

Background: HVR1 spans 27 residues at the N terminus of the HCV envelope glycoprotein E2 and is the most variable region within the HCV polyprotein.
Results: Three independent functional microdomains were identified in HCV HVR1.
Conclusion: Different microdomains in HVR1 cooperate to mediate HCV cell entry and immune evasion.
Significance: The data provide novel insights into understanding the mechanisms of HCV infection and immune evasion.
Keywords: Cell Surface Receptor, Hepatitis C Virus, Viral Immunology, Viral Protein, Virus Entry, Hypervariable Region 1, Immune Evasion
Abstract
High genetic heterogeneity is an important characteristic of hepatitis C virus (HCV) that contributes to its ability to establish persistent infection. The hypervariable region 1 (HVR1) that includes the first 27 amino acid residues of the E2 envelope glycoprotein is the most variable region within the HCV polyprotein. HVR1 plays a major role in both HCV cell entry and immune evasion, but the respective contribution of specific amino acid residues is still unclear. Our mutagenesis analyses of HCV pseudoparticles and cell culture-derived HCV using the H77 isolate indicate that five residues at positions 14, 15, and 25–27 mediate binding of the E2 protein to the scavenger receptor class B, type I receptor, and any residue herein is indispensable for HCV cell entry. The region spanning positions 16–24 contains the sole neutralizing epitope and is dispensable for HCV entry, but it is involved in heparan binding. More importantly, this region is necessary for the enhancement of HCV entry by high density lipoprotein and interferes with virus neutralization by E2-neutralizing antibodies. Residues at positions 1–13 are also dispensable for HCV entry, but they can affect HCV infectivity by modulating binding of the envelope protein to scavenger receptor class B, type I. Mutations occurring at this site may confer resistance to HVR1 antibodies. These findings further our understanding about the mechanisms of HCV cell entry and the significance of HVR1 variation in HCV immune evasion. They have major implications for the development of HCV entry inhibitors and prophylactic vaccines.
Introduction
Hepatitis C virus (HCV)6 is an enveloped single-stranded positive-sense RNA virus in the Flaviviridae family that infects more than 180 million individuals worldwide (1) and is a major cause of chronic liver disease, cirrhosis, and hepatocellular carcinoma (2). HCV infection is the most common indication for liver transplantation in developed countries (3). Antiviral treatment is limited by resistance and adverse effects, and a vaccine is not currently available (4). Patients with acute HCV infection are generally asymptomatic, and ∼75–85% of the HCV-infected persons progress toward chronic infection (5). HCV has evolved highly successful mechanisms for evading host immune responses, among which mutation is an important strategy that contributes to the development of chronic infection (6–8). Indeed, HCV has a high mutation rate that results from an error-prone RNA polymerase that yields genetically heterogeneous quasispecies that are characteristic of most infections (9, 10). These sequence variations are concentrated in specific regions of the genome, and the best characterized hypervariable region is the 27-amino acid residue component located at the N-terminal part of the envelope protein E2 (hypervariable region 1 (HVR1)) (11–13). HVR1 contains dominant neutralizing epitopes, and its variation may lead to virus escape from pre-existing neutralizing antibodies (14–18). Mutation of HVR1 has been proposed to be driven by immune pressure and to play an important role in the establishment of persistent infection and disease progression (19–21).
HCV cell entry involves the initial attachment of the virus onto the cell surface through interaction of the virion envelope and associated low density lipoproteins (LDL) with cell surface glycosaminoglycans (such as heparan sulfate) (22, 23) and the LDL receptor (LDLr) (24, 25). This is followed by more specific interaction with several cellular surface molecules as follows: scavenger receptor class B, type I (SR-BI) (26–28); CD81 (29); Claudin 1 (CLDN1) (30), and occludin (OCLN) (31). SR-BI, a major receptor of high density lipoprotein (HDL), is a key host factor for HCV infection that is required for an entry step (26–28). HVR1 is responsible for the interaction between the HCV envelope glycoproteins and SR-BI and plays an important role in HCV cell entry (26, 32, 33), but it is dispensable for RNA replication, virion assembly, and release (34). In line with this interpretation, removal of HVR1 markedly decreased the infectivity of HCV pseudoparticles (HCVpp) and cell culture-produced HCV (HCVcc) (32, 35), and an HCV mutant lacking HVR1 was shown to be infectious in chimpanzees, but its infectivity was significantly attenuated (36). Two recent reports have shown that HVR1 is critical for the infectivity of low density HCV particles (34, 37). Moreover, human HDL enhances HCV infection and protects HCV against neutralizing antibodies via its interplay with HVR1 and SR-BI, which further highlights the importance of HVR1 in the HCV life cycle (33, 38–40). In addition, HVR1 is involved in the binding of HCV envelope proteins to cell surface glycosaminoglycans, and it facilitates viral particle-host cell interaction (23, 24). Although HVR1 is highly variable, its importance in virus entry suggests that structural constraints might be present within this region. In this context, it has been suggested that the variability of HVR1 is not random, as its chemical and physical properties and its conformation are conserved (41, 42). Despite these observations, the relationship between the HVR1 structure and its function remains to be defined.
HCVpp bearing functional envelope proteins represents a valid model for the study of HCV cell entry and antibody-mediated neutralization (43, 44). Using this model, we investigated the functional role of amino acid stretches or single residues within the HVR1 domain of the HCV H77 isolate in cell entry and antibody-mediated neutralization, and we confirmed the findings by using the HCVcc model system. Our data show that HCV HVR1 contains three different functional microdomains that cooperate to confer HCV cell entry and immune evasion.
EXPERIMENTAL PROCEDURES
Preparation of Rabbit and Mouse Antibodies to HVR1
The DNA sequences encoding the 27 residues of HVR1 of the H77 isolate (1a genotype), aa 1–13 (N13), aa 8–20 (M13), and aa 14–27 (C14) of H77 HVR1, or the 27 residues of HVR1 of Con1 isolate (1b genotype) were fused to the 3′ terminus of the thioredoxin (TRX) gene in the vector pET32a (Novagen, Madison, WI). Protein expression and antibody preparation were as we described recently (45). Briefly, the TRX and HVR1 peptide fusion proteins were expressed using Escherichia coli BL21/DE3 under induction by isopropyl β-d-thiogalactopyranoside and purified using nickel-chelating Sepharose resin (Qiagen, Hilden, Germany). The proteins were emulsified with Freund's adjuvant (Sigma) and used to immunize New Zealand White rabbits for a total of four times over a 2-week interval. Sera were collected 1 week after the last immunization. Total IgG was purified using protein A resin (GE Healthcare).
The DNA sequence encoding H77 HVR1 was spliced to the 5′- or 3′-terminal of the HBsAg gene. The resulting fusion genes HVR1-HBsAg and HBsAg-HVR1 were inserted into the pcDNA3.1 vector (Invitrogen), respectively, and then the expression plasmids were used to immunize BALB/c mice (50 μg/mouse) by intramuscular injection for a total of three times at a 2-week interval. Sera were collected at 2 weeks after the third immunization, and their binding to H77 envelope proteins was assayed by ELISA. The procedures used in the handling and care of the animals were approved by the Animal Ethical Committee of the Second Military Medical University, Shanghai, China.
Plasmid Constructs
The plasmid phCMV-E1E2 carrying the HCV E1E2 sequence of the H77 isolate was kindly provided by Cosset and co-workers (43). This plasmid was used as a template to prepare HVR1 deletion mutants using standard fusion PCR, followed by insertion into phCMV vector. The plasmid containing full-length cDNA of the Con1 isolate was kindly provided by Rice and co-workers (46). This plasmid was used as a template to amplify the E1E2 sequence by PCR, and the E1E2 sequences with HVR1 deletion mutations using fusion PCR and the resulting fragments were inserted into the phCMV vector. The 77-Con1 chimeric E1E2 expression plasmid was constructed by replacement of the HVR1 16–24-aa encoding sequence in the context of the H77 E1E2 backbone with corresponding sequence in HVR1 of Con1 isolate using fusion PCR. Similarly, Con1-H77 plasmid was constructed by replacement of the HVR1 16–24-aa sequence in the Con1 envelope backbone for that of H77 HVR1. HJ3/QL H77/JFH1 chimeric genome was kindly provided by Lemon and co-workers (47). HVR1 deletion mutants were generated by deleting the indicated sequences in the genomic cDNA backbone using fusion PCR together with endonuclease digestion and ligation. All the envelope encoding sequences were confirmed by DNA sequencing.
Generation, Infection, and Neutralization of HCVpp
HCVpp was generated as described (45, 48). Briefly, HEK 293T cells were co-transfected with expression plasmids encoding HCV envelope glycoproteins, Gag/Pol (pLP1), Rev (pLP2) and the transfer vector, pLenti6 (Invitrogen) containing the green fluorescent protein (GFP) gene. Cell culture supernatants containing pseudoparticles were harvested at 48 h after transfection and filtered through 0.45-μm membranes. To confirm incorporation of HCV envelope glycoproteins into pseudotyped particles, pseudoparticles in cell culture supernatants were pelleted by centrifugation through a 20% sucrose cushion and examined for the E1, E2, and HIV Gag proteins by Western blot assay as described previously (42). Briefly, proteins separated by SDS-PAGE were electrotransferred onto Hybond-ECL nitrocellulose membranes (Amersham Biosciences) and probed with the appropriate antibodies (E1 mAb A4 clone, goat anti-E2 pAb and HIV Gag mAb). HCV E1 mAb A4 was described previously (42). Goat anti-E2 pAb was purchased from Biodesign International (Memphis, TN). Anti-HIV Gag mAb was obtained from Wantai Biological Pharmacy (Beijing, China). The immunoreactive bands were visualized using horseradish peroxidase (HRP)-conjugated anti-goat or mouse IgG (Sigma) followed by enhanced chemiluminescence (Pierce).
Target Huh7.5 cells (kindly provided by C. M. Rice) were seeded into 96-well plates at a density of 8 × 103 cells/well and incubated overnight at 37 °C. HCVpp normalized to equal quantities of HCV E2 and HIV Gag proteins were added to each well, incubated for 5 h, and removed. The cells were further incubated in regular medium at 37 °C for 72 h. HCVpp entry into Huh7.5 cells was analyzed by flow cytometry (43). In indicated cases, the medium containing HCVpp was added to Huh7.5 cell cultures together with HDL (Calbiochem) at a concentration of 6 μg/ml, and the supernatants were replaced with regular medium 5 h later as described previously (33, 38, 39). For neutralization assay, HCVpp supernatants were preincubated with total IgG purified from rabbit immune sera or anti-HCV E2 mAb H48 (49) or IgG purified from sera of HCV-infected individuals by fractionation on protein A/G columns (30 min at 37 °C) prior to infection of Huh7.5 cells. Pseudoparticles generated with VSV-G glycoprotein (VSV-GP) (Invitrogen) were used as control.
E2 Binding to Human SR-BI and CD81
Lentivirus vectors for human SR-BI or CD81 and mouse immune sera against human SR-BI were prepared as described (45). CHO cells were infected with the SR-BI or CD-81 vectors using the empty lentivirus vector as control, and SR-BI and CD81 expression was confirmed by FACS using mouse immune sera against SR-BI and anti-CD81 mAb clone JS81 (BD Biosciences), respectively. HEK 293T cells were transfected with prototype E1E2 or a panel of mutant E1E2 expression plasmids or the empty vector, and 72 h later the cells were detached from the dishes in phosphate-buffered saline (PBS)/EDTA, centrifuged, resuspended in PBS supplemented with a proteinase inhibitor mixture (Roche Applied Science), and lysed by ultrasonication. The cell lysates were centrifuged, and the supernatants were concentrated 5-fold using a Centricon ultrafiltration tube (Millipore, Bedford, MA). E2 protein in crude cell extract was normalized using Galanthus nivalis agglutinin (GNA) capture ELISA with conformation-dependent anti-E2 mAb H53 or H48 as described (50, 51).
The binding of normalized envelope protein to cell surface-associated SR-BI or CD81 was determined using a FACS-based assay as described previously (45, 52). Briefly, 4 × 105 CHO cells expressing human SR-BI or CD81 or transfected with the empty vector were incubated with crude cell extract that contained equivalent amounts of E2 protein for 1 h at room temperature (RT), washed twice, and incubated with anti-E2 mAb H53 (1 h at RT). After labeling with Alexa Fluor 488-conjugated goat anti-mouse IgG (Invitrogen), the mean fluorescence intensity (MFI) was quantified by a flow cytometry (Cell Lab QuantaTM SC Instrument, Beckman Coulter, Fullerton, CA). Background MFI was defined as E2 binding to CHO cells infected with empty vector and subtracted from the MFI values for the mutant and prototype E2 binding to SR-BI or CD81-expressing cells. The binding activity of each mutant envelope protein was expressed as the percentages of subtracted MFI relative to the MFI of H77 prototype envelope protein. The extract from 293T cells given the empty vector (containing equivalent amounts of total cellular protein) was used as control to probe the potential binding of 293T cell-derived proteins with SR-BI or CD81.
Envelope Protein Binding to HVR1 Antibodies
The binding of envelope proteins to HVR1 antibodies was assayed by GNA capture ELISA as described previously (50, 51). In brief, ELISA plates were coated with GNA (Sigma) and incubated overnight at 4 °C. The plates were washed with PBS containing 0.05% Tween 20 (PBST), and nonspecific binding sites were saturated with blocking buffer (3% BSA and 0.05% Tween 20 in PBS). The lysates of HEK 293T cells transfected with vectors expressing the various mutant HCV envelope proteins or empty vector were added to the plates and incubated at RT for 2 h. After extensive washing, rabbit anti-HVR1 IgG (20–500 μg/ml) and sera collected from mice immunized with HBsAg and HVR1 fusion gene expression plasmids or anti-E2 mAb H53 (1:1000) were added at RT for 40 min. The plates were washed and incubated (40 min at RT) with horseradish peroxidase-conjugated goat anti-rabbit or goat anti-mouse IgG (diluted 1:2,000). Then the plates were washed, and color was developed using 3,3′,5,5′-tetramethylbenzidine as the substrate. The absorbance at 450 nm wavelength was measured.
Envelope Protein-Heparan Interaction
The binding of HCV envelope protein to heparan and inhibition of envelope-heparan binding by HVR1 antibodies was assayed as described previously (22, 23). Briefly, 96-well ELISA plates were coated with sulfated heparan (Merck) in PBS (1 μg/well) and incubated overnight at 4 °C. They were blocked with blocking buffer (3% BSA and 0.05% Tween 20 in PBS) (1 h at RT). Cell extracts containing equal quantities of HCV envelope protein were preincubated (1 h at RT) with different concentrations of IgG fractionated from sera collected from rabbits immunized with TRX-HVR1 fusion protein. Subsequently, envelope protein-antibody complexes were added to the wells precoated with heparan and incubated for 1 h at 37 °C. After washing, heparan-bound envelope protein was detected by the addition of anti-E2 mAb H53 and horseradish peroxidase-conjugated goat anti-mouse IgG. Protein-bound antibodies were detected by colorimetry as described above.
Production, Infection, and Neutralization of HCVcc
The generation of H77/JFH1 chimeric HCVcc was as described previously (35, 53, 54). Briefly, the plasmids harboring chimeric genomic cDNA were linearized and used as transcription templates with an in vitro MEGAscript kit (Promega, Madison, WI). CD81-deficient Huh7 cells were kindly provided by Dr. J. Zhong (Institut Pasteur of Shanghai, Chinese Academy of Sciences, Shanghai, China) (55). The in vitro-transcribed RNA was delivered to Huh7.5 cells or CD81-deficient Huh7 cells by electroporation. HCV core protein in transfected Huh7.5 or Huh7 cells and the respective culture supernatants were determined using a commercial ELISA kit according to the manufacturer's instructions (Kangyun, Yueyang, China). Culture supernatants from Huh7.5 cells were collected on day 5 after transfection and were used to infect Huh7.5 cells in the absence or presence of HDL (6 μg/ml). After 2 days, HCVcc-infected cells were examined for NS5A expression by immunofluorescent staining with mAb 9E10 (53). Stained foci were counted in quadruplicate wells, and virus titers are expressed as foci forming units (FFU)/ml. HCVcc neutralization was assayed as described previously (56, 57). Briefly, normalized HCVcc were preincubated (37 °C for 1 h) with the total IgG fraction from sera collected from rabbits immunized with TRX-HVR1 fusion protein or E2 mAb H48 at the indicated concentrations and added to Huh7.5 cells in 96-well plates. After 2 days, the cells were immunostained for NS5A, and the results were expressed as percent neutralization calculated as the mean percentages of infectious titer inhibition relative to incubation with medium devoid of antibody.
Statistical Analysis
Data are expressed as means ± S.D. Statistical significance is analyzed by the Student's t test. p values below 5% are considered significant.
RESULTS
HVR1 Residues at Positions 14, 15, and 25–27 Are Required for H77 HCVpp Cell Entry (Five Key Residues)
A panel of E1E2 expression plasmids containing deletion mutations within the HVR1 of HCV H77 isolate were constructed and used to prepare HCV pseudoparticles by co-transfection of 293T cells with lentivirus capsid and transfer plasmids. The amino acid sequence of HVR1 is shown in Fig. 1A. GNA capture ELISA of the expression products from 293T cells transfected with the prototype or mutant envelope plasmids using anti-E2 polyclonal antibodies and conformation-dependent E2 mAb H53 and H48 indicated that none of the single residue deletions affected E2 protein expression or conformation (data not shown). Western blotting of purified pseudoparticles in the cell supernatants showed that none of the deletions affected the HCV envelope proteins incorporated into pseudoparticles (Fig. 1B). Examination of the infectivity of the corresponding pseudoparticles in Huh7.5 cells indicated that HCVpp infectivity was decreased to less than 5% of the unmodified pseudoparticles by deletion of the entire HVR1 (ΔHVR1) and deletion of a single residue at positions 14, 15, 25, 26, or 27 reduced the infectivity by more than 85%. By contrast, deletion of any single residue within position 1–13 or 16–24 only moderately affected the pseudoparticle infectivity (Fig. 1C).
FIGURE 1.

Residues at positions 14, 15, and 25–27 in HVR1 are indispensable for HCVpp cell entry. A, amino acid sequence of the H77 isolate HVR1 used for functional microdomain identification. B, HEK 293T cells were transfected with lentivirus vectors containing the GFP reporter gene, HIV capsid (Gag/Pol) plasmid, Rev plasmid, and HCV envelope protein (H77 isolate) expression plasmids harboring single residue deletion in HVR1. Co-transfection of 293T cells devoid of HCV envelope protein (−env) plasmid was performed as a negative control. Forty eight hours later, pseudoparticles in the culture media were pelleted through 20% sucrose cushions and analyzed by Western blotting. HCV envelope proteins and HIV Gag protein were probed with goat anti-E2 pAb, anti-E1 mAb (A4), and anti-HIV Gag (anti-Gag) mAb, respectively. Results are shown for one representative experiment out of three independent assays. C, plasmids encoding different deletion mutants of HVR1 in the context of a full-length E1E2 glycoprotein were used to generate HCVpp. Co-transfection of 293T cells devoid of the HCV envelope protein (−env) plasmid was performed as a negative control. Pseudoparticles normalized to equal quantities of HCV E2 and HIV Gag proteins were used to infect Huh7.5 target cells, and HCVpp cell entry was determined 3 days later by quantification of GFP-positive cells by flow cytometry. The infectivity of H77 prototype pseudoparticles was ∼6 × 104 FFU/ml, the infectivity of pseudoparticles with mutation in HVR1 is expressed as percentage of the infectivity of H77 prototype. Data are the means of four independent experiments ± S.D.
Regions Spanning HVR1 Amino Acids 1–13 and 16–24 Are Not Necessary for HCVpp Infection, but the Latter Stretch Is Necessary for Infection Enhancement by HDL
Having seen that deletion of any residue in aa stretches 1–13 and 16–24 did not attenuate HCVpp infectivity, we wanted to better understand the relevance of these regions with respect to HCVpp infectivity. Deletion of either one of the two stretches did not affect the binding of conformation-dependent mAb H53 and H48 clones to E2 protein (data not shown) nor pseudoparticle production (Fig. 2B). When infection was done in the absence of human HDL, deletion of the residues across aa 1–13 (Δ1–13) reduced HCVpp infectivity by about 20%. Interestingly, deletion of nine residues across aa 16–24 (Δ16–24) did not reduce HCVpp infectivity, but instead it increased the infectivity by up to 40% (Fig. 2A). Pseudoparticles bearing HCV envelope proteins with deletion of both aa 1–13 and 16–24 in HVR1 (five key, containing only the five residues at position 14, 15, and 25–27) retained the infectivity by about 34% (Fig. 2A). Mutation of alanine at position 14 to glycine and alanine scanning mutagenesis of residues 15 and 25–27 reduced pseudoparticle infectivity by less than 30%, and the combined mutations of both aa 14 and 15 (A14G/G15A) or aa 25–27 (C3A, all of the three residues were replaced with alanine) decreased the infectivity by about 60 and 75%, respectively, indicating that residues at these positions cooperate to mediate HCV cell entry, and they are not specifically selected during viral evolution (Fig. 2C).
FIGURE 2.

Regions spanning aa 1–13 and 16–24 are dispensable for HCVpp cell entry. A, 293T cells were co-transfected with lentivirus transfer plasmid, HIV capsid plasmid, Rev plasmid, and plasmids encoding H77 envelope glycoproteins with deletion in HVR1. Pseudoparticles normalized to equal quantities of HCV E2 and HIV Gag proteins were used to infect Huh7.5 target cells, and infectivity was determined in the presence or absence of human HDL at a concentration of 6 μg/ml. The results are expressed as percentage of infectivity of wild-type H77. 5 key refers to envelope mutants preserving only aa 14, 15. and 25–27 in HVR1. Results are the means ± S.D. of four independent experiments. *, p < 0.001; **, p < 0.01 compared with H77 prototype pseudoparticles in the absence of HDL. B, pseudoparticles in culture media of 293T cells were pelleted through 20% sucrose cushions, and HCV envelope proteins and HIV capsid protein in pseudoparticle preparations were assayed by Western blotting. C, envelope expression plasmids containing various mutations were used to generate HCVpp, and the infectivity of the respective pseudoparticles was measured as described above. Co-transfection of 293T cells devoid of HCV envelope proteins (−env) plasmid was performed as a negative control. C3A represents a construct in which all three residues located at positions 25–27 were replaced with alanine. Data are means ± S.D. of three independent experiments. D, CHO cells were infected with lentivirus containing human SR-BI cDNA or CD81 cDNA or empty lentivirus, respectively, and the expression of human SR-BI and CD81 was determined by flow cytometry with mAb to CD81 or mouse polyclonal antibodies to SR-BI. CHO cells infected with empty lentivirus were used as a negative control. E, crude extracts of 293T cells transfected with HCV envelope protein expression plasmid were normalized by GNA capture ELISA, and the extracts containing equivalent amounts of prototype or mutant E2 protein were used to measure cell surface-associated SR-BI binding using a FACS-based assay. The extract of mock plasmid-transfected 293T cells that contained equivalent amounts of cellular protein as the E2 protein containing extract was used as a negative control (−env). The binding activity is expressed as the percentage of MFI relative to that of H77 prototype envelope proteins. Results are the means ± S.D. of three independent experiments. *, p < 0.05 compared with H77 prototype envelope proteins. F, binding of mutant E2 proteins to cell surface-associated CD81 was measured using a FACS-based assay. Results are the means ± S.D. of three independent experiments. *, p < 0.05 compared with H77 prototype envelope proteins.
HVR1 acts as an essential viral component that modulates HDL-mediated enhancement of HCV infection (38–40). To identify the potential regions involved in this function, we examined the effect of HDL on the infectivity of these pseudoparticles. As shown in Fig. 2A, the enhancement of HCVpp infectivity by HDL is dependent on the presence of the 16–24-aa stretch.
Previous studies have indicated that HVR1 plays an important role in modulating E2 binding to SR-BI and CD81 (26, 58). To investigate the role of the identified amino acid stretches in the interaction with SR-BI and CD81, we assessed the binding of the mutant envelope proteins to CHO cells that express human SR-BI or CD81 (Fig. 2D). The infectivity of the pseudoparticles positively correlated with the SR-BI binding activity of the corresponding envelope proteins (Fig. 2E). A single deletion within aa 14, 15, and 25–27 reduced SR-BI binding activity by more than 85%, whereas a single point substitution mutation within these five residues or deletion of aa 1–13 reduced SR-BI binding activity by less than 25% (Fig. 2E). Furthermore, deletion of aa 16–24 increased binding of the envelope protein to SR-BI (Fig. 2E). Among the four stretch deletion mutants (Δ1–13, Δ16–24, five key, and ΔHVR1), only deletion of the entire HVR1 showed enhanced CD81 binding activity compared with the prototype protein (Fig. 2F).
HVR1 Residues 16–24 Contain a Neutralization Epitope
To identify neutralization epitopes in HVR1, rabbits were immunized with fusion proteins containing TRX and H77 HVR1, aa 1–13 (N13), aa 8–20 (M13), or aa 14–27 (C14) (Fig. 3A), and IgG fractions from the resulting sera were characterized for their ability to recognize epitopes in HVR1. As shown in Fig. 3B, serum IgG purified from a rabbit immunized with the TRX-HVR1 fusion protein (RH104) bound to the Δ1–13 protein as well as to the unmodified envelope protein. However, it did not bind to Δ16–24 (Fig. 3B). Similarly, IgG from rabbits immunized with TRX-C14 (RC201 and RC202) recognized the Δ1–13 and prototype proteins but not the Δ16–24 protein. IgG from rabbits immunized with TRX-N13 fusion protein (RN203 and RN204) or with the TRX-M13 protein (RM205) was unable to recognize the prototype or any one of the deletion mutant envelope proteins. IgG from rabbit RH104 and RC202 did not bind the Δ25 protein but bound the Δ15 protein, whereas IgG from rabbit RC201 was unable to bind Δ15, but it could bind to Δ25 (Fig. 3B). The IgG preparations (20–500 μg/ml) had similar binding profiles. The data suggest that aa 16–24 contain an antigenic epitope, and epitope recognition at flanking residues may differ among different immunized animals. The neutralizing activity of these antibodies for HCVpp was determined. As shown in Fig. 3C, both anti-HVR1 (RH104) and anti-C14 antibodies (RC201) efficiently neutralized the prototype and Δ1–13 pseudoparticles. However, these antibodies did not affect the infectivity of Δ16–24 or five key pseudoparticles. As expected, IgG from TRX-N13-immunized rabbit (RN203) did not neutralize any of the indicated pseudoparticles.
FIGURE 3.

Region comprising aa 16–24 contains a neutralization epitope. A, amino acid sequence of H77 HVR1 and the peptide expressed as a thioredoxin-peptide fusion protein that was used for rabbit immunization. B, HCV envelope proteins in lysates of 293T cells transfected with a panel of plasmids were coated onto microplates using GNA, and then the binding of the envelope proteins to IgG purified from six rabbits immunized with TRX-HVR1 peptide fusion proteins was measured by ELISA. The anti-HVR1 peptide IgG preparations were used at a concentration of 100 μg/ml. Conformation-dependent anti-E2 monoclonal antibody H53 (1:1,000 dilution) was used to assay E2 protein in cell lysate. The lysate of empty plasmid-transfected 293T cells was used as a negative control (Mock). C, similar inputs of various viral particles normalized for HCV E2 and HIV Gag proteins were preincubated with IgG (100 μg/ml) fractionated from sera collected from immunized or unimmunized rabbits at RT for 30 min before infection of Huh7.5 target cells. The infectivity of HCVpp was determined after 3 days as described above. VSV-GP was used as control. Results are the means ± S.D. of four independent experiments. D, total of 12 mice were immunized with HBsAg and H77 HVR1 fusion gene expression plasmids. Six mice (NS1–NS6) received HVR1-HBsAg expression plasmid and other six mice (SC1–SC6) received HBsAg-HVR1 expression plasmid. The mouse sera were collected at 2 weeks after the third immunization and were used as anti-HVR1 antibodies in ELISA. The data are the values for mouse sera at a dilution of 1:500. The mAb H53 diluted at 1:1,000 was used as control to assay envelope protein captured onto microplates. Mock means the lysate of 293T cells transfected with the empty vector.
Similar results were obtained with immune sera collected from mice that received HBsAg and H77 HVR1 fusion gene vaccination (Fig. 3D). All 12 serum samples recognized parental E1E2 and Δ1–13 proteins, but none could recognize Δ16–24 proteins. The residue at position 15 was involved in an antigenic epitope in four mice (NS2, NS6, SC3, and SC4) and that at position 25 was involved in antigenic epitope in two mice (NS5 and SC6). Both aa 15 and 25 were involved in the antigenic epitope in mouse NS3.
Stretch Comprising HVR1 Amino Acids 16–24 Inhibits Antibody Neutralization in the Presence of HDL
To investigate whether aa 16–24 in HVR1 contains a neutralization epitope in other HCV isolates, plasmids that express chimeric H77-Con1 and Con1-H77 envelope proteins were constructed. In these constructs, the 16–24-aa encoding sequence of HVR1 in H77 was swapped with the corresponding sequence of Con1 isolate (1b subtype) and vice versa (Fig. 4A). The amino acid sequences of parental and chimeric HVR1 are shown in Fig. 4B. The chimeric protein expressed in 293T cells could bind to SR-BI and CD81 at a similar efficiency as that shown by prototype protein (data not shown). Rabbit anti-H77 HVR1 IgG could bind to the H77 prototype and Con1-H77 chimeric proteins, whereas rabbit anti-Con1 HVR1 IgG could bind to Con1 prototype and H77-Con1 chimeric proteins (Fig. 4C). The H77-Con1 and Con1-H77 chimeric pseudoparticles demonstrated about 78 and 83% of the infectivity that was observed for the respective parental pseudoparticles in media devoid of HDL (Fig. 4D). Deletion of aa 16–24 in Con1 HVR1 did not affect pseudoparticle infectivity, but it ablated the ability of HDL to stimulate HCVpp infection, and the 16–24-aa stretch in the H77 or Con1 HVR1 isolates restored the pseudoparticle response to HDL when transferred into heterogeneous envelope proteins (Fig. 4D). In neutralization assays with HVR1 antibodies, the infectivity of H77-Con1 chimeric virus was not inhibited by antibodies to H77 HVR1, whereas it was markedly reduced by antibodies to Con1 HVR1 (Fig. 4E). Antibodies to Con1 HVR1 efficiently neutralized both autologous Con1 and chimeric H77-Con1 pseudoparticles, but it did not inhibit the infectivity of autologous pseudoparticles with 16–24-aa deletion (Fig. 4E).
FIGURE 4.

Stretch across HVR1 aa 16–24 protects HCVpp against neutralization in the presence of HDL. A, two chimeric HCV envelope protein expression plasmids were constructed. In H77-Con1 plasmid, HVR1 16–24-aa stretch in H77 envelope backbone was swapped for the corresponding sequence in the Con1 isolate (1b subtype). In Con1-H77 plasmid, HVR1 16–24-aa stretch in the Con1 envelope backbone was swapped for that of H77 HVR1. B, amino acid sequences of parental and chimeric HVR1. C, HCV envelope proteins in lysates of 293T cells transfected with H77, Con1, or chimeric plasmids were coated onto microplates using GNA, and then the binding of the envelope proteins to IgG fractionated from sera of rabbits immunized with TRX-H77 HVR1 or TRX-Con1 HVR1 (100 μg/ml) was analyzed by ELISA. The lysate of mock-transfected 293T cells was used as a control. Results are the means of three independent experiments. D, pseudoparticles were prepared as described. After filtration, equal volumes of culture supernatants were used to infect Huh7.5 cells in the absence or presence of HDL (6 μg/ml), and infectivity was determined 3 days later. Results are the means ± S.D. of three independent experiments. *, p < 0.001. E, pseudoparticles bearing prototype and chimeric envelope proteins were normalized to equal infectivity levels and used to assess the neutralizing activity of rabbit antibodies to H77 HVR1 or Con1 HVR1. VSV-GP was used as a control. Results are the means ± S.D. of four independent experiments with antibodies at a concentration of 100 μg/ml. F, pseudoparticles normalized to equal infectivity were incubated (30 min at RT) with E2 mAb H48 at the indicated concentrations in the absence or presence of 6 μg/ml HDL and then added to Huh7.5 cells. Infectivity was determined 3 days later. Data are the means ± S.D. of three independent experiments. *, p < 0.001. G, IgG preparations (A–C) were fractionated from sera of three confirmed HCV-infected individuals (positive for serum HCV RNA and anti-HCV antibodies) using protein A/G column. H77 and Con1 pseudoparticles (prototype and aa 16–24 deleted) normalized for equal infectivity were incubated with IgG preparations at a concentration of 100 μg/ml in presence of 6 μg/ml HDL for 30 min at RT and then added to Huh7.5 cells. Infectivity was determined after 3 days. Data are the means ± S.D. of three independent experiments. *, p < 0.001. H, pseudoparticles bearing prototype or various envelope proteins deletion mutants were normalized to equal infectivity and then used to assess the neutralization activity of E2 mAb H48 at a concentration of 0.5 μg/ml in presence of 6 μg/ml HDL as described above. Data are the means ± S.D. of three independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001 compared with H77 prototype pseudoparticles.
H77 E2-specific mAb H48 is a conformation-dependent neutralizing antibody that disturbs interaction between HCV envelope protein and the HCV secondary receptor CD81 (49). This antibody efficiently reduced both H77 and H77-Con1 pseudoparticle infectivity, but the neutralizing activity was notably inhibited when HDL was present (Fig. 4F). For pseudoparticles with a 16–24-aa deletion, the neutralizing activity of mAb H48 was not affected by HDL (Fig. 4F). Moreover, in the presence of HDL, neutralization of H77 and Con1 pseudoparticles with deletion of aa 16–24 by IgG preparations from three individuals chronically infected with HCV (1b genotype) was much more robust than that seen for the prototype pseudoparticles (Fig. 4G). To identify key residues in aa 16–24 that confer HCVpp resistance to E2 antibodies, HCVpp harboring single residue deletions in aa 16–24 were used in neutralization assay. In the presence of HDL, removal of any residue significantly increased pseudoparticle neutralization by mAb H48, and pseudoparticles with deletion of the entire 16–24-aa neutralization epitope showed maximum sensitivity to this antibody (Fig. 4H).
Region across Amino Acids 16–24 Plays an Important Role in Envelope-Heparan Binding
The glycosaminoglycan heparan sulfate acts as a primary docking site for HCV, and HVR1 is one of the important viral determinants for HCV-heparan interaction (23, 59). To identify potential peptide fragments involved in heparan binding, the binding of envelope protein to heparan and the inhibition of envelope-heparan binding by HVR1 antibodies were assayed by ELISA. Equal quantities of envelope proteins from 293T cell extracts were subjected to heparan binding analysis (Fig. 5A). Deletion of the entire HVR1 or of residues at the HVR1 C terminus markedly decreased envelope-heparan binding; however, deletion of aa 1–13 did not affect envelope-heparan binding (Fig. 5, B and C). Preincubation of unmodified E1E2 and Δ1–13 proteins with HVR1 antibody (Fig. 5B) or C14 antibody (Fig. 5C) resulted in a concentration-dependent inhibition of envelope-heparan binding. In addition, neither IgG from rabbits immunized with TRX-N13 nor that from rabbits immunized with TRX-M13 fusion protein could inhibit HCV envelope-heparan binding (data not shown). These data suggest that the 16–24-aa region plays an important role in mediating the interaction of HCV envelope protein to heparan.
FIGURE 5.
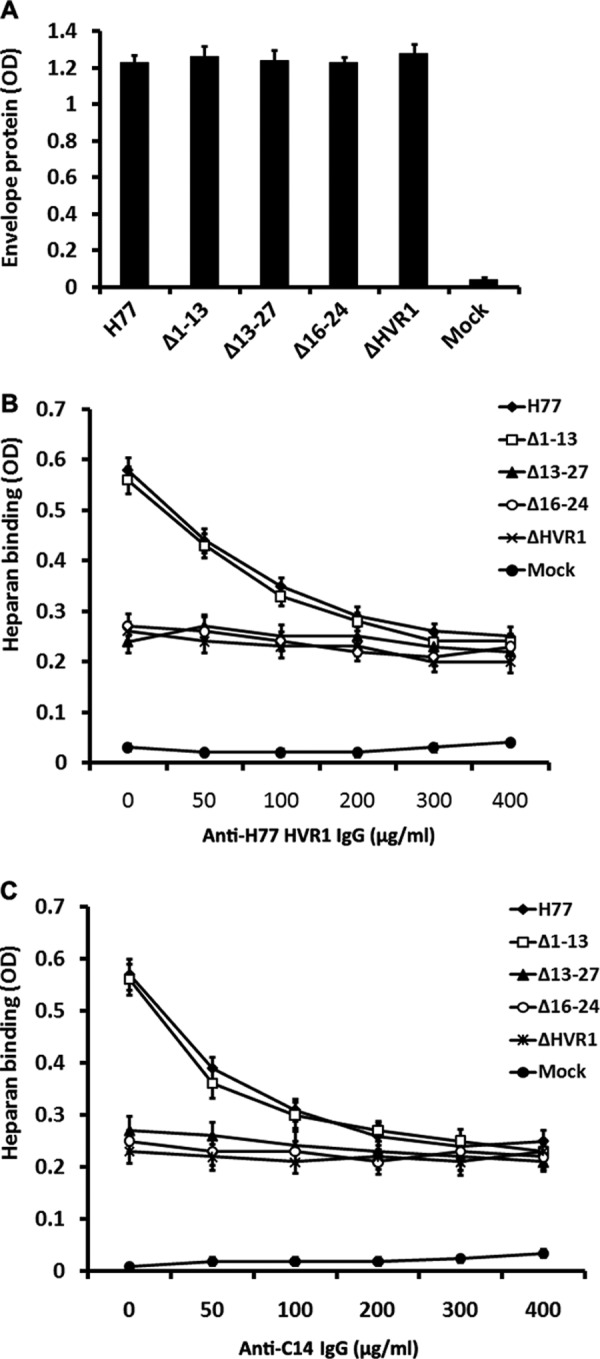
Stretch across HVR1 aa 16–24 plays an important role in the binding of HCV envelope proteins to heparan. HCV envelope proteins in 293T cells extracts were normalized by GNA capture ELISA, and equal quantities were preincubated with IgG from sera of rabbits immunized with TRX-HVR1 fusion protein for 1 h at RT. Subsequently, envelope protein/antibody mixtures were added to ELISA plates coated with heparan. Heparan-bound E2 was detected using mAb H53. Data are shown as mean OD ± S.D. obtained from a representative experiment performed in triplicate. The extract from mock plasmid-transfected 293T cells that contained equivalent amounts of total cellular protein as the E2 protein containing extracts was used as a negative control (Mock). A, assay of HCV envelope protein in the extracts of plasmid transfected 293T cells by GNA capture ELISA. B, inhibition of envelope-heparan binding by IgG from TRX-HVR1 immunized rabbit RH104. C, inhibition of envelope-heparan binding by IgG from TRX-C14 immunized rabbit RC202.
Combined Mutation in HVR1 N Terminus Enhances HCVpp Infectivity and Reduces the Neutralizing Activity of HVR1 Antibodies
Having seen that the region across residues 1–13 is not necessary for HCVpp infectivity, experiments were performed to investigate the potential significance of mutation in this region. The envelope sequence of the H77 isolate used here corresponds to that deposited in GenBankTM under accession number AF011752. Another envelope sequence with accession number AF011753 was obtained from the same patient at the same time. The clones differ in two residues. In AF011752, a serine and a histidine are located at positions 8 and 11 in HVR1, whereas the residues at corresponding positions in AF011753 are, respectively, asparagine and arginine (Fig. 6A). To determine whether two point mutations are functionally significant, Ser-8 and His-11 in the original H77 sequence were replaced by an Asn and an Arg (S8N and H11R), and the infectivity of the corresponding pseudoparticles was determined. As shown in Fig. 6B, the double mutant showed a 1.9-fold increase in HCVpp infectivity, whereas both single residue mutations did not affect pseudoparticles infectivity. Examination of the binding potential of the mutant envelope proteins to SR-BI and CD81 indicated that the double mutant but not the single mutants had a much higher binding efficiency to SR-BI than that of prototype E2 protein (Fig. 6C). However, the CD81 binding profiles of the three mutants were similar to that of prototype E2 protein (Fig. 6D).
FIGURE 6.

Residues within HVR1 N terminus modulate HCVpp infectivity. A, amino acid sequences of H77 HVR1 and its mutants. B, plasmids encoding H77/S8N+H11R, H77/S8N, and H77/H11R cDNAs were used to generate HCVpp that were used in infectivity assays. Results are expressed as the mean percentage of infectivity relative to that of the H77 prototype ± S.D. of five independent experiments. *, p < 0.001; **, p > 0.05 relative to H77 prototype pseudoparticles. C, binding of E2 protein containing S8N and/or H11R mutation to cell surface-associated SR-BI was measured using a FACS-based assay. Results are means ± S.D. of three independent experiments. *, p < 0.001; **, p > 0.05 compared with H77 prototype envelope proteins. D, binding of E2 protein containing S8N and/or H11R mutation with cell surface-associated CD81 was measured using a FACS-based assay. Results are means ± S.D. of three independent experiments. *, p > 0.05 relative to H77 prototype envelope proteins. E, pseudoparticles bearing the H77 prototype or the H77/S8N + H11R mutation were normalized to equal quantities of particles (by assaying HCV envelope proteins and HIV Gag protein incorporated into pseudoparticles) (left panel) or to equal infectivity (right panel) and then treated with H77 HVR1 antibodies at indicated concentrations. The infectivity was determined 3 days after infection. VSV-GP was used as a control. Data are the means ± S.D. of quadruple measurements and are from one representative out of four independent experiments.
Pseudoparticles normalized with equivalent particles or infectivity were used to analyze the effect of the double mutations on HVR1 antibody neutralization. When anti-HVR1 was preincubated with equal amounts of pseudoparticles (by assaying HCV envelope and HIV Gag proteins incorporated into pseudoparticles), the neutralization of the pseudoparticles containing double mutations was less efficient than that of the unmodified pseudoparticles (Fig. 6E, left panel). When pseudoparticles with equal infectivity (by assaying pseudoparticles infectivity using Huh7.5 cells as target cells) were used, HVR1 antibodies showed similar neutralizing activities to both pseudoparticles (Fig. 6E, right panel).
HCVcc Model Confirms the Functional Impact of the Identified HVR1 Regions on HCV Infection
To examine the relevance of these HVR1 regions in authentic HCV particles, deletions were introduced into H77/JFH1 chimeric cDNA plasmid, and the transcribed RNAs were used to transfect Huh7.5 cells or CD81-deficient Huh7 cells by electroporation. Intracellular and extracellular core protein levels were determined by ELISA. At 24 h post-transfection, the amount of accumulated core in Huh7.5 cells or the culture medium was similar among the different transfectants (Fig. 7A, left panel). Removal of aa 13–27 or the entire HVR1 led to a marked decrease in the levels of both intracellular and extracellular core protein when assayed at 48 h post-transfection, but core protein levels were not affected by the removal of aa 1–13 or 16–24 (Fig. 7A, right panel). To exclude the effect caused by infection of progeny virus, the core protein assays were done with CD81-deficient Huh7 cells, which cannot not be infected by free HCVcc virions but support HCV RNA replication (55). When assayed at 24 or 48 h post-transfection, intracellular and extracellular core protein levels for all the CD81-deficient Huh7 transfectants were comparable (Fig. 7B).
FIGURE 7.

Effect of HVR1 deletions on infectivity of HCVcc and neutralization by HVR1 and E2 antibodies. A, Huh7.5 cells were transfected with transcribed HJ3/QL chimeric genomic RNA or each of the indicated deletion mutants. HCV core protein in transfected Huh7.5 cells and culture supernatant was determined at 24 h (left panel) and 48 h (right panel) post-transfection using a commercial ELISA kit. The cell lysate or culture medium of sham-transfected Huh7.5 cells devoid of transcribed RNA were used as negative control (Mock). Results are the means ± S.D. of three independent transfection and ELISA experiments. *, p > 0.05; **, p < 0.001 relative to intracellular or extracellular core protein of Huh7.5 cells transfected with H77-JFH1 prototype genomic RNA. B, CD81-deficient Huh7 cells were transfected with transcribed HJ3/QL chimeric genomic RNA, and intracellular and extracellular HCV core protein was determined at 24 h (left panel) and 48 h (right panel) as described. *, p > 0.05 relative to intracellular or extracellular core protein of cells transfected with H77-JFH1 prototype genomic RNA. C, Huh7.5 cells were transfected with transcribed HJ3/QL chimeric genomic RNA or each of the indicated deletion mutants. The culture supernatants collected at day 5 post-transfection were incubated with Huh7.5 cells in the presence or absence of 6 μg/ml HDL, and HCVcc infectivity was determined by FFU assays 2 days later. Control represents the infectivity in supernatants of sham-transfected Huh7.5 cells. The cutoff value of the assay was 20 FFU/ml. Results are the means ± S.D. of three independent transfection and infection experiments. *, p < 0.001. D, HCVcc supernatants were concentrated 5-fold by ultrafiltration and normalized to an equal infectivity of 2,000 FFU/ml. Virus stock (100 μl) was preincubated with total IgG fractionated from sera collected from rabbits immunized with TRX fused to H77 or Con1 HVR1 peptide (100 μg/ml) and added to Huh7.5 cells in 96-well plates. After 2 days, the cells were immunostained for NS5A, and virus neutralization was expressed as percent inhibition of infectious titers relative to antibody-free medium. Results are expressed as means ± S.D. of quadruplicate measurements and are representative of three independent experiments. E, virus stock with a normalized infectivity of 2000 FFU/ml (100 μl) was incubated (30 min at RT) with E2 mAb H48 at indicated concentrations in the absence or presence of 6 μg/ml HDL and added to Huh7.5 cells in 96-well plates. After 2 days, the cells were immunostained for NS5A, and virus neutralization was calculated. Results are the means ± S.D. of quadruplicate measurements and are representative of three independent experiments. *, p < 0.01; **, p < 0.001. F, normalized HCVcc harboring deletion mutations in the HVR1 neutralization epitope were used to assess the neutralization activity of E2 mAb H48 at a concentration of 5 μg/ml in the presence of 6 μg/ml HDL as described above. Data are the means ± S.D. of three independent experiments. *, p < 0.01; **, p < 0.001 relative to H77-JFH1 HCVcc.
Finally, we assayed the infectivity of Huh7.5 culture supernatants at day 5 post-transfection. As shown in Fig. 7C, when infection was done in the absence of human HDL, deletion of the entire HVR1 or the C-terminal 15-aa residues (Δ13–27) decreased the infectivity below the assay detection limit of 20 FFU/ml, although deletion of aa 1–13 (Δ1–13) decreased the infectious titers to about 76% of the parental virus titers. Deletion of aa 16–24 (Δ16–24) did not affect infectivity with detected virus titers similar to those of the parental virus (Fig. 7C). Furthermore, just as in the case of the pseudoparticle model, both the HDL-mediated infectivity enhancement and the HVR1 antibody neutralizing potentials were dependent on the 16–24-aa region (Fig. 7, C and D). The HCVcc neutralizing potential of the mAb H48 was inhibited by HDL, and it is clear that aa 16–24 conferred virus resistance to this antibody (Fig. 7E). HCVcc with deletions of single residues within aa 16–24 showed comparable infectivity to that seen for the prototype virus (data not shown). As seen for HCVpp, deletion of any residue within aa 16–24 up-regulated HCVcc sensitivity to mAb H48, and HCVcc with deletion of aa 16–24 were most easily neutralized by mAb H48 (Fig. 7F).
DISCUSSION
In this study, we identified three independent functional regions in the HVR1 component of the H77 isolate. The data herein show the relevance of these microdomains for mediating virus cell entry, antibody-mediated neutralization, and immune evasion.
Using deletion mutagenesis and function analyses, we found that five aa residues (Ala-14, Gly-15, Lys-25, Gln-26, and Asn-27) within the HVR1 of the H77 isolate play a key role in HCVpp entry. Deletion of any one of these five residues caused a significant loss of pseudoparticle infectivity, whereas deletion of any residue in other regions had only a slight effect on their infectivity. Despite the requirement of these five residues for HCVpp infectivity, a single residue substitution at each one of these five positions only moderately decreased the pseudoparticle infectivity. However, the combined mutation of Ala-14 and Gly-15 (A14G/G15A) or Lys-25, Gln-26, and Asn-27 (C3A) decreased HCVpp infectivity to about 40 and 25%, respectively. The importance of these five residues is further supported by the findings that deletion of aa 1–13 (Δ1–13) attenuated HCVpp infectivity by only about 20%, whereas deletion of aa 16–24 (Δ16–24) did not decrease but instead increased the infectivity of the resulting HCVpp by about 40%. These findings extend a previous report that the basic residue at position 25 in H77 HVR1 is important for the infectivity of HCVpp (42), but they differ from that report in that they suggest that the basic residues at positions 3 and 11 are dispensable for HCV cell entry.
HCV envelope proteins are heavily glycosylated, and some of the glycans have been shown to play an essential role in protein folding and HCV entry (42, 60). Bankwitz et al. (34) recently reported that deletion of HVR1 does not influence the glycosylation of the HCV envelope. Because HVR1 does not contain glycosylation sites, variable infectivity of HCVpp carrying HVR1 deletions cannot be due to the defective glycosylation of envelope proteins. It has been suggested that there is a complex interplay between HVR1 and the SR-BI and CD81 receptors (34, 58). We found that binding of envelope mutants to SR-BI directly correlates with the cell entry efficiency of the corresponding pseudoparticles, suggesting that the five identified key residues mediate entry predominantly through their ability to regulate interaction between envelope proteins and SR-BI. Because of the poor conservation of these five residues in different HCV isolates, and the limited effect that their individual mutation has on HCVpp infectivity, we cannot exclude the possibility that their role in interaction with SR-BI is indirect. It is possible that deletion of residues within HVR1 could create local conformational changes or affect the interaction between HVR1 and other domains outside of HVR1. In this case, these residues may cooperate to regulate the E2-SR-BI interaction through modulating the triaxial conformation of the SR-BI binding domain in the E2 protein. Deletion of aa 16–24 enhanced the E2-SR-BI interaction and pseudoparticle infectivity, indicating that this region may increase the steric hindrance around key residues of the SR-BI binding domain and in turn can interfere with the accessibility of these residues to SR-BI.
To identify antigenic epitopes within HVR1, HVR1-specific antibodies were raised by immunizing rabbits with thioredoxin-HVR1 peptide fusion proteins. At least in our experimental conditions, the N-terminal 13 amino acids and middle 13 amino acids do not contain a neutralization epitope. It is worth noting that non-neutralizing monoclonal antibodies recognizing the N terminus of HVR1 have been described (61), suggesting that the region across the N-terminal 13 residues may comprise an epitope that interacts with non-neutralizing antibodies. The failure to generate antibodies against this region in our immunization experiments suggests that this epitope may need to be recognized within the context of E2 to induce an appropriate immune response.
Mapping of the epitopes that are recognized by antibodies elicited during a natural infection to HVR1 requires sera from HCV-infected patients. However, our data demonstrate that a neutralizing epitope in H77 HVR1 is located in a region across aa 16–24. The E2 protein deleted in HVR1 aa 16–24 was unable to bind antibodies from any immunized rabbit, and the presence of aa 16–24 was required for HCVpp neutralization by HVR1 antibodies. The data indicate that the region spanning aa 16–24 includes a neutralization epitope, a conclusion that is further supported by the results of the antibody binding assay using sera from mice vaccinated with HBsAg and HVR1 fusion gene DNA vaccines. To our knowledge, there are no current reports about the definite mapping of neutralizing epitopes in HVR1, although some reports suggested that the residues at the C terminus are important for inducing immune responses (62–64). Vieyres et al. (65) reported that the neutralizing antibodies induced by immunization with H77c and Gla HVR1 peptides reacted with the C-terminal portion of HVR1, but the exact epitopes were not mapped. The infectivity and neutralization assays using H77-Con1 chimeric pseudoparticles and Con1 pseudoparticles with deletions in the HVR1 aa 16–24 suggest that aa 16–24 also constitute a sole neutralization epitope in Con1 HVR1. Considering the important role played by this region in eliciting a humoral response, it may be reasonable to suggest that the residues in this region may have a higher variation frequency than those in other regions of HVR1. Supporting this interpretation, the mutations in HVR1 mainly occurred in this region, when comparing isolate H77 that represents HCV in the acute-phase plasma and H90, which was isolated from a chronic-phase plasma obtained from the same patient 13 years after the acute infection (66).
HDL enhances HCV infectivity and inhibits the neutralizing activity of antibodies against the E2 protein by involving the lipid-transfer function of SR-BI, which is the receptor for both HDL and HCV (33, 38, 39). It was demonstrated that apoC-I, an exchangeable apolipoprotein that predominantly resides in HDL, specifically enhances HCV infectivity and increases the rates of fusion between viral and target membranes via a direct interaction with HVR1 (39). Our data indicate that different amino acid stretches in HVR1 are responsible for E2-SR-BI interaction and infection enhancement by HDL. In the H77 and Con1 HVR1, the 16–24-aa stretch acts as the exact functional region for HDL interplay, with the nine residues in this region cooperating to reach maximal activity. Indeed, the 16–24-aa stretch robustly decreased the neutralizing activity of the HCV E2 mAb H48 and IgG from HCV-infected individuals in the presence of HDL. This observation may account for the significance of the neutralizing epitope for HCV survival in vivo, although it is dispensable for HCV entry. Together with a recent report that HCVcc that lack HVR1 have fewer particles with low density and are more prone to neutralization by E2-specific monoclonal antibodies and patient sera (34), our data suggest that aa 16–24 in HVR1 may be involved in the association of lipoprotein with HCV particles in vivo and in turn may modulate HCV infectivity and immune evasion.
Interaction with glycosaminoglycan heparan sulfate is considered to be the first event of HCV cell attachment that represents an important step for the initiation of HCV infection. Some studies have identified HVR1 as a key component of HCV interaction with heparan (23, 59). In this study, we provide evidence that the neutralization epitope at aa 16–24 plays an important role in HCV envelope-heparan binding. Removal of this region led to markedly decreased heparan binding by envelope protein, but it did not impede HCVpp infectivity, which may be due to increased SR-BI binding of envelope protein. Our data also substantiate that HVR1 antibodies neutralize HCV infection partly by disturbing the envelope-heparan interaction (23), because the envelope binding to heparan was inhibited by antibodies to the 16–24-aa region.
There are two residue differences (at HVR1 positions 8 and 11) between the sequence used for functional mapping (GenBankTM accession number AF011752) and another sequence obtained from the same patient at the same time (GenBankTM accession number AF011753). Replacement of both residues in the former HVR1 with those in the latter HVR1 enhanced HCVpp infectivity by 1.9-fold. The neutralizing efficiency of anti-HVR1 antibody to an equivalent amount of pseudoparticles harboring dual mutations in HVR1 was markedly decreased. This may be related to enhanced SR-BI binding of the E2 protein carrying the double mutation. Collectively, the data suggest that the N-terminal region of HVR1 regulates the infectivity of the virus and in turn protects it from neutralizing antibodies directed to the epitope within the HVR1 C terminus. Unlike classical immune evasion through mutation in a neutralization epitope, mutations in the HVR1 N terminus may represent an alternative strategy that protects HCV from HVR1 neutralizing antibodies.
Cell culture- and serum-derived HCV is associated with lipoproteins, which contributes to the heterogeneous density of HCV particles (67). The interaction with lipoproteins is important for HCV infectivity (68). HCVpp was created in 293T cells, a human embryonic kidney cell line that is unlikely to produce lipoprotein, suggesting that this model may not fully recapitulate the infectivity of authentic HCV particles. However, we confirmed the findings in the HCVcc system, the state-of-the-art model for HCV infection. Intracellular and extracellular core protein levels could be used as an indicator of RNA replication and virus production (34, 35, 53). The core assay of Huh7.5 cells transfected with HCV genomic RNA at an early stage post-transfection (24 h) indicated that removal of the entire HVR1 or amino acid stretches in the HVR1 N terminus does not affect HCV RNA replication and virus release. The decreased intracellular and extracellular core protein levels at 48 h post-transfection caused by deletion of the entire HVR1 or aa 13–27 are likely due to impaired virus infectivity. Indeed, CD81-deficient Huh7 cells transfected with RNA deleted at different HVR1 sites produced similar amounts of core protein at 24 or 48 h post-transfection.
Kawaguchi et al. (69) have recently reported that a quasispecies could be reproduced in vitro from plasma collected in 1977 from patient H (the same patient investigated herein), but serial passage of the quasispecies resulted in replacement of the initially dominant species with a new HVR1 species. Together with our data, this may be due to the evolution of HVR1 functional microdomains that increase virus cell entry in culture.
Consistent with our findings for the H77 and Con1 isolates, the residues at the C terminus in the HVR1 of the J6 isolate (2a genotype) are important for HCVpp infectivity, whereas those located at the N terminus are nonessential (data not shown). Furthermore, residues across position 16–24 were dispensable for HCVpp infectivity and served as a neutralization epitope in the HVR1 of the J6 isolates, and this peptide is indispensable for enhancement of HCV infectivity by HDL. These data suggest that our observation is also relevant for other HCV isolates and genotypes, and it is possible that HVR1 evolves in a similar manner inside a given genotype to optimize both cell entry and immune evasion.
In conclusion, our data indicate that the HCV HVR1 contains independent functional microdomains. For the H77 HVR1, the first microdomain includes five residues at positions 14, 15, and 25–27 and plays a key role in binding of the HCV envelope protein to the SR-BI receptor. Any residue in this microdomain is indispensable for HCV cell entry. The nine residues across positions 16–24 constitute the second microdomain, which contains the neutralization epitope and appears to be dispensable for cell entry. This microdomain is necessary for the enhancement of HCV infectivity by HDL and confers HCV resistance to neutralizing antibodies targeted to an epitope outside of HVR1. It also plays an important role in HCV envelope-heparan binding. The third HVR1 microdomain across aa 1–13 is dispensable for HCV cell entry, but it can affect HCV infectivity by modulating the binding of the envelope protein to SR-BI. Mutations in this microdomain may confer HCV resistance to HVR1 antibodies. These findings provide novel insights into our understanding of the role played by HVR1 in mediating HCV cell entry, immune evasion, and antibody-mediated neutralization and will thus contribute to the development of entry inhibitors or a protective vaccine that targets the interaction between HVR1 and cell entry factors.
Acknowledgments
We are grateful to F. L. Cosset, C. M. Rice, S. M. Lemon, and J. Zhong for providing us with research materials. We thank Dr. Qiang Deng for review of the manuscript.
This work was supported by 973 Programme of China Research Grants 2009CB522501 and 2009CB522503, Important National Science and Technology Special Projects for Prevention and Treatment of Major Infectious Diseases Grant 2012ZX10002003-004-010, Natural Science Foundation of China Grants 81071364 and 31170150, Science Fund for Creative Research Groups, NSFC Grant 30921006, and Shanghai Leading Academic Discipline Project B901.
- HCV
- hepatitis C virus
- HVR1
- hypervariable region 1
- HCVpp
- HCV pseudoparticles
- HCVcc
- cell culture produced HCV
- SR-BI
- scavenger receptor class B, type I
- aa
- amino acid
- GNA
- G. nivalis agglutinin
- TRX
- thioredoxin
- FFU
- foci forming unit
- MFI
- mean fluorescence intensity
- GP
- glycoprotein.
REFERENCES
- 1. Alter M. J. (2007) Epidemiology of hepatitis C virus infection. World J. Gastroenterol. 13, 2436–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Seeff L. B. (2002) Natural history of chronic hepatitis C. Hepatology 36, S35–S46 [DOI] [PubMed] [Google Scholar]
- 3. Brown R. S. (2005) Hepatitis C and liver transplantation. Nature 436, 973–978 [DOI] [PubMed] [Google Scholar]
- 4. Pawlotsky J. M. (2011) Treatment failure and resistance with direct-acting antiviral drugs against hepatitis C virus. Hepatology 53, 1742–1751 [DOI] [PubMed] [Google Scholar]
- 5. Hoofnagle J. H. (2002) Course and outcome of hepatitis C. Hepatology 36, S21–S29 [DOI] [PubMed] [Google Scholar]
- 6. Dustin L. B., Rice C. M. (2007) Flying under the radar. The immunobiology of hepatitis C. Annu. Rev. Immunol. 25, 71–99 [DOI] [PubMed] [Google Scholar]
- 7. Rehermann B. (2009) Hepatitis C virus versus innate and adaptive immune responses. A tale of co-evolution and co-existence. J. Clin. Invest. 119, 1745–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Uebelhoer L., Han J. H., Callendret B., Mateu G., Shoukry N. H., Hanson H. L., Rice C. M., Walker C. M., Grakoui A. (2008) Stable cytotoxic T cell escape mutation in hepatitis C virus is linked to maintenance of viral fitness. PLoS Pathog. 4, e1000143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gómez J., Martell M., Quer J., Cabot B., Esteban J. I. (1999) Hepatitis C viral quasispecies. J. Viral. Hepat. 6, 3–16 [DOI] [PubMed] [Google Scholar]
- 10. Simmonds P., Bukh J., Combet C., Deléage G., Enomoto N., Feinstone S., Halfon P., Inchauspé G., Kuiken C., Maertens G., Mizokami M., Murphy D. G., Okamoto H., Pawlotsky J. M., Penin F., Sablon E., Shin-I T., Stuyver L. J., Thiel H. J., Viazov S., Weiner A. J., Widell A. (2005) Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology 42, 962–973 [DOI] [PubMed] [Google Scholar]
- 11. Hijikata M., Kato N., Ootsuyama Y., Nakagawa M., Ohkoshi S., Shimotohno K. (1991) Hypervariable regions in the putative glycoprotein of hepatitis C virus. Biochem. Biophys. Res. Commun. 175, 220–228 [DOI] [PubMed] [Google Scholar]
- 12. Weiner A. J., Brauer M. J., Rosenblatt J., Richman K. H., Tung J., Crawford K., Bonino F., Saracco G., Choo Q. L., Houghton M., et al. (1991) Variable and hypervariable domains are found in the regions of HCV corresponding to the flavivirus envelope and NS1 proteins and the pestivirus envelope glycoproteins. Virology 180, 842–848 [DOI] [PubMed] [Google Scholar]
- 13. Wang G. P., Sherrill-Mix S. A., Chang K. M., Quince C., Bushman F. D. (2010) Hepatitis C virus transmission bottlenecks analyzed by deep sequencing. J. Virol. 84, 6218–6228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kato N., Sekiya H., Ootsuyama Y., Nakazawa T., Hijikata M., Ohkoshi S., Shimotohno K. (1993) Humoral immune response to hypervariable region 1 of the putative envelope glycoprotein (GP70) of hepatitis C virus. J. Virol. 67, 3923–3930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Farci P., Shimoda A., Coiana A., Diaz G., Peddis G., Melpolder J. C., Strazzera A., Chien D. Y., Munoz S. J., Balestrieri A., Purcell R. H., Alter H. J. (2000) The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies. Science 288, 339–344 [DOI] [PubMed] [Google Scholar]
- 16. van Doorn L. J., Capriles I., Maertens G., DeLeys R., Murray K., Kos T., Schellekens H., Quint W. (1995) Sequence evolution of the hypervariable region in the putative envelope region E2/NS1 of hepatitis C virus is correlated with specific humoral immune responses. J. Virol. 69, 773–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Farci P., Shimoda A., Wong D., Cabezon T., De Gioannis D., Strazzera A., Shimizu Y., Shapiro M., Alter H. J., Purcell R. H. (1996) Prevention of hepatitis C virus infection in chimpanzees by hyperimmune serum against the hypervariable region 1 of the envelope 2 protein. Proc. Natl. Acad. Sci. U.S.A. 93, 15394–15399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dowd K. A., Netski D. M., Wang X. H., Cox A. L., Ray S. C. (2009) Selection pressure from neutralizing antibodies drives sequence evolution during acute infection with hepatitis C virus. Gastroenterology 136, 2377–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Weiner A. J., Geysen H. M., Christopherson C., Hall J. E., Mason T. J., Saracco G., Bonino F., Crawford K., Marion C. D., Crawford K. A., et al. (1992) Evidence for immune selection of hepatitis C virus (HCV) putative envelope glycoprotein variants. Potential role in chronic HCV infections. Proc. Natl. Acad. Sci. U.S.A. 89, 3468–3472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Laskus T., Wilkinson J., Gallegos-Orozco J. F., Radkowski M., Adair D. M., Nowicki M., Operskalski E., Buskell Z., Seeff L. B., Vargas H., Rakela J. (2004) Analysis of hepatitis C virus quasispecies transmission and evolution in patients infected through blood transfusion. Gastroenterology 127, 764–776 [DOI] [PubMed] [Google Scholar]
- 21. Liu L., Fisher B. E., Dowd K. A., Astemborski J., Cox A. L., Ray S. C. (2010) Acceleration of hepatitis C virus envelope evolution in humans is consistent with progressive humoral immune selection during the transition from acute to chronic infection. J. Virol. 84, 5067–5077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barth H., Schnober E. K., Zhang F., Linhardt R. J., Depla E., Boson B., Cosset F. L., Patel A. H., Blum H. E., Baumert T. F. (2006) Viral and cellular determinants of the hepatitis C virus envelope-heparan sulfate interaction. J. Virol. 80, 10579–10590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barth H., Schafer C., Adah M. I., Zhang F., Linhardt R. J., Toyoda H., Kinoshita-Toyoda A., Toida T., Van Kuppevelt T. H., Depla E., Von Weizsacker F., Blum H. E., Baumert T. F. (2003) Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J. Biol. Chem. 278, 41003–41012 [DOI] [PubMed] [Google Scholar]
- 24. Molina S., Castet V., Fournier-Wirth C., Pichard-Garcia L., Avner R., Harats D., Roitelman J., Barbaras R., Graber P., Ghersa P., Smolarsky M., Funaro A., Malavasi F., Larrey D., Coste J., Fabre J. M., Sa-Cunha A., Maurel P. (2007) The low density lipoprotein receptor plays a role in the infection of primary human hepatocytes by hepatitis C virus. J. Hepatol. 46, 411–419 [DOI] [PubMed] [Google Scholar]
- 25. Owen D. M., Huang H., Ye J., Gale M., Jr. (2009) Apolipoprotein E on hepatitis C virion facilitates infection through interaction with low density lipoprotein receptor. Virology 394, 99–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Scarselli E., Ansuini H., Cerino R., Roccasecca R. M., Acali S., Filocamo G., Traboni C., Nicosia A., Cortese R., Vitelli A. (2002) The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 21, 5017–5025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zeisel M. B., Koutsoudakis G., Schnober E. K., Haberstroh A., Blum H. E., Cosset F. L., Wakita T., Jaeck D., Doffoel M., Royer C., Soulier E., Schvoerer E., Schuster C., Stoll-Keller F., Bartenschlager R., Pietschmann T., Barth H., Baumert T. F. (2007) Scavenger receptor class B type I is a key host factor for hepatitis C virus infection required for an entry step closely linked to CD81. Hepatology 46, 1722–1731 [DOI] [PubMed] [Google Scholar]
- 28. Dreux M., Dao Thi V. L., Fresquet J., Guérin M., Julia Z., Verney G., Durantel D., Zoulim F., Lavillette D., Cosset F. L., Bartosch B. (2009) Receptor complementation and mutagenesis reveal SR-BI as an essential HCV entry factor and functionally imply its intra- and extracellular domains. PLoS Pathog. 5, e1000310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pileri P., Uematsu Y., Campagnoli S., Galli G., Falugi F., Petracca R., Weiner A. J., Houghton M., Rosa D., Grandi G., Abrignani S. (1998) Binding of hepatitis C virus to CD81. Science 282, 938–941 [DOI] [PubMed] [Google Scholar]
- 30. Evans M. J., von Hahn T., Tscherne D. M., Syder A. J., Panis M., Wölk B., Hatziioannou T., McKeating J. A., Bieniasz P. D., Rice C. M. (2007) Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446, 801–805 [DOI] [PubMed] [Google Scholar]
- 31. Ploss A., Evans M. J., Gaysinskaya V. A., Panis M., You H., de Jong Y. P., Rice C. M. (2009) Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457, 882–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bartosch B., Vitelli A., Granier C., Goujon C., Dubuisson J., Pascale S., Scarselli E., Cortese R., Nicosia A., Cosset F. L. (2003) Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-BI scavenger receptor. J. Biol. Chem. 278, 41624–41630 [DOI] [PubMed] [Google Scholar]
- 33. Bartosch B., Verney G., Dreux M., Donot P., Morice Y., Penin F., Pawlotsky J. M., Lavillette D., Cosset F. L. (2005) An interplay between hypervariable region 1 of the hepatitis C virus E2 glycoprotein, the scavenger receptor BI, and high density lipoprotein promotes both enhancement of infection and protection against neutralizing antibodies. J. Virol. 79, 8217–8229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bankwitz D., Steinmann E., Bitzegeio J., Ciesek S., Friesland M., Herrmann E., Zeisel M. B., Baumert T. F., Keck Z. Y., Foung S. K., Pécheur E. I., Pietschmann T. (2010) Hepatitis C virus hypervariable region 1 modulates receptor interactions, conceals the CD81-binding site, and protects conserved neutralizing epitopes. J. Virol. 84, 5751–5763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wakita T., Pietschmann T., Kato T., Date T., Miyamoto M., Zhao Z., Murthy K., Habermann A., Kräusslich H. G., Mizokami M., Bartenschlager R., Liang T. J. (2005) Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11, 791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Forns X., Thimme R., Govindarajan S., Emerson S. U., Purcell R. H., Chisari F. V., Bukh J. (2000) Hepatitis C virus lacking the hypervariable region 1 of the second envelope protein is infectious and causes acute resolving or persistent infection in chimpanzees. Proc. Natl. Acad. Sci. U.S.A. 97, 13318–13323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Prentoe J., Jensen T. B., Meuleman P., Serre S. B., Scheel T. K., Leroux-Roels G., Gottwein J. M., Bukh J. (2011) Hypervariable region 1 differentially impacts viability of hepatitis C virus strains of genotypes 1–6 and impairs virus neutralization. J. Virol. 85, 2224–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dreux M., Pietschmann T., Granier C., Voisset C., Ricard-Blum S., Mangeot P. E., Keck Z., Foung S., Vu-Dac N., Dubuisson J., Bartenschlager R., Lavillette D., Cosset F. L. (2006) High density lipoprotein inhibits hepatitis C virus-neutralizing antibodies by stimulating cell entry via activation of the scavenger receptor BI. J. Biol. Chem. 281, 18285–18295 [DOI] [PubMed] [Google Scholar]
- 39. Dreux M., Boson B., Ricard-Blum S., Molle J., Lavillette D., Bartosch B., Pécheur E. I., Cosset F. L. (2007) The exchangeable apolipoprotein apoC-I promotes membrane fusion of hepatitis C virus. J. Biol. Chem. 282, 32357–32369 [DOI] [PubMed] [Google Scholar]
- 40. Meunier J. C., Engle R. E., Faulk K., Zhao M., Bartosch B., Alter H., Emerson S. U., Cosset F. L., Purcell R. H., Bukh J. (2005) Evidence for cross-genotype neutralization of hepatitis C virus pseudo-particles and enhancement of infectivity by apolipoprotein C1. Proc. Natl. Acad. Sci. U.S.A. 102, 4560–4565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Penin F., Combet C., Germanidis G., Frainais P. O., Deléage G., Pawlotsky J. M. (2001) Conservation of the conformation and positive charges of hepatitis C virus E2 envelope glycoprotein hypervariable region 1 points to a role in cell attachment. J. Virol. 75, 5703–5710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Callens N., Ciczora Y., Bartosch B., Vu-Dac N., Cosset F. L., Pawlotsky J. M., Penin F., Dubuisson J. (2005) Basic residues in hypervariable region 1 of hepatitis C virus envelope glycoprotein E2 contribute to virus entry. J. Virol. 79, 15331–15341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bartosch B., Dubuisson J., Cosset F. L. (2003) Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 197, 633–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fafi-Kremer S., Fofana I., Soulier E., Carolla P., Meuleman P., Leroux-Roels G., Patel A. H., Cosset F. L., Pessaux P., Doffoël M., Wolf P., Stoll-Keller F., Baumert T. F. (2010) Viral entry and escape from antibody-mediated neutralization influence hepatitis C virus reinfection in liver transplantation. J. Exp. Med. 207, 2019–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tong Y., Zhu Y., Xia X., Liu Y., Feng Y., Hua X., Chen Z., Ding H., Gao L., Wang Y., Feitelson M. A., Zhao P., Qi Z. T. (2011) Tupaia CD81, SR-BI, claudin-1, and occludin support hepatitis C virus infection. J. Virol. 85, 2793–2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Blight K. J., McKeating J. A., Rice C. M. (2002) Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76, 13001–13014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yi M., Ma Y., Yates J., Lemon S. M. (2007) Compensatory mutations in E1, p7, NS2, and NS3 enhance yields of cell culture-infectious intergenotypic chimeric hepatitis C virus. J. Virol. 81, 629–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pestka J. M., Zeisel M. B., Bläser E., Schürmann P., Bartosch B., Cosset F. L., Patel A. H., Meisel H., Baumert J., Viazov S., Rispeter K., Blum H. E., Roggendorf M., Baumert T. F. (2007) Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc. Natl. Acad. Sci. U.S.A. 104, 6025–6030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Op De Beeck A., Voisset C., Bartosch B., Ciczora Y., Cocquerel L., Keck Z., Foung S., Cosset F. L., Dubuisson J. (2004) Characterization of functional hepatitis C virus envelope glycoproteins. J. Virol. 78, 2994–3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lucas M., Tsitoura E., Montoya M., Laliotou B., Aslanoglou E., Kouvatsis V., Entwisle C., Miller J., Klenerman P., Hadziyannis A., Hadziyannis S., Borrow P., Mavromara P. (2003) Characterization of secreted and intracellular forms of a truncated hepatitis C virus E2 protein expressed by a recombinant herpes simplex virus. J. Gen. Virol. 84, 545–554 [DOI] [PubMed] [Google Scholar]
- 51. Flint M., Dubuisson J., Maidens C., Harrop R., Guile G. R., Borrow P., McKeating J. A. (2000) Functional characterization of intracellular and secreted forms of a truncated hepatitis C virus E2 glycoprotein. J. Virol. 74, 702–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Flint M., Maidens C., Loomis-Price L. D., Shotton C., Dubuisson J., Monk P., Higginbottom A., Levy S., McKeating J. A. (1999) Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. J. Virol. 73, 6235–6244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lindenbach B. D., Evans M. J., Syder A. J., Wölk B., Tellinghuisen T. L., Liu C. C., Maruyama T., Hynes R. O., Burton D. R., McKeating J. A., Rice C. M. (2005) Complete replication of hepatitis C virus in cell culture. Science 309, 623–626 [DOI] [PubMed] [Google Scholar]
- 54. Zhong J., Gastaminza P., Cheng G., Kapadia S., Kato T., Burton D. R., Wieland S. F., Uprichard S. L., Wakita T., Chisari F. V. (2005) Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U.S.A. 102, 9294–9299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhong J., Gastaminza P., Chung J., Stamataki Z., Isogawa M., Cheng G., McKeating J. A., Chisari F. V. (2006) Persistent hepatitis C virus infection in vitro. Co-evolution of virus and host. J. Virol. 80, 11082–11093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhang P., Wu C. G., Mihalik K., Virata-Theimer M. L., Yu M. Y., Alter H. J., Feinstone S. M. (2007) Hepatitis C virus epitope-specific neutralizing antibodies in IGs prepared from human plasma. Proc. Natl. Acad. Sci. U.S.A. 104, 8449–8454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cao J., Chen Z., Ren Y., Luo Y., Cao M., Lu W., Zhao P., Qi Z. (2011) Oral immunization with attenuated Salmonella carrying a co-expression plasmid encoding the core and E2 proteins of hepatitis C virus capable of inducing cellular immune responses and neutralizing antibodies in mice. Vaccine 29, 3714–3723 [DOI] [PubMed] [Google Scholar]
- 58. Roccasecca R., Ansuini H., Vitelli A., Meola A., Scarselli E., Acali S., Pezzanera M., Ercole B. B., McKeating J., Yagnik A., Lahm A., Tramontano A., Cortese R., Nicosia A. (2003) Binding of the hepatitis C virus E2 glycoprotein to CD81 is strain-specific and is modulated by a complex interplay between hypervariable regions 1 and 2. J. Virol. 77, 1856–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Basu A., Kanda T., Beyene A., Saito K., Meyer K., Ray R. (2007) Sulfated homologues of heparin inhibit hepatitis C virus entry into mammalian cells. J. Virol. 81, 3933–3941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Goffard A., Callens N., Bartosch B., Wychowski C., Cosset F. L., Montpellier C., Dubuisson J. (2005) Role of N-linked glycans in the functions of hepatitis C virus envelope glycoproteins. J. Virol. 79, 8400–8409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hsu M., Zhang J., Flint M., Logvinoff C., Cheng-Mayer C., Rice C. M., McKeating J. A. (2003) Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. U.S.A. 100, 7271–7276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Scarselli E., Cerino A., Esposito G., Silini E., Mondelli M. U., Traboni C. (1995) Occurrence of antibodies reactive with more than one variant of the putative envelope glycoprotein (GP70) hypervariable region 1 in viremic hepatitis C virus-infected patients. J. Virol. 69, 4407–4412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zibert A., Kraas W., Meisel H., Jung G., Roggendorf M. (1997) Epitope mapping of antibodies directed against hypervariable region 1 in acute self-limiting and chronic infections due to hepatitis C virus. J. Virol. 71, 4123–4127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mondelli M. U., Cerino A., Lisa A., Brambilla S., Segagni L., Cividini A., Bissolati M., Missale G., Bellati G., Meola A., Bruniercole B., Nicosia A., Galfrè G., Silini E. (1999) Antibody responses to hepatitis C virus hypervariable region 1. Evidence for cross-reactivity and immune-mediated sequence variation. Hepatology 30, 537–545 [DOI] [PubMed] [Google Scholar]
- 65. Vieyres G., Dubuisson J., Patel A. H. (2011) Characterization of antibody-mediated neutralization directed against the hypervariable region 1 of hepatitis C virus E2 glycoprotein. J. Gen. Virol. 92, 494–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ogata N., Alter H. J., Miller R. H., Purcell R. H. (1991) Nucleotide sequence and mutation rate of the H strain of hepatitis C virus. Proc. Natl. Acad. Sci. U.S.A. 88, 3392–3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. André P., Komurian-Pradel F., Deforges S., Perret M., Berland J. L., Sodoyer M., Pol S., Bréchot C., Paranhos-Baccalà G., Lotteau V. (2002) Characterization of low and very low density hepatitis C virus RNA-containing particles. J. Virol. 76, 6919–6928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chang K. S., Jiang J., Cai Z., Luo G. (2007) Human apolipoprotein E is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 81, 13783–13793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kawaguchi K., Faulk K., Purcell R. H., Emerson S. U. (2011) Reproduction in vitro of a quasispecies from a hepatitis C virus-infected patient and determination of factors that influence selection of a dominant species. J. Virol. 85, 3408–3414 [DOI] [PMC free article] [PubMed] [Google Scholar]