. C., Fogo A. B., Zent R., Pozzi A. (2004) Lack of integrin α1β1 leads to severe glomerulosclerosis after glomerular injury. Am. J. Pathol. 165, 617–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Borza C. M., Su Y., Chen X., Yu L., Mont S., Chetyrkin S., Voziyan P., Hudson B. G., Billings P. C., Jo H., Bennett J. S., Degrado W. F., Eckes B., Zent R., Pozzi A. (2012) Inhibition of integrin α2β1 ameliorates glomerular injury. J. Am. Soc. Nephrol. 23, 1027–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ivaska J., Reunanen H., Westermarck J., Koivisto L., Kähäri V. M., Heino J. (1999) Integrin α2β1 mediates isoform-specific activation of p38 and up-regulation of collagen gene transcription by a mechanism involving the α2 cytoplasmic tail. J. Cell Biol. 147, 401–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zent R., Yan X., Su Y., Hudson B. G., Borza D. B., Moeckel G. W., Qi Z., Sado Y., Breyer M. D., Voziyan P., Pozzi A. (2006) Glomerular injury is exacerbated in diabetic integrin α1-null mice. Kidney Int. 70, 460–470 [DOI] [PubMed] [Google Scholar]
- 8. Chen X., Abair T. D., Ibanez M. R., Su Y., Frey M. R., Dise R. S., Polk D. B., Singh A. B., Harris R. C., Zent R., Pozzi A. (2007) Integrin α1β1 controls reactive oxygen species synthesis by negatively regulating epidermal growth factor receptor-mediated Rac activation. Mol. Cell. Biol. 27, 3313–3326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen X., Whiting C., Borza C., Hu W., Mont S., Bulus N., Zhang M. Z., Harris R. C., Zent R., Pozzi A. (2010) Integrin α1β1 regulates epidermal growth factor receptor activation by controlling peroxisome proliferator-activated receptor γ-dependent caveolin-1 expression. Mol. Cell. Biol. 30, 3048–3058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Borza C. M., Chen X., Mathew S., Mont S., Sanders C. R., Zent R., Pozzi A. (2010) Integrin α1β1 promotes caveolin-1 dephosphorylation by activating T cell protein-tyrosine phosphatase. J. Biol. Chem. 285, 40114–40124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Emsley J., King S. L., Bergelson J. M., Liddington R. C. (1997) Crystal structure of the I domain from integrin α2β1. J. Biol. Chem. 272, 28512–28517 [DOI] [PubMed] [Google Scholar]
- 12. Emsley J., Knight C. G., Farndale R. W., Barnes M. J., Liddington R. C. (2000) Structural basis of collagen recognition by integrin α2β1. Cell 101, 47–56 [DOI] [PubMed] [Google Scholar]
- 13. Bahou W. F., Potter C. L., Mirza H. (1994) The VLA-2 (α2β1) I domain functions as a ligand-specific recognition sequence for endothelial cell attachment and spreading: molecular and functional characterization. Blood 84, 3734–3741 [PubMed] [Google Scholar]
- 14. Calderwood D. A., Tuckwell D. S., Eble J., Kühn K., Humphries M. J. (1997) The integrin α1 A-domain is a ligand binding site for collagens and laminin. J. Biol. Chem. 272, 12311–12317 [DOI] [PubMed] [Google Scholar]
- 15. Kern A., Eble J., Golbik R., Kühn K. (1993) Interaction of type IV collagen with the isolated integrins α1β1 and α2β1. Eur. J. Biochem. 215, 151–159 [DOI] [PubMed] [Google Scholar]
- 16. Dickeson S. K., Mathis N. L., Rahman M., Bergelson J. M., Santoro S. A. (1999) Determinants of ligand binding specificity of the α(1)β(1) and α(2)β(1) integrins. J. Biol. Chem. 274, 32182–32191 [DOI] [PubMed] [Google Scholar]
- 17. Tulla M., Lahti M., Puranen J. S., Brandt A. M., Käpylä J., Domogatskaya A., Salminen T. A., Tryggvason K., Johnson M. S., Heino J. (2008) Effects of conformational activation of integrin α 1I and α 2I domains on selective recognition of laminin and collagen subtypes. Exp. Cell Res. 314, 1734–1743 [DOI] [PubMed] [Google Scholar]
- 18. Tulla M., Pentikäinen O. T., Viitasalo T., Käpylä J., Impola U., Nykvist P., Nissinen L., Johnson M. S., Heino J. (2001) Selective binding of collagen subtypes by integrin α 1I, α 2I, and α 10I domains. J. Biol. Chem. 276, 48206–48212 [DOI] [PubMed] [Google Scholar]
- 19. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 20. Vandenberg P., Kern A., Ries A., Luckenbill-Edds L., Mann K., Kühn K. (1991) Characterization of a type IV collagen major cell binding site with affinity to the α1β1 and the α2β1 integrins. J. Cell Biol. 113, 1475–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eble J. A., Golbik R., Mann K., Kühn K. (1993) The α1β1 integrin recognition site of the basement membrane collagen molecule [α1(IV)]2α2(IV). EMBO J. 12, 4795–4802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gunwar S., Noelken M. E., Hudson B. G. (1991) Properties of the collagenous domain of the α3(IV) chain, the Goodpasture antigen, of lens basement membrane collagen. Selective cleavage of α(IV) chains with retention of their triple helical structure and noncollagenous domain. J. Biol. Chem. 266, 14088–14094 [PubMed] [Google Scholar]
- 23. Rich R. L., Deivanayagam C. C., Owens R. T., Carson M., Höök A., Moore D., Symersky J., Yang V. W., Narayana S. V., Höök M. (1999) Trench-shaped binding sites promote multiple classes of interactions between collagen and the adherence receptors, α(1)β(1) integrin and Staphylococcus aureus cna MSCRAMM. J. Biol. Chem. 274, 24906–24913 [DOI] [PubMed] [Google Scholar]
- 24. Fleming F. E., Graham K. L., Takada Y., Coulson B. S. (2011) Determinants of the specificity of rotavirus interactions with the α2β1 integrin. J. Biol. Chem. 286, 6165–6174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pozzi A., Wary K. K., Giancotti F. G., Gardner H. A. (1998) Integrin α1β1 mediates a unique collagen-dependent proliferation pathway in vivo. J. Cell Biol. 142, 587–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fu G., Wang W., Luo B. H. (2012) Overview: structural biology of integrins. Methods Mol. Biol. 757, 81–99 [DOI] [PubMed] [Google Scholar]
- 27. Lee J. O., Rieu P., Arnaout M. A., Liddington R. (1995) Crystal structure of the A domain from the α subunit of integrin CR3 (CD11b/CD18). Cell 80, 631–638 [DOI] [PubMed] [Google Scholar]
- 28. Michishita M., Videm V., Arnaout M. A. (1993) A novel divalent cation-binding site in the A domain of the β2 integrin CR3 (CD11b/CD18) is essential for ligand binding. Cell 72, 857–867 [DOI] [PubMed] [Google Scholar]
- 29. Kamata T., Puzon W., Takada Y. (1995) Identification of putative ligand-binding sites of the integrin α4β1 (VLA-4, CD49d/CD29) Biochem. J. 305, 945–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kern A., Briesewitz R., Bank I., Marcantonio E. E. (1994) The role of the I domain in ligand binding of the human integrin α1β1. J. Biol. Chem. 269, 22811–22816 [PubMed] [Google Scholar]
- 31. Zhang W. M., Kapyla J., Puranen J. S., Knight C. G., Tiger C. F., Pentikainen O. T., Johnson M. S., Farndale R. W., Heino J., Gullberg D. (2003) α11β1 integrin recognizes the GFOGER sequence in interstitial collagens. J. Biol. Chem. 278, 7270–7277 [DOI] [PubMed] [Google Scholar]
- 32. Lahti M., Bligt E., Niskanen H., Parkash V., Brandt A. M., Jokinen J., Patrikainen P., Käpylä J., Heino J., Salminen T. A. (2011) Structure of collagen receptor integrin α(1)I domain carrying the activating mutation E317A. J. Biol. Chem. 286, 43343–43351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Käpylä J., Ivaska J., Riikonen R., Nykvist P., Pentikäinen O., Johnson M., Heino J. (2000) Integrin alpha(2)I domain recognizes type I and type IV collagens by different mechanisms. J. Biol. Chem. 275, 3348–3354 [DOI] [PubMed] [Google Scholar]
- 34. Kamata T., Takada Y. (1994) Direct binding of collagen to the I domain of integrin α2β1 (VLA-2, CD49b/CD29) in a divalent cation-independent manner. J. Biol. Chem. 269, 26006–26010 [PubMed] [Google Scholar]
- 35. Abair T. D., Bulus N., Borza C., Sundaramoorthy M., Zent R., Pozzi A. (2008) Functional analysis of the cytoplasmic domain of the integrin (α)1 subunit in endothelial cells. Blood 112, 3242–3254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mattila E., Pellinen T., Nevo J., Vuoriluoto K., Arjonen A., Ivaska J. (2005) Negative regulation of EGFR signalling through integrin-α1β1-mediated activation of protein tyrosine phosphatase TCPTP. Nat. Cell Biol. 7, 78–85 [DOI] [PubMed] [Google Scholar]
- 37. Abair T. D., Sundaramoorthy M., Chen D., Heino J., Ivaska J., Hudson B. G., Sanders C. R., Pozzi A., Zent R. (2008) Cross-talk between integrins α1β1 and α2β1 in renal epithelial cells. Exp. Cell Res. 314, 3593–3604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhu J., Carman C. V., Kim M., Shimaoka M., Springer T. A., Luo B. H. (2007) Requirement of α and β subunit transmembrane helix separation for integrin outside-in signaling. Blood 110, 2475–2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Papakrivopoulou J., Lindahl G. E., Bishop J. E., Laurent G. J. (2004) Differential roles of extracellular signal-regulated kinase 1/2 and p38MAPK in mechanical load-induced procollagen α1(I) gene expression in cardiac fibroblasts. Cardiovasc. Res. 61, 736–744 [DOI] [PubMed] [Google Scholar]
- 40. Dzobo K., Leaner V. D., Parker M. I. (2012) Feedback regulation of the α2(1) collagen gene via the Mek-Erk signaling pathway. IUBMB Life 64, 87–98 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.