

Background: Mutations on the KIAA1530 gene cause UV sensitivity syndrome (UVsS).
Results: KIAA1530 is recruited by the CSA protein to UV-damaged chromatin. CSA patient mutant W361C was unable to bind KIAA1530 and failed to recruit KIAA1530.
Conclusion: UVsS is caused by defective KIAA1530 recruitment, either due to CSA or KIAA1530 mutations.
Significance: This study provides new insights into TCR and UVsS.
Keywords: DNA Damage, DNA Damage Response, DNA Nucleotide Excision Repair, DNA Repair, Protein-Protein Interactions
Abstract
Transcription-coupled repair (TCR) is the major pathway involved in the removal of UV-induced photolesions from the transcribed strand of active genes. Two Cockayne syndrome (CS) complementation group proteins, CSA and CSB, are important for TCR repair. The molecular mechanisms by which CS proteins regulate TCR remain elusive. Here, we report the characterization of KIAA1530, an evolutionarily conserved protein that participates in this pathway through its interaction with CSA and the TFIIH complex. We found that UV irradiation led to the recruitment of KIAA1530 onto chromatin in a CSA-dependent manner. Cells lacking KIAA1530 were highly sensitive to UV irradiation and displayed deficiency in TCR. In addition, KIAA1530 depletion abrogated stability of the CSB protein following UV irradiation. More excitingly, we found that a unique CSA mutant (W361C), which was previously identified in a patient with UVsS syndrome, showed defective KIAA1530 binding and resulted in a failure of recruiting KIAA1530 and stabilizing CSB after UV treatment. Together, our data not only reveal that KIAA1530 is an important player in TCR but also lead to a better understanding of the molecular mechanism underlying UVsS syndrome.
Introduction
The nucleotide excision repair (NER)3 pathway recognizes and removes a wide spectrum of DNA damages, primarily UV-induced cyclobutane pyrimidine dimers (CPDs) and pyrimidine-pyrimidone photoproducts (6-4PPs) (1). The basic NER process includes damage recognition and verification, opening of the DNA helix around damage sites, dual excision, and replacing the damaged strand and ligation (2). So far, four NER-impaired genetic defects in humans have been identified, including xeroderma pigmentosum (XP), Cockayne syndrome (CS), trichothiodystrophy, and the mild ultraviolet (UV) light-sensitive syndrome (UVSS) (3–6).
NER can be subdivided into two distinct pathways based on the genomic location of the repair event. The first is called transcript-coupled repair (TCR), which efficiently detects and removes damages only on the transcribed strands of active genes. The other subpathway is global genomic repair, which repairs damages located anywhere in the whole genome. TCR is presumably initiated when RNA polymerase II (RNAP II) stalls (7), whereas global genomic repair is triggered by direct binding of the UV-DDB and XPC-HR23b complex onto photolesions (8, 9). In the TCR pathway, stalled RNAP II is in turn supposed to be removed or backtracked (10–12). The subsequent recruitment of ERCC8/CSA and ERCC6/CSB factors may facilitate assembly of downstream repair machinery. However, how CSA and CSB operate during this procedure is not clear (7, 13).
Mutations in the CSA or CSB gene cause Cockayne syndrome, which is characterized by UV hypersensitivity, premature aging, and severe developmental and neurological dysfunctions (14). Cells derived from CSA and CSB patients have elevated sensitivity to both UV irradiation and oxidative damage and are defective on the recovery of RNA synthesis. CSB is an ATPase that belongs to the SWI2/SNF2 family and has been shown remodeling nucleosomes in vitro (15, 16). Besides the role as a chromatin remodeler, CSB interacts constitutively with RNAP II and is essential for the assembly of pre-incision TCR complex at arrested RNAP II (17). CSB is also important for the recruitment of histone acetyltransferase p300 and CSA to the nuclear matrix upon exposure to UV irradiation (18, 19). Unlike CSB, CSA is a WD40 motif-containing protein that interacts with Cullin4A (Cul4A) and ROC1/Rbx1 ubiquitin E3 ligase (20, 21). This ubiquitin E3 ligase is initially inhibited after UV irradiation via its association with COP9 signalosome and later becomes activated to ubiquitinate and degrade CSB (20, 22, 23). Besides its role in degrading CSB, CSA has been shown to be important in the recruitment of other factors, including XAB2, HMGN1, and TFIIS, that are essential for TCR repair and resumption of transcription (17).
Eighteen distinct CSA mutations have been reported to date (24). Most of these point mutations and premature terminations lead to severe CS clinical manifestations, as described above (25). However, a point mutation, CSA (p.W361C) identified in patient UVSS1VI, causes mild UVSS syndrome, which is manifested by hypersensitivity to UV irradiation and a failure to resume RNA synthesis. This patient does not have any neurological or developmental abnormalities, and the cellular sensitivity to oxidative stress is normal, indicating that this mutant may uncouple the roles of CSA in UV response versus its function in oxidative damage repair (26). So far, the molecular pathology of CS remains poorly understood because of relative heterogeneous cellular and clinical phenotypes of patients carrying mutations on the same gene (14, 24). In this study, we identify and characterize KIAA1530, a protein that is recruited to sites of DNA damage by CSA. We showed that this protein is essential for transcription recovery and cell survival after UV irradiation. Moreover, we demonstrated a link between KIAA1530 and the pathogenic CSA mutation (p.W361C) in the patient with UVSS and thereby shed light on the molecular mechanism of TCR repair.
EXPERIMENTAL PROCEDURES
Plasmids and Cloning
The full-length KIAA1530 and CSA cDNAs were derived from KIAA1530-TOP1 (purchased from Harvard Plasmid) and CSA-PDONR221 plasmids. cDNAs were generated by PCR and subcloned into pDONR 201 vector using Gateway recombination technology (Invitrogen). For the generation of the Gateway destination vector containing the C-terminal triple-tag (S-protein, FLAG and a streptavidin-binding peptide) or N-terminal hemagglutinin (HA) epitope tag, KIAA1530 and CSA cDNAs were transferred from the pDONR201 vector into the corresponding tagging vector by the LR recombination reaction. KIAA1530 point mutations, truncations, and internal deletions used in this study were generated using the QuikChange site-directed mutagenesis kit (Stratagene). All constructs were confirmed by sequencing.
Antibodies
Primary antibodies against p89/XPB (s-19), CSA (c-18), and CSB (E-18) were obtained from Santa Cruz Biotechnology. Anti-hemagglutinin (HA) antigen (3F10) and anti-FLAG (M2) antibodies were purchased from Sigma. Anti-USP7 was from Bethyl Laboratories Inc. Anti-CPDs (TDM-2) and anti-6-4PPs (64M-2) antibodies were obtained from Cosmo Bio USA. Anti-KIAA1530 rabbit polyclonal antibody was raised by immunizing rabbit with the recombinant GST-KIAA1530 fragment (residues 1–311), which was expressed and purified from Escherichia coli. KIAA1530 antibody was then affinity-purified using AminoLink plus immobilization and purification kit (Pierce) according to the manufacturer's protocol. Horseradish peroxidase (HRP)-conjugated secondary antibodies against rabbit or mouse IgG and HRP-conjugated protein A were obtained from Sigma.
Cell Culture and Transfection
Human CS-A (CS3BE), CSA complemented CS3BE, and XP-A fibroblasts were obtained from Dr. Lei Li, University of Texas MD Anderson Cancer Center. XP-A, HeLa, and HEK293T cells were cultured using Dulbecco's modified Eagle's medium (DMEM) containing 10% (v/v) fetal bovine serum (FBS; Invitrogen), 100 units/ml penicillin, and 0.1 mg/ml streptomycin. CS3BE, CS3BE-CSA, CS3BE-CSA-HA-FLAG, and CS3BE-CSA (p.W361C)-HA-FLAG were cultured using DMEM/F-12 medium containing 15% (v/v) FBS. Cells were maintained in a humidified incubator atmosphere at 37 °C with 5% CO2. CSA-complemented CS3BE cells were cultured in the presence of 75 μg/ml hygromycin. Plasmid transfection was performed using polyethyleneimine reagent. pGIPZ lentiviral control silencing shRNA and KIAA1530 shRNAs were purchased from Open Biosystems. The KIAA1530 targeting sequences were as follows: KIAA1530#1, 5′-CCCGACTAAATCCTGAGAA-3′; KIAA1530#2, 5′-CAGTTTAACTACGCACTGA-3′; KIAA1530#4, 5′-CGGATGCTGGTTGTTTCCA-3′; and KIAA1530#5, 5′-CCGACTAAATCCTGAGAAA-3′. The KIAA1530#2 shRNA-resistant wild-type and mutant KIAA1530 constructs were generated by six nucleotide substitutions (2109G→A, 2112T→C, 2115C→T, 2118C→T, 2121A→C, and 2122C→T). The lentiviral packaging was performed by co-transfecting pMD2G and pSPAX2 packaging plasmids together with shRNA constructs into HEK293T cells. Two sequential infections to HeLa cells were performed 48 and 72 h after the initial transfection to enhance the infection efficiency. Infected cells were selected with DMEM complete medium containing 2 μg/ml puromycin (Sigma).
Tandem Affinity Purification (TAP) of Protein Complexes
HEK-293T cells were transfected with KIAA1530-SFB plasmid by using polyethyleneimine transfection reagents. The stable clone was selected with medium containing 2 μg/ml puromycin. Immunofluorescence and Western blots were performed to verify the expression and subcellular localization of tagged proteins. 200 ml of HEK293T cells stably expressing recombinant protein were collected and lysed in NETN buffer (20 mm Tris-HCl, pH 8.0, 100 mm NaCl, 1 mm EDTA, 1 mm MgCl2, 0.5% Nonidet P-40, and 1 μg/ml each pepstatin A and aprotinin) for 30 min at 4 °C. The crude lysates were sonicated for 10 s, three times at 4 °C to release chromatin-bound proteins, and recovered by centrifugation (15,000 × g, 30 min, 4 °C). The whole lysate was then incubated with streptavidin-conjugated beads (Amersham Biosciences) for 2 h at 4 °C with gentle agitation. To release beads bound protein complexes, streptavidin beads were then washed four times with NETN buffer and incubated in NETN buffer containing 2 mg/ml biotin (Sigma) for 2 h at 4 °C. The eluate was then incubated with S-protein-agarose beads (Novagen) overnight at 4 °C with gentle agitation. The beads were then washed three times with NETN buffer, resuspended in Laemmli sample buffer, resolved by denaturing PAGE, and analyzed by the Taplin Mass Spectrometry Facility at Harvard Medical School.
Preparation of Cell Lysate under Denaturing Conditions
After designated treatment, cells were washed two times with PBS, lysed in denaturing buffer (50 mm Tris-HCl, pH 7.5, 5 mm DTT, and 1% SDS), and boiled for 10 min. Samples then were diluted with 4 volumes of NETN buffer before immunoprecipitation.
Preparation of Cell Fractions
After indicated UV post-irradiation time, cells were washed twice with ice-cold PBS and lysed in 0.5 ml of NETN buffer containing protease inhibitors. Crude lysate was cleared by centrifugation at 15,000 × g for 10 min at 4 °C. The supernatant was marked as a soluble fraction. The pellet was washed twice with ice-cold NETN buffer, briefly sonicated in 100 μl of denaturing buffer, and boiled for 10 min before 5 volumes of NETN buffer were added. This was the chromatin fraction used in this study.
Precipitation and Western Blot
For precipitation of SFB-tagged proteins, cell extracts were incubated with S-beads under rotation for 2 h at 4 °C. For immunoprecipitation of endogenous protein complexes, cell extracts were incubated with antibody against KIAA1530 for 4 h and another 4 h with protein A beads (RepliGen) at 4 °C. After four washes with ice-cold NETN buffer, immunoprecipitates were released from the beads by boiling in 1× Laemmli buffer and resolved by SDS-PAGE. The electrophoretically resolved samples were transferred to a polyvinylidene fluoride (PVDF) membrane (Bio-Rad). The membrane was blocked by incubation with Tris-buffered saline with 0.05% (v/v) Tween 20 (TBST) buffer containing 5% (w/v) nonfat dry milk for 2 h at room temperature. The membrane was then probed with antibodies as indicated.
Immunocytochemistry
HeLa cells or KIAA1530 stable knockdown cells cultured on coverslips were washed with PBS and irradiated with 10 J/m2 UV light. After the designated incubation time after irradiation, the medium was aspirated, and cells were washed with PBS and fixed with 3% paraformaldehyde in PBS for 15 min at room temperature. After permeabilization with 0.5% Triton solution, DNA was denatured with 0.07 m NaOH for 8 min. Cells were incubated with 20% FBS in PBS to block nonspecific binding. Samples were then incubated with primary antibodies against CPDs (1:1000) or 6-4PPs (1:700) for 1 h at 37 °C. Coverslips were washed and incubated with rhodamine-conjugated secondary antibody for 30 min at 37 °C. The nuclei were stained with DAPI. The samples were analyzed using a Nikon ECLIPSE 90i fluorescence microscope with a Nikon Plan Fluor 10× objective lens. Images were acquired under the same setting with a CCD camera. Quantification of the UV lesions was carried out with ImageJ software with a minimum of 35 cells per experiment.
Survival Assay
Approximately 500 cells were seeded in triplicate onto 60-mm dishes. For UV-induced cell death assay, 24 h after seeding, cells were washed two times with PBS and irradiated with the indicated dose of UV light. Cells were then incubated with fresh medium for 10–14 days. Resulting colonies were fixed and stained with Coomassie Blue and counted. Results were normalized with plating efficiency. For H2O2 sensitivity assay, cells were plated onto 6-well plates 24 h before the assay. 30% H2O2 (w/w, Sigma) was diluted in DMEM complete medium to reach the indicated concentration. Treatment was done by incubating cells with H2O2-containing complete medium at 37 °C for 2 h. After treatment, cells attached on the plate were washed gently once with 1× PBS before trypsinization. Cell number was determined using a standard cell counter. Results were normalized with plating efficiency.
Recovery of RNA Synthesis after UV Irradiation
After the designated time of incubation after UV irradiation, cells were incubated with [3H]uridine (NET-174; 1.0 mCi/ml, specific activity 22.6 Ci/mmol; PerkinElmer Life Sciences) at 37 °C for 1 h to quantify the RNA synthesis rate. The cells were washed twice with PBS, lysed with 0.4 m perchloric acid, and dissolved in 0.5 m KOH. The [3H]uridine incorporated into RNA was quantified by counting radioactive intensity of 3H over the remaining pellet by liquid scintillation counting.
RESULTS
KIAA1530 Interacts with CSA and TFIIH Complex
To study the exact roles of CSA in TCR, we performed TAP of CSA-containing protein complexes and identified the Cullin4-DDB ubiquitin ligase complex, the COP9 signalsome complex, and an uncharacterized protein KIAA1530 as CSA-associated proteins (Fig. 1A). To decipher the pathway in which KIAA1530 may participate, we performed reversal TAP using KIAA1530-SFB as the bait protein. Mass spectrometry analysis revealed many putative KIAA1530-interacting proteins, including the deubiquitinating enzyme USP7 and several NER proteins, such as DDB1, CSA, and the TFIIH complex subunits XPB, XPD, p62, and CDK7 (Fig. 1B). Using the overexpression system, we confirmed the interaction between KIAA1530 and CSA (Fig. 1C). The endogenous TFIIH complex subunit XPB also co-precipitated with KIAA1530-SFB in this experiment (Fig. 1C). Conversely, KIAA1530 was co-immunoprecipitated with SFB-tagged CSA and endogenous XPB (Fig. 1, D and E). Moreover, endogenous CSA and XPB also associated with endogenous KIAA1530 (Fig. 1, F and G). Together, these data indicate that KIAA1530 interacts with CSA and XPB and may play a role in TCR.
FIGURE 1.

KIAA1530 associates with CSA and TFIIH complex. A, mass spectrometry analysis results of CSA-containing protein complexes purified from HEK293T cells stably expressing CSA-SFB. “# of peptides identified by mass-spec analysis” is provided. Red indicates the bait protein, and blue indicates the known NER factors or putative interaction partners. B, TAP purification coupled with mass-spectrometry analysis using KIAA1530-SFB as the bait protein. C, full-length KIAA1530-SFB interacted with CSA-HA but not Rad51-HA. HEK293T cells were transfected as indicated. The lysates were incubated with S-protein beads, and the resulting precipitates were analyzed by Western blotting (WB) using anti-FLAG and anti-HA antibodies. D, co-precipitation of CSA-SFB with KIAA1530-GFP was carried out using S-beads, and KIAA1530-GFP was detected using anti-GFP antibody. E, co-immunoprecipitation of endogenous XPB with ectopically expressed KIAA1530-SFB. 0.5 μg of XPB antibody was used to pull down the endogenous XPB complex. KIAA1530-SFB was detected using FLAG antibody. F, HeLa cell lysate was subjected to immunoprecipitation (IP) using KIAA1530 polyclonal antibody or rabbit IgG. Immunoprecipitates were analyzed by Western blotting using KIAA1530 antibody or pre-bleed serum. G, detecting the endogenous KIAA1530 complex. HEK293T cell lysate was incubated with KIAA1530 antibody or IgG. Western blot was performed using indicated antibodies. The asterisk in CSA immunoprecipitates denotes a nonspecific band.
Recruitment of KIAA1530 to UV-damaged Chromatin Is CSA-dependent
Because KIAA1530 interacts with many NER proteins, we wanted to know whether KIAA1530 is also an NER protein. To this end, we first investigated whether KIAA1530 accumulates on UV lesions. As shown in Fig. 2A, we detected an accumulation of KIAA1530 in chromatin fraction, where CSA, CSB, and XPB were also enriched after induction of UV lesions, suggesting that KIAA1530 is recruited to UV lesions on the chromatin.
FIGURE 2.

Chromatin translocation of KIAA1530 upon UV irradiation. A, HeLa cells were mock- or UV-treated (30 J/m2) and incubated for 1 h. Cells were lysed with Nonidet P-40 buffer containing 0.3 m salt. The distribution of KIAA1530, CSB, XPB, and histone H3 were analyzed by Western blotting (WB) using the indicated antibodies. B, distribution of KIAA1530 in UV-irradiated xeroderma pigmentosum complementation group A cells. C, CS3BE and CS3BE complemented with wild-type CSA were irradiated with 30 J/m2 UV and recovered for 1 h. Lysates were fractionated and analyzed by immunoprecipitation and Western blot using indicated antibodies.
To delineate where KIAA1530 fits with the current TC-NER model, we used XP-A-deficient cells and showed that the absence of functional XPA protein did not affect the chromatin recruitment of KIAA1530 after UV irradiation (Fig. 2B). However, when we used the CSA patient cell line (CS3BE) that harbors two point mutations on the CSA gene, which severely impairs the WD40 motif of CSA protein (18, 20, 24), we found that the UV-induced chromatin accumulation of KIAA1530 was abolished (Fig. 2C). This defect in KIAA1530 recruitment was corrected by complementation of CS3BE cells with wild-type CSA (Fig. 2C), suggesting that KIAA1530 acts downstream of CSA in the TCR.
Knockdown of KIAA1530 Sensitizes Cells to UV Irradiation and Abrogates Transcription Recovery
To reveal the roles of KIAA1530 in UV damage response, we generated KIAA1530 stable knockdown cells by infecting HeLa cells with lentivirus carrying specific KIAA1530 shRNA constructs (Fig. 3A). Knockdown of KIAA1530 by three different shRNAs elevated their sensitivity to UV irradiation compared with HeLa cells transfected with a control shRNA (Fig. 3B). Immunostaining of UV-induced lesions in these cells revealed that the efficiencies of removing CPDs and 6-4PPs in KIAA1530 knockdown cells were similar to those in control cells as well as those in CSA-deficient CS3BE cells (Fig. 3C), suggesting that KIAA1530 is dispensable for global genomic repair.
FIGURE 3.

KIAA1530 deficiency results in UV hypersensitivity. A, immunoblot analysis of HeLa whole cell extract indicating the degree of KIAA1530 knockdown by infection of cells with the indicated shRNA lentivirus constructs. shCtrl, control shRNA. B, UV survival of control and KIAA1530 stable knockdown cells. 500 cells were seeded onto 60-mm dishes and exposed to UV light. Cells were then incubated for 10–14 days, fixed, and stained with Coomassie Blue. C, detection and quantification of CPDs and 6,4-PPs lesions in shRNA control cells, KIAA1530 knockdown cells, CS3BE cells, and repair-deficient XP-A cells at various time points after UV irradiation (10 J/m2). Images were taken with equal exposure time and with the identical microscopy settings. Fluorescence intensities of UV lesions from at least 20 cells were quantified. D, incorporation of [3H]uridine in different cell lines before and after UV irradiation (10 J/m2). All radioactive intensities were normalized to cell count. Two independent experiments were performed in duplicates shown with S.D. error bars.
We further tested whether KIAA1530 is a TCR factor. The essential function of TCR is to repair UV lesions on the transcribed strand of the gene and thus allow rapid resumption of post-UV RNA synthesis. Therefore, in KIAA5130 knockdown cells, we measured the RNA synthesis recovery rate by measuring [3H]uridine incorporation. As shown in Fig. 3D, 24 h after UV irradiation, the transcription level recovered to about 80% of the normal level in control cells and in CSA-complemented CS3BE cells. However, this recovery was not observed in KIAA1530 knockdown cells or in CSA-deficient CS3BE cells (Fig. 3D). These data indicate that KIAA1530 is involved in TCR.
KIAA1530 Binds to CSA and XPB through Different Regions
To further investigate how KIAA1530 participates in UV-induced DNA damage response, we generated a series of N- and C-terminal truncations and internal deletion mutants of KIAA1530 (Fig. 4A) and examined their associations with CSA, XPB, or USP7 by co-immunoprecipitation experiments. As shown in Fig. 4B, deletion mutants D4 (Δ1–100 amino acids) and D5 (Δ100–200 amino acids) failed to interact with CSA, whereas the interaction of KIAA1530 with XPB was not affected by these deletions. Although deletion D7 (Δ400–500 amino acids) abolished the interaction between KIAA1530 and XPB, it did not change the binding to CSA. We also mapped the interaction domain with USP7 to the N terminus of KIAA1530 (residues 100–300). Expression of shRNA-resistant KIAA1530 (D5)-SFB or KIAA1530 (D7)-SFB mutants in KIAA1530-depleted cells did not rescue cell viability after UV irradiation (Fig. 4C), suggesting that both XPB and CSA binding are required for KIAA1530 function in DNA damage response.
FIGURE 4.

Mapping of the interactions between KIAA1530 and its associated proteins. A, schematic representation of the truncation and deletion mutants of KIAA1530 used in domain mapping. B, HEK293T cells ectopically expressing indicated the KIAA1530-SFB and CSA-HA proteins were subjected to S-beads pulldown and analyzed by Western blotting (WB) using the indicated antibodies. C, KIAA1530 stable knockdown cells were reconstituted with either full-length KIAA1530 or KIAA1530 with internal deletion mutants as indicated. Colony formation survival assay was performed as indicated. D, interaction between CSA mutant and KIAA1530-SFB in HEK293T cells. Constructs were transiently transfected into HEK293T cells, and co-precipitation was performed using S-beads and analyzed by Western blotting. E, recruitment of KIAA1530 in CS3BE (CS-A) cells complemented with wild-type CSA or CSA mutant W361C. Cells were incubated with complete medium for 1 h after 30 J/m2 UV irradiation. Cells were lysed with Nonidet P-40 buffer containing 0.3 m NaCl. The distribution of KIAA1530, CSB, and H3 were analyzed by Western blots. F, CS3BE-CSA-HA-FLAG and CS3BE-CSA(W361C)-HA-FLAG stable cells were UV-irradiated (10 J/m2) and incubated for indicated time period. Whole cell extracts were used for Western blot analysis. IP, immunoprecipitation.
CSA W361C Mutant Is Defective in KIAA1530 Binding
Because CSA is a prerequisite for KIAA1530 recruitment after UV irradiation, we tested whether CSA mutants identified in patients would affect KIAA1530 recruitment. So far, all identified CSA mutations severely disrupt protein folding, except the unique W361C point mutation, which leads to only limited structural change and causes a mild UVsS syndrome (23, 26). We therefore tested this mutant of CSA and found that its interaction with KIAA1530 was greatly reduced (Fig. 4D). We further examined the recruitment of endogenous KIAA1530 in CSA-deficient CS3BE cells stably expressing wild-type or the W361C mutant of CSA-HA-FLAG. In cells expressing wild-type CSA-HA-FLAG, we could observe UV-induced chromatin recruitment of KIAA1530 and CSB. However, this accumulation of KIAA1530 and CSB in chromatin fraction was greatly reduced in cells stably expressing CSA (W361C)-HA-FLAG following UV irradiation (Fig. 4E). Immunoblotting experiments showed that whereas KIAA1530 was relatively stable in these cell lines, CSB was destabilized upon UV irradiation in CSA-deficient or mutant cells but was stable in CS3BE cells expressing wild-type CSA (Fig. 4F). These data suggest that the CSA mutation (p.W361C) identified in a UVSS patient was defective in both KIAA1530 recruitment and CSB stabilization after UV irradiation.
Knockdown of KIAA1530 Destabilizes CSB upon UV Irradiation
To test whether the UV-dependent degradation of CSB observed in cells expressing CSA mutant (p.W361C) was due to its failure to recruit KIAA1530, we compared the UV-induced recruitment and stabilization of CSB in KIAA1530 knockdown cells with that in control cells. Although we observed a modest decrease of XPB chromatin recruitment following UV irradiation in KIAA1530 knockdown cells, the amount of CSB on UV-damaged chromatin was greatly reduced (Fig. 5A). Moreover, in whole cell lysate, we observed a rapid UV-induced turnover of CSB only in KIAA1530 knockdown cells but not in control cells (Fig. 5B), indicating that KIAA1530 acts to stabilize CSB upon UV irradiation.
FIGURE 5.
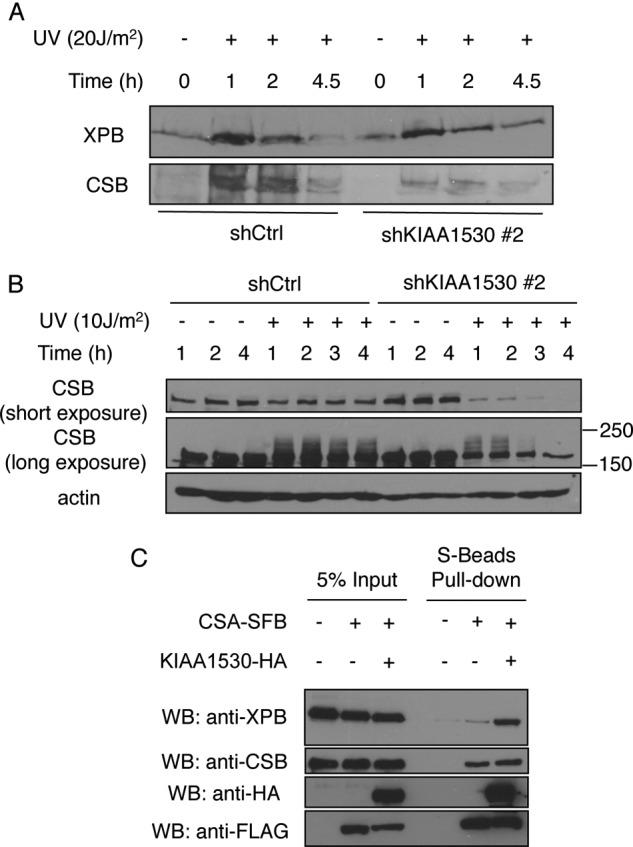
A, XPB and CSB on UV (20 J/m2)-damaged chromatin from shCtrl cells and shKIAA1530#2 cells were analyzed by Western blotting. B, Western blot (WB) analysis showing accelerated proteolysis of CSB in the absence of KIAA1530 after UV irradiation (10 J/m2). Control and KIAA1530 knockdown cells were seeded in 6-well plates and irradiated as indicated. Whole cell extracts were used for Western blot analysis. C, KIAA1530-HA enhances the interaction between CSA and XPB. Constructs encoding CSA-SFB and KIAA1530-HA were transfected into HEK293T cells as indicated. Cell extracts were incubated with S-beads for 1 h. Protein bounds to the beads were Western analyzed by blotting using indicated antibodies.
KIAA1530 Enhances the Interaction between CSA and XPB
Because KIAA1530 interacts with both CSA and TFIIH complex, we hypothesized that KIAA1530 may also have a secondary function to enhance the recruitment of the TFIIH complex following UV irradiation. As mentioned above, we observed a modest reduction of UV-induced XPB chromatin association in KIAA1530 knockdown cells (Fig. 5A). Here, we tested whether KIAA1530 could enhance the interaction between CSA and XPB. As shown in Fig. 5C, although overexpression of KIAA1530 did not affect the interaction between CSA and CSB, it greatly enhanced the binding of XPB to CSA.
DISCUSSION
Based on the literature and the findings presented in this study, we propose the following working model for KIAA1530 function in TCR. In the absence of UV damage, KIAA1530 forms a complex with CSA-DDB1 ubiquitin E3 ligase and TFIIH complex. Upon DNA damage, RNAP II is stalled by the presence of UV lesions on the transcribed stand. CSB is then recruited to damaged chromatin via its association with RNAP II. CSB plays a major role in recruiting CSA and the pre-incision complex (17). CSA then recruits KIAA1530, XAB2, HMGN1, and TFIIS (17). At damage sites, KIAA1530 stabilizes ubiquitinated CSB and thus permits successful chromatin remodeling and the recruitment of the pre-incision complex. The amino acid Trp-361 of CSA is important for its binding to KIAA1530. Besides stabilizing CSB, KIAA1530 may act together with XAB2 and help position the TFIIH complex and XPA to correctly form the pre-incision complex.
In this study, we show that CSA binds to the N-terminal region, which contains the VHS domain of KIAA1530. This interaction is important for the loading of KIAA1530 onto chromatin. A single amino acid substitution (Cys for Trp at position 361) of CSA identified in a patient with UVSS syndrome significantly inhibits its binding to KIAA1530. This mutant is unable to recruit KIAA1530 to the stalled RNAP II complex. A recent structural study predicted that mutation of this residue, which is situated at blade 7 of CSA, only mildly disrupts the integrity of the WD40 fold (23). Because no other mutations had been found in this UVSS patient (26), our findings strongly suggest that the pathological defects observed in this patient are due to its impaired ability to bind to and recruit KIAA1530 following UV irradiation.
CSA is part of the DDB1-CUL4A ubiquitin E3 ligase and is structurally similar to UV-DDB-CUL4 (20, 21, 23). Unlike UV-DDB-CUL4, whose E3 activity is stimulated by UV, the E3 ligase activity of CSA-DDB1-Cul4A is inhibited by UV irradiation through its association with COP9 signalosome at the initial steps of TCR. Binding of CSB then replaces COP9 signalosome, which leads to the activation of the E3 ligase and in turn ubiquitinates and degrades CSB at later steps of TCR (22, 23). The degradation of CSB is believed to be important for the recovery of RNA synthesis (22). However, it was not clear until recently as to how CSB is initially stabilized at UV damage sites to allow the initial steps of TCR to occur. During the preparation of this manuscript, three different groups reported that KIAA1530 regulates the stability of CSB in collaboration with its interaction partner USP7 (27–29). In agreement with these studies, we also found USP7 is one of the major KIAA1530-binding proteins (Fig. 1B). Thus, it is likely that KIAA1530 brings in USP7 and thus stabilizes CSB at damage sites.
It is known that ubiquitination of CSB is CSA-dependent (22). In CSA patient cells (CS3BE), we found the ubiquitination of CSB could be rescued by introducing even the CSA-W361C mutant, although not to the extent as those observed in wild-type CSA complemented cells. This is likely due to the rapid ubiquitination-dependent degradation of CSB in the absence of KIAA1530 and USP7. In cells reconstituted with wild-type CSA, CSA would recruit KIAA1530 and USP7 to chromatin and prevent CSB degradation. Therefore, we observed a dramatic increase of CSB modification following UV treatment.
KIAA1530 is the gene responsible for UVSS syndrome (27–29). Thus, KIAA1530 was renamed as UVSSA (encoding UV-stimulated scaffold protein A). In the report by Schwertman et al. (28), it was shown that KIAA1530-GFP formed 266-nm UV-C laser foci in both TCR-proficient and TCR-deficient CS-A and CS-B cells. In the study from Zhang et al. (27), however, KIAA1530-HA-FLAG was unable to form a complex with CSB and RNAP II in the absence of CSA protein, suggesting the loading of KIAA1530 to stalled RNAP II is CSA-dependent. Our studies agree with that of Zhang et al. (27). Moreover, we showed that the only other mutation that associates with UVSS syndrome, CSA-W361C, is defective in binding to and recruiting KIAA1530 following UV damage. Thus, our study unified our understanding of the UVSS syndrome, suggesting that this syndrome is caused by defective KIAA1530 function at damage sites.
To date, all identified UVSS syndrome patient cells are not sensitive to oxidative stress (4, 5, 26). However, in our study we found KIAA1530 knockdown cells are highly sensitive to an acute oxidative stress generated by short term treatment of H2O2 (supplemental Fig. S1). KIAA1530 knockdown cells expressing D5 or D6 mutants, which do not bind CSA or USP7, respectively, show hypersensitivity to such a stress as well (supplemental Fig. S1). This result adds another possible phenotype to UVSS syndrome cells, and if proven to be true, it would be useful for the diagnosis of patients with UVSS syndrome. Of course, it is also possible that sensitivity to oxidative stress may be regulated by other mechanisms rather than TCR.
As concluded in supplemental Fig. S2, KIAA1530 uses different domains to interact with CSA, USP7, TFIIH, and CSB, respectively. Nakazawa et al. (29) used site-directed mutagenesis to map the residues on KIAA1530 that are important for its function. They concluded that the VHS domain of KIAA1530 supports a direct contact of KIAA1530 with the TFIIH and CSB only after UV radiation. In our study, we used a different approach and found the VHS domain of KIAA1530 is required for its interaction with USP7 and CSA with or without UV damage. Moreover, we determined the TFIIH-interacting domain of KIAA1530 to a region containing residues 400–500. Taken together, we propose that CSA and USP7 bind to KIAA1530 via its VHS domain even in the absence of UV damage. Following UV irradiation, CSA recruits KIAA1530 to chromatin and thus brings ubiquitinated CSB at close proximity to USP7, because USP7 binds directly to KIAA1530. This allows USP7 to stabilize CSB at UV-damaged chromatin regions and facilitate UV damage repair. Further crystal structure studies would be extremely useful to confirm our hypothesis and extend our understanding of KIAA1530 functions at molecular details.
Searching the NCBI database, we found homologues of KIAA1530 in many higher eukaryotes, but not in yeast or Drosophila. Drosophila seems not to have functional TCR (30). The CSA ortholog Rad28 in Saccharomyces cerevisiae has been shown previously not to be required for TCR (31). Perhaps KIAA1530 evolves together with a functional CSA and represents a recent evolutionary adaptation to provide a further regulation of TCR.
Supplementary Material
Acknowledgments
We thank all the members of Chen laboratory for many discussions; J. Yuan, G. Ghosal, and W. Wang for technical assistance; L. Li, Y. Wang, and Y. Xuan for NER-deficient patient cell lines; and V. Gandhi for help in performing uridine incorporation assay.

This article contains supplemental Figs. 1 and 2.
- NER
- nucleotide excision repair
- TCR
- transcription-coupled repair
- CS
- Cockayne syndrome
- CPD
- cyclobutane pyrimidine dimer
- 6-4PP
- 6-4 pyrimidine-pyrimidone photoproduct (6-(1,2)-dihydro-2-oxo-4-pyrimidinyl-5-methyl-2,4-(1H,3H)-pyrimidinedione)
- XP
- xeroderma pigmentosum
- RNAP II
- RNA polymerase II
- TAP
- tandem affinity purification.
REFERENCES
- 1. Mitchell D. L., Nguyen T. D., Cleaver J. E. (1990) Nonrandom induction of pyrimidine(6-4) pyrimidone photoproducts in ultraviolet-irradiated human chromatin. J. Biol. Chem. 265, 5353–5356 [PubMed] [Google Scholar]
- 2. Hoeijmakers J. H. (2009) DNA damage, aging, and cancer. N. Engl. J. Med. 361, 1475–1485 [DOI] [PubMed] [Google Scholar]
- 3. Lehmann A. R. (2003) DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. Biochimie 85, 1101–1111 [DOI] [PubMed] [Google Scholar]
- 4. Itoh T., Ono T., Yamaizumi M. (1994) A new UV-sensitive syndrome not belonging to any complementation groups of xeroderma pigmentosum or Cockayne syndrome. Siblings showing biochemical characteristics of Cockayne syndrome without typical clinical manifestations. Mutat. Res. 314, 233–248 [DOI] [PubMed] [Google Scholar]
- 5. Itoh T., Fujiwara Y., Ono T., Yamaizumi M. (1995) UVs syndrome, a new general category of photosensitive disorder with defective DNA repair, is distinct from xeroderma pigmentosum variant and rodent complementation group I. Am. J. Hum. Genet. 56, 1267–1276 [PMC free article] [PubMed] [Google Scholar]
- 6. Fujiwara Y., Ichihashi M., Kano Y., Goto K., Shimizu K. (1981) A new human photosensitive subject with a defect in the recovery of DNA synthesis after ultraviolet-light irradiation. J. Invest. Dermatol. 77, 256–263 [DOI] [PubMed] [Google Scholar]
- 7. Hanawalt P. C., Spivak G. (2008) Transcription-coupled DNA repair. Two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 9, 958–970 [DOI] [PubMed] [Google Scholar]
- 8. Volker M., Moné M. J., Karmakar P., van Hoffen A., Schul W., Vermeulen W., Hoeijmakers J. H., van Driel R., van Zeeland A. A., Mullenders L. H. (2001) Sequential assembly of the nucleotide excision repair factors in vivo. Mol. Cell 8, 213–224 [DOI] [PubMed] [Google Scholar]
- 9. Sugasawa K., Okamoto T., Shimizu Y., Masutani C., Iwai S., Hanaoka F. (2001) A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev. 15, 507–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Verma R., Oania R., Fang R., Smith G. T., Deshaies R. J. (2011) Cdc48/p97 mediates UV-dependent turnover of RNA pol II. Mol. Cell 41, 82–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Donahue B. A., Yin S., Taylor J. S., Reines D., Hanawalt P. C. (1994) Transcript cleavage by RNA polymerase II arrested by a cyclobutane pyrimidine dimer in the DNA template. Proc. Natl. Acad. Sci. U.S.A. 91, 8502–8506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tornaletti S., Hanawalt P. C. (1999) Effect of DNA lesions on transcription elongation. Biochimie 81, 139–146 [DOI] [PubMed] [Google Scholar]
- 13. Lagerwerf S., Vrouwe M. G., Overmeer R. M., Fousteri M. I., Mullenders L. H. (2011) DNA damage response and transcription. DNA Repair 10, 743–750 [DOI] [PubMed] [Google Scholar]
- 14. Weidenheim K. M., Dickson D. W., Rapin I. (2009) Neuropathology of Cockayne syndrome. Evidence for impaired development, premature aging, and neurodegeneration. Mech. Ageing Dev. 130, 619–636 [DOI] [PubMed] [Google Scholar]
- 15. Citterio E., Van Den Boom V., Schnitzler G., Kanaar R., Bonte E., Kingston R. E., Hoeijmakers J. H., Vermeulen W. (2000) ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol. Cell. Biol. 20, 7643–7653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Beerens N., Hoeijmakers J. H., Kanaar R., Vermeulen W., Wyman C. (2005) The CSB protein actively wraps DNA. J. Biol. Chem. 280, 4722–4729 [DOI] [PubMed] [Google Scholar]
- 17. Fousteri M., Vermeulen W., van Zeeland A. A., Mullenders L. H. (2006) Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol. Cell 23, 471–482 [DOI] [PubMed] [Google Scholar]
- 18. Kamiuchi S., Saijo M., Citterio E., de Jager M., Hoeijmakers J. H., Tanaka K. (2002) Translocation of Cockayne syndrome group A protein to the nuclear matrix. Possible relevance to transcription-coupled DNA repair. Proc. Natl. Acad. Sci. U.S.A. 99, 201–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Koehler D. R., Hanawalt P. C. (1996) Recruitment of damaged DNA to the nuclear matrix in hamster cells following ultraviolet irradiation. Nucleic Acids Res. 24, 2877–2884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Groisman R., Polanowska J., Kuraoka I., Sawada J., Saijo M., Drapkin R., Kisselev A. F., Tanaka K., Nakatani Y. (2003) The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell 113, 357–367 [DOI] [PubMed] [Google Scholar]
- 21. Henning K. A., Li L., Iyer N., McDaniel L. D., Reagan M. S., Legerski R., Schultz R. A., Stefanini M., Lehmann A. R., Mayne L. V., Friedberg E. C. (1995) The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell 82, 555–564 [DOI] [PubMed] [Google Scholar]
- 22. Groisman R., Kuraoka I., Chevallier O., Gaye N., Magnaldo T., Tanaka K., Kisselev A. F., Harel-Bellan A., Nakatani Y. (2006) CSA-dependent degradation of CSB by the ubiquitin-proteasome pathway establishes a link between complementation factors of the Cockayne syndrome. Genes Dev. 20, 1429–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fischer E. S., Scrima A., Böhm K., Matsumoto S., Lingaraju G. M., Faty M., Yasuda T., Cavadini S., Wakasugi M., Hanaoka F., Iwai S., Gut H., Sugasawa K., Thomä N. H. (2011) The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell 147, 1024–1039 [DOI] [PubMed] [Google Scholar]
- 24. Laugel V., Dalloz C., Durand M., Sauvanaud F., Kristensen U., Vincent M. C., Pasquier L., Odent S., Cormier-Daire V., Gener B., Tobias E. S., Tolmie J. L., Martin-Coignard D., Drouin-Garraud V., Heron D., Journel H., Raffo E., Vigneron J., Lyonnet S., Murday V., Gubser-Mercati D., Funalot B., Brueton L., Sanchez Del Pozo J., Muñoz E., Gennery A. R., Salih M., Noruzinia M., Prescott K., Ramos L., Stark Z., Fieggen K., Chabrol B., Sarda P., Edery P., Bloch-Zupan A., Fawcett H., Pham D., Egly J. M., Lehmann A. R., Sarasin A., Dollfus H. (2010) Mutation update for the CSB/ERCC6 and CSA/ERCC8 genes involved in Cockayne syndrome. Hum. Mutat. 31, 113–126 [DOI] [PubMed] [Google Scholar]
- 25. Nance M. A., Berry S. A. (1992) Cockayne syndrome. Review of 140 cases. Am. J. Med. Genet. 42, 68–84 [DOI] [PubMed] [Google Scholar]
- 26. Nardo T., Oneda R., Spivak G., Vaz B., Mortier L., Thomas P., Orioli D., Laugel V., Stary A., Hanawalt P. C., Sarasin A., Stefanini M. (2009) A UV-sensitive syndrome patient with a specific CSA mutation reveals separable roles for CSA in response to UV and oxidative DNA damage. Proc. Natl. Acad. Sci. U.S.A. 106, 6209–6214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang X., Horibata K., Saijo M., Ishigami C., Ukai A., Kanno S., Tahara H., Neilan E. G., Honma M., Nohmi T., Yasui A., Tanaka K. (2012) Mutations in UVSSA cause UV-sensitive syndrome and destabilize ERCC6 in transcription-coupled DNA repair. Nat. Genet. 44, 593–597 [DOI] [PubMed] [Google Scholar]
- 28. Schwertman P., Lagarou A., Dekkers D. H., Raams A., van der Hoek A. C., Laffeber C., Hoeijmakers J. H., Demmers J. A., Fousteri M., Vermeulen W., Marteijn J. A. (2012) UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair. Nat. Genet. 44, 598–602 [DOI] [PubMed] [Google Scholar]
- 29. Nakazawa Y., Sasaki K., Mitsutake N., Matsuse M., Shimada M., Nardo T., Takahashi Y., Ohyama K., Ito K., Mishima H., Nomura M., Kinoshita A., Ono S., Takenaka K., Masuyama R., Kudo T., Slor H., Utani A., Tateishi S., Yamashita S., Stefanini M., Lehmann A. R., Yoshiura K., Ogi T. (2012) Mutations in UVSSA cause UV-sensitive syndrome and impair RNA polymerase IIo processing in transcription-coupled nucleotide-excision repair. Nat. Genet. 44, 586–592 [DOI] [PubMed] [Google Scholar]
- 30. Sekelsky J. J., Brodsky M. H., Burtis K. C. (2000) DNA repair in Drosophila. insights from the Drosophila genome sequence. J. Cell Biol. 150, F31–F36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bhatia P. K., Verhage R. A., Brouwer J., Friedberg E. C. (1996) Molecular cloning and characterization of Saccharomyces cerevisiae RAD28, the yeast homolog of the human Cockayne syndrome A (CSA) gene. J. Bacteriol. 178, 5977–5988 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.