

Abstract
Complex II couples oxidoreduction of succinate and fumarate at one active site with that of quinol/quinone at a second distinct active site over 40 Å away. This process links the Krebs cycle to oxidative phosphorylation and ATP synthesis. The pathogenic mutation or inhibition of human complex II or its assembly factors is often associated with neurodegeneration or tumor formation in tissues derived from the neural crest. This brief overview of complex II correlates the clinical presentations of a large number of symptom-associated alterations in human complex II activity and assembly with the biochemical manifestations of similar alterations in the complex II homologs from Escherichia coli. These analyses provide clues to the molecular basis for diseases associated with aberrant complex II function.
Keywords: Electron Transport, Membrane Enzymes, Metabolic Diseases, Mitochondria, Respiration, Tricarboxylic Acid (TCA) Cycle, Tumor Metabolism
Introduction
Over the past 6 decades, complex II (succinate dehydrogenase, succinate:quinone oxidoreductase (SQR)3) has been at the forefront in the discovery of redox cofactors involved in bioenergetics. Helmut Beinert noted that in the 1950s–1960s, complex II was instrumental in the discovery of covalently bound flavin, tightly bound iron, and acid-labile sulfur associated with Fe-S clusters and bound ubiquinone of the mitochondrial respiratory chain (1). In 1995, a mutation of human complex II was the first report of a nuclear gene mutation shown to cause a mitochondrial respiratory chain deficiency (2). More recently, germ-line mutations in complex II have been found to be associated with hereditary tumors, suggesting that complex II genes may act as tumor suppressors (3).
Complex II enzymes (Fig. 1A) are heterotetramers containing two soluble subunits (SdhA (flavoprotein) and SdhB (Fe-S protein)) and two integral membrane subunits (SdhC and SdhD). This architecture houses two distinct active sites, and the coordinated catalysis at these sites links two key biological pathways, i.e. succinate oxidation to the Krebs cycle and quinone reduction to the electron transport chain. The SdhA subunit harbors a covalently attached FAD redox moiety and the dicarboxylate-binding site (Fig. 1B), where succinate is oxidized to fumarate. The product protons of this oxidation are transferred to bulk solvent, and fumarate acts as the next substrate in the Krebs cycle. The product electrons are transferred over 40 Å via three Fe-S clusters in the SdhB protein. These electrons act as co-substrates at the second active site, located at the interface of the integral membrane subunits. At this quinone-reducing site (Fig. 1C), 2H+ and 2e− reduce ubiquinone to ubiquinol. The resultant quinol pool supports ATP synthesis by oxidative phosphorylation.
FIGURE 1.

Overview of the structure and active sites of porcine mitochondrial complex II. A–C, the SdhA chain (flavoprotein) is colored slate, the SdhB chain (iron protein) is colored purple, the SdhC chain (cytochrome bL) is colored green, and the SdhD chain (cytochrome bS) is colored brown. The FAD cofactor (yellow carbon atoms and bonds) is shown as a bond representation. The Fe-S clusters are highlighted with the irons in orange and the sulfurs in yellow. The heme and ubiquinone are highlighted with yellow bonds. Amino acid numbering is for the porcine complex. A, the x-ray structure of the porcine complex II (Protein Data Bank code 1ZOY (11)) is colored by subunit. B, the active site catalyzing succinate oxidation is located near the covalently bound FAD. The view is the same as shown in A. Active site residues analogous to those demonstrated to be important for catalysis in the E. coli homologs (SdhA His-254, Thr-266, Arg-298, His-365, and Arg-409) are shown as sticks, with carbons colored slate and nitrogens colored blue. The succinate is modeled into the active site based upon the placement of fumarate in E. coli QFR (Protein Data Bank code 3P4P (32)), and the rotamer of SdhA His-254 has been modeled with a 180° rotation to promote hydrogen bond formation. Putative hydrogen-bonding interactions are depicted with gray spheres. The proposed routes of proton and hydride transfer to the C2–C3 bond of succinate are shown by green spheres. C, the active site catalyzing quinone reduction with ubiquinone in the Q1 position (27, 28) is located in a pocket within the membrane subunits. The view is a 90° rotation from A and is looking from the bottom of the membrane up through the complex.
The complex II superfamily contains both SQRs and quinol:fumarate reductases (QFRs). QFRs are enzymes that are kinetically poised to catalyze quinol oxidation and fumarate reduction as part of anaerobic electron transport chains. Both enzymes have evolved to function optimally in the physiological niche in which they are expressed. SQR and QFR are capable of functionally replacing each other in vivo in Escherichia coli and also can function with naphtho- or ubiquinones (4–6). The overall structural and functional similarity of E. coli SQR and QFR to mammalian complex II has made them a useful model system.
Structure-based Alignments to Correlate Biochemical Changes with Clinical Symptoms
Sequence comparisons of each subunit of the complex II enzymes (supplemental Figs. S1–S4 and Table 1) strongly suggest a common evolutionary ancestor and similar structure for the soluble domains (SdhA/FrdA and SdhB/FrdB), with ∼50% sequence identity between mitochondrial and bacterial complex II family members and >90% sequence identity between mitochondrial complex II proteins of different species. Confirming this, a similar three-dimensional architecture of the soluble domains of complex II enzymes is observed in x-ray structures of six different complex II superfamily members (7–12).
The availability of these structures supports the proposal that divergent or independent evolution of the membrane subunits (SdhC/FrdC and SdhD/FrdD) has occurred within this superfamily. Of the bacterial complex II enzymes, the membrane subunits of E. coli SQR are closely related to the mitochondrial counterparts in structure despite limited sequence similarity (supplemental Figs. S3 and S4). It is notable that the location of the quinone-binding site and the identity of the amino acids forming hydrogen bonds to the quinone are conserved in the E. coli and mitochondrial SQR enzymes. This supports the contention that E. coli SQR is a useful model for assessment of the biochemical function of complex II.
In the past decade, researchers have identified numerous human complex II mutations associated with a variety of diseases. These diseases can result from deletions or missense or nonsense mutations in the genes encoding complex II or its assembly factors. The deletions and nonsense mutations are presumed to result in a complete loss of complex II folding, assembly, and function. The biochemical consequences of the missense mutations are, however, often unclear because of the difficulty in obtaining sufficient material and the lack of appropriate cellular models to mimic the mutations. The mutations associated with human disease are annotated in the TCA cycle gene mutation database (13), and the results for the missense mutations are summarized in supplemental Tables 2–4. As shown in these tables, the majority of the mutations are found in the SdhB and SdhD subunits. In this minireview, the missense mutations (supplemental Tables 2–4) (13) were mapped onto the structure of porcine complex II (Fig. 2 and supplemental Fig. S5) to identify the overall location of the mutation and to relate this to the corresponding region in the E. coli counterparts. Because in vitro characterization of E. coli SQR and QFR has been extensive, the consequences of mutation of a particular region could be used to predict the structural and biochemical consequences of mutations on the human enzyme. Single nucleotide polymorphisms not associated with a clinical phenotype are not described here.
FIGURE 2.

Three-dimensional mapping of disease-associated missense mutations. The structure of porcine mitochondrial complex II (Protein Data Bank code 1ZOY (11)) is used as a model for the human enzyme, and numbering is that of the human enzyme. The polypeptide chains of complex II are lightly colored with the same scheme described in the legend to Fig. 1. The locations of mutations associated with Leigh disease are highlighted, with the Cα atom shown as a green sphere. Mutations or inhibition associated with respiratory and neurodegenerative disease states are highlighted in blue. Mutations associated with tumors are highlighted with the Cα atom as a red sphere. The dicarboxylate inhibitor oxaloacetate is shown as blue sticks. Three major groups of disease-associated mutations are observed outside of the immediately vicinity of the cofactors. Group 1 is at the N terminus of the SdhB subunit; Group 2 is within the bacterial ferredoxin domain of the SdhB subunit; and Group 3 is located at the distal side of the membrane, near the lipid-binding site, and is predominated by mutations of the SdhD subunit. A version of this figure with each amino acid labeled is provided in supplemental Fig. S5. UQ, ubiquinone.
Aberrant Complex II Activity Associated with Neurodegeneration
Either mutation of amino acids within the SdhA subunit or inhibition of the succinate-binding site by small molecules can result in a range of clinical symptoms hallmarked by neurodegeneration. The associated symptoms are reported to range from moderate to severe.
Point Mutations Resulting in Leigh Disease
Among the most severe clinical manifestations of missense or nonsense mutations of complex II is Leigh disease (14). Leigh disease generally presents at an early age, and patients usually die within 1 year of the onset of symptoms. At the molecular level, Leigh disease can be associated with mutations in 1 of ∼30 different genes, with many of these mutations found in proteins that, like complex II, are involved in oxidative respiration.
Three independent missense mutations in SdhA are associated with Leigh disease. Sequence analysis indicates that these three amino acids are poorly conserved between the mammalian and bacterial enzymes (supplemental Table 2), and structural mapping shows that these substitutions are not immediately adjacent to the succinate-oxidizing active site in SdhA. Nevertheless, one can predict how these mutations reduce the succinate oxidation activity of the enzyme.
SdhA mutant A83V was found in a compound heterozygous patient with one copy of sdhA having a nonsense mutation and the second having the missense mutation (15). The patient had a reduced quantity of complex II, presumably from loss of one copy of the gene. Patient tissues exhibited ∼23% of the activity expected for controls, suggesting that A83V reduces enzyme activity to about half of wild-type levels. SdhA Ala-83 is located near the surface of SdhA within a hydrophobic pocket. Although the role of the corresponding residue has not yet been investigated in the bacterial homologs, one might predict a deficit in folding or stability of the SdhA subunit that causes a decrease in enzyme activity. In conjunction with the nonsense mutation in the heterozygous patient, this may reduce overall complex II activity to below a threshold level, resulting in the disease phenotype.
A second Leigh disease-associated substitution, SdhA A524V, occurred in a compound heterozygous patient with one copy of sdhA having a nonsense mutation and the second having the missense mutation (16). Although the identity of this amino acid is not well conserved in the superfamily, Ala-524 is located at the interface between two domains of SdhA known as the “FAD-binding” and “capping” domains. These domains have been proposed to move with respect to each other during cofactor insertion and catalysis. Investigation of the residue corresponding to Ala-524 has not been investigated in bacterial systems, although mutations within E. coli QFR that influence the interaction between these domains lower enzyme turnover to ∼13% of wild-type levels (17).
The third missense mutation (SdhA R554W) was the first Leigh disease mutation discovered in this subunit (2). The equivalent mutation in yeast shows increased sensitivity to inhibition by oxaloacetate (2), a normal intermediate of the Krebs cycle and a potent inhibitor of complex II (18, 19). The enhanced sensitivity of complex II to oxaloacetate likely reduces enzyme activity to <50% of wild-type levels in patients harboring the R554W mutation (2). The location of SdhA Arg-554 is near the surface of the SdhA subunit, and although near the interface of SdhA and SdhB, this residue does not appear to contribute directly to intersubunit interaction.
Recently, a number of substitutions within the SdhA subunit of complex II have been described with different clinical presentations. One of these, SdhA R451C, has a clinical presentation suggesting neurodegeneration at a more gradual pace than in Leigh syndrome. The mutation of human SdhA Arg-451 to Cys results in late-onset optical atrophy and myopathy and acts in a dominant fashion (20). The influence of this mutation on the activity of complex II is readily apparent from structural and biochemical analyses (Fig. 2 and supplemental Fig. S5 and Table 2). SdhA Arg-451 is an active site residue important for substrate affinity and is also known to be essential for formation of the covalent flavin linkage. Using E. coli QFR as a model system, it was shown that loss of the covalent flavin linkage results in an enzyme incapable of succinate oxidation (21). In patients harboring a single copy of the SdhA R451C mutation, only 50% of the complex II-associated succinate oxidase activity is anticipated, which is consistent with what is observed (20).
The SdhA G555E variant is observed in a number of patients, albeit with wide phenotypic variability (22–24). Symptoms range from minor muscle weakness to moderate Leigh disease to severe mitochondrial deficiency or neonatal cardiomyopathy and death. Immunoblotting of patient tissue suggests decreased assembly of the SdhA and SdhB subunits compared with healthy patients (23, 24). Consistent with this observation, SdhA Gly-555 maps to the interdomain interface between SdhA and SdhB and would be predicted to disrupt complex assembly.
Complex II Inhibition Associated with Neurodegeneration
Competitive inhibition of succinate oxidation in human and animal models results in the specific loss of striatal neurons and symptoms reminiscent of a large subset of those associated with Huntington disease (25, 26). It is well established that molecules that resemble the dicarboxylate substrate succinate (Fig. 3) may act as inhibitors of complex II. These dicarboxylates can be either normal metabolic intermediates (18, 19, 29) or toxins (30). For example, fumarate, malate, oxaloacetate, and citrate are all Krebs cycle intermediates with a broad range of Ki values that may regulate complex II activity through feedback inhibition (18, 19).
FIGURE 3.
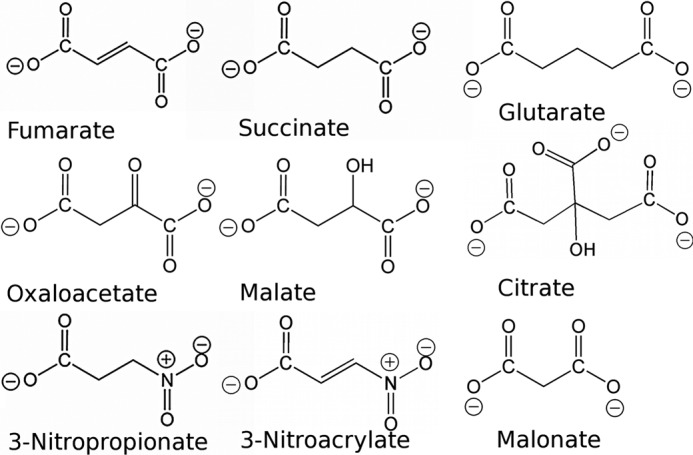
Comparison of chemical structures of dicarboxylate substrates, products, and inhibitors of complex II. Fumarate and succinate are the natural dicarboxylate substrates and products of complex II homologs. The remaining small molecules are demonstrated to be inhibitors. Inhibition of complex II by any of these small molecules, except citrate, is associated with neurodegenerative symptoms in humans or in animal models, and malate and 3-nitropropionate are commonly used in animal models of Huntington disease (25, 26). This figure was originally published in Ref. 32.
Each of these small molecules has been evaluated to determine the influence on the activity of the E. coli complex II homologs, and all act as either competitive or suicide inhibitors. Co-crystal structures of mitochondrial complex II (10, 11, 31) and E. coli QFR (32) with these small molecules show that each binds at the active site, thus blocking the binding pocket, explaining how they inhibit enzyme activity.
Succinate Dehydrogenase Assembly Factor 1 (SdhAF1) and Neurodegeneration
Sequencing of DNA from patients with infantile leukoencephalopathy identified two distinct mutations, G57R and R55P, in a previously uncharacterized protein now called SdhAF1 (33). SdhAF1 is a 115-amino acid soluble protein that targets to mitochondria and contains a distinct LYR sequence motif similar to other proteins involved in Fe-S cluster assembly. Prior to this, complex II assembly was believed to require only nonspecific accessory proteins, including chaperones and a putative mitochondrial FAD transporter (34–36). Because mutation of SdhAF1 influences solely complex II activity and not the activity of other Fe-S-containing mitochondrial proteins, it was suggested that this is the first identified specific assembly factor for complex II (33). Disruption of the yeast ortholog of SdhAF1 results in deficiency in oxidative phosphorylation. However, there is little other molecular information available on the biochemical function of SdhAF1. To date, there is no structure of any homolog of this protein. As a result, the molecular basis for how mutation of SdhAF1 leads to the observed clinical manifestation is not understood.
Aberrant Complex II Activity Associated with Tumor Formation
Many years ago, Otto Warburg and co-workers demonstrated a glycolytic shift in the metabolism of tumors (37). One hypothesis to explain this phenomenon is that tumor cells may have altered mitochondrial metabolism affecting oxidative phosphorylation. In the past decade, a number of mutations of complex II and other TCA cycle enzymes, such as fumarase and isocitrate dehydrogenase, have been associated with the formation of tumors (3, 38–41). The tissue distribution and physiologically consequences are highly varied, although complex II-associated tumors are most often pheochromocytomas or paragangliomas (3). It has been observed that loss of function of complex II and fumarase can lead to accumulation of their dicarboxylate substrates succinate and fumarate and generation of reactive oxygen species (38, 40, 41). This leads to stabilization of the transcription factor hypoxia-inducible factor-1α due to inhibition of hypoxia-inducible factor prolyl hydroxylases (42, 43). Accumulation of TCA cycle intermediates is also implicated in inhibition of α-ketoglutarate-dependent enzymes involved in sulfur metabolism and histone demethylation (44, 45).
Tumorigenic mutations of human complex II were first identified within the sdhD gene (39), and the majority of complex II-associated mutations map to the sdhD and sdhB genes, with a lesser number in sdhC and rarely in sdhA (13). Recently, however, a second assembly factor, SdhAF2, has been identified (46). A G78R substitution in this small mitochondrial matrix protein leads to paraganglioma-pheochromocytoma syndrome.
It is anticipated that a nonsense mutation within the coding sequence of the sdhB, sdhC, or sdhD gene would result in the loss of complex II assembly (13), which would perturb TCA cycle metabolism. Numerous missense mutations have also been identified as tumorigenic. Interestingly, these substitutions are distributed throughout the sequence, but most can be grouped spatially in the folded protein.
Pheochromocytoma and paraganglioma tumors tend to present between ages 30 and 40, are more commonly benign than malignant, and are generally recessive, but may be sporadic. A majority of the complex II tumor-associated missense mutations are predicted to have major consequences for enzyme activity. These include the mutations of the ligands to the electron transfer cofactors of complex II and residues immediately surrounding any of these cofactors or the quinone-reducing active site (13).
The biochemical consequences of these mutations have rarely been defined in human tissues because of the paucity of available material. However, numerous site-directed substitutions of many of these amino acids have been studied in both bacterial and lower eukaryotic systems. For example, the absolutely conserved Cys ligands to the three Fe-S clusters in the B subunit of complex II have been mutated in E. coli QFR (47–49). These studies demonstrated that the [2Fe-2S] cluster domain (B subunit Cys-93, Cys-98, Cys-101, and Cys-113 in the human sequence) is more tolerant to amino acid substitutions than the [4Fe-4S]/[3Fe-4S] domain. Nevertheless, in the case of the pheochromocytoma or paraganglioma patients, in which mutations of the [2Fe-2S]-binding Cys residues have been reported (13, 50–55), the substitutions are either an Arg or a Tyr residue, which would be expected to prevent assembly of the Fe-S cluster and perturb assembly of complex II. Missense mutation of any Cys residue ligating either the [4Fe-4S] or [3Fe-4S] cluster to Arg or Tyr residues (50–58) also prevents proper assembly of the membrane-bound form of complex II (50–58). Informative is one human mutation (SdhB C243S, a ligand of the [3Fe-4S] cluster) that results in a pathogenic germ-line malignant paraganglioma (58). The same substitution in E. coli QFR resulted in a catalytically inactive enzyme that was found in the cytoplasm and that was unable to assemble properly (47). Numerous studies have shown that failure of the [3Fe-4S] cluster to assemble results in the SdhA/SdhB subunits being unable to associate with the membrane domain (1, 59). In addition, residues proximal to the Cys ligands of the Fe-S clusters have been mutated and shown to affect the assembly and catalytic properties of complex II. For example, SdhB Pro-197 is located adjacent to one of the Cys ligands of the [3Fe-4S] cluster and has been associated with paragangliomas in several studies (13, 54, 55, 57). When the equivalent residue was mutated in E. coli QFR (60) or in Saccharomyces cerevisiae (61) or Caenorhabditis elegans (62) complex II, increased reactive oxygen species were produced, quinone reduction was significantly perturbed, and C. elegans had a shortened life span phenotypically similar to the SdhC mev-1 mutant, in which the [3Fe-4S]/quinone-binding domain is perturbed (63).
A large number of site-directed mutations of bacterial and lower eukaryotic complex II enzymes have been constructed to understand the role and properties of the quinone and heme redox centers. Some of these substitutions have involved residues directly associated with tumor formation, and all of them inform as to consequences of perturbation of this domain of complex II. An example is SdhC Arg-72 (57), which is associated with paragangliomas. Mutation of the equivalent residue in E. coli or S. cerevisiae SQR (64, 65) showed severe impairment of quinone reduction, although each enzyme was stable. Interestingly, in yeast, the mutants secreted succinate (65), consistent with reduced complex II activity elevating succinate levels and stabilizing hypoxia-inducible factor-1α, leading to a pseudohypoxic phenotype (40, 42). Similar findings were made with SdhD Asp-113 mimics, although in E. coli SQR (64), the catalytic activity was less severely affected than in yeast (65). For SdhD His-102-associated tumor formation (39, 66–68) and Tyr-114-associated tumor formation (67, 69–72), there is relevant information supplied by study of yeast and bacterial complex II. Detailed analysis shows that mutation of any of these residues (4, 73–75) results in reduced enzyme activity, although the enzyme is stably associated with the membrane. This suggests that it is the loss of enzyme activity and the resultant increased levels of cellular succinate, rather than reactive oxygen species generation, that are related to tumor formation in this class of mutant complex II.
Tumorigenesis-associated mutations are also observed outside of the immediate vicinity of the cofactors and active sites. Biochemical analyses of these missense mutations have been rarely performed because they were not predicted to be important for function prior to the identification of the clinical variation. The vast majority of these fall into three distinct spatial groups (Fig. 2 and supplemental Fig. S5). Because extrapolation of the biochemical consequences of mutations near the cofactors from the E. coli and lower eukaryotic complexes II predicts major impact on enzyme activity in the human enzyme with similar mutations, it strongly suggests that mutations within the three groups similarly have a dramatic influence on activity. One explanation consistent with the spatial grouping is that they could influence folding or assembly of complex II, disrupting enzyme stability. This possibility requires experimental verification.
The first group of mutations (Group 1; see Fig. 2 and supplemental Fig. S5 and Table 3 for residues included in Group 1) is spatially located near the N terminus of SdhB, in a domain that ligates the [2Fe-2S] cluster. One could speculate that these mutations prevent the proper folding of the SdhB subunit by destabilizing the overall fold of the Fe-S protein, altering the conformation of this domain of SdhB (with the consequence of influencing the interaction between the SdhB and SdhA subunits), or reducing the insertion of the [2Fe-2S] cluster.
The second group of mutations (Group 2) is also located within the SdhB subunit, but in the domain that ligates both the [4Fe-4S] and [3Fe-4S] clusters. This group contains fewer amino acid substitutions (Fig. 2 and supplemental Fig. S5 and Table 3), consistent with the core of this bacterial ferredoxin-like domain being smaller than the plant-type ferredoxin domain. The possible molecular influences of each of these mutations are similar to those of the mutations in Group 1.
The third group of mutations (Group 3) is located at the distal side of the membrane and is predominated by substitutions of the SdhD subunit (Fig. 2 and supplemental Fig. S5 and Table 3). Investigations of the molecular basis for complex II deficiency in patients with mutations of Group 3 are limited, with evaluation of the SdhD Y93C substitution suggesting reduced transcription of sdhD mRNA (76). Interestingly, the mutations in Group 3 are all located immediately adjacent to a binding site for lipid that is conserved between the avian and porcine enzymes and similar in location in E. coli SQR. This lipid (identified as cardiolipin in E. coli SQR (9)) is fully integrated within the membrane subunits and has been proposed to be important for folding and stability. The distinct group of mutations within SdhD is consistent with this location being key for complex folding or assembly, and mutations may act in part by altering the affinity for bound lipid or by influencing membrane insertion of SdhD.
A disproportionately large number of the point mutations of complex II associated with tumorigenesis are within the SdhD subunit of the enzyme. Indeed, 20% of the amino acids in the mature SdhD polypeptide chain are associated with tumor formation. One possibility to explain this apparent skewing could be that the transcription and translation of sdhD or the folding or membrane insertion of the SdhD subunit is more challenging than that of the other subunits, and that even small perturbations in the sequence could reduce complex quantity or assembly.
Alteration of the SdhA Subunit of Complex II and Tumorigenesis
Until 2010, alterations of the SdhA subunit of complex II, whether missense or nonsense, manifested in reduced mitochondrial respiration and were exclusively linked with neurodegeneration. Two missense mutations and one nonsense mutation have recently been identified where the patient's clinical presentation instead matches that of patients with mutations that cause tumorigenesis (77–79). Interestingly, the two missense mutations, SdhA R585W (78) and SdhA R589W (77, 79), are located adjacent to each other in a conserved region of the flavoprotein subunit. However, they are located away from the electron transfer pathway and near the surface of the protein (Fig. 2 and supplemental Fig. S5). Considering that other tumorigenic mutations of complex II have been either demonstrated or hypothesized to influence electron transfer between FAD and quinone, it is surprising to find these mutations positioned distal to the electron transfer pathway.
SdhAF2 and Paraganglioma
Around the same time as the discovery of SdhAF1, a second complex II assembly factor, SdhAF2, was identified (46). SdhAF2 was found to have a G78R mutation in four familial paraganglioma patients (46). The SdhAF2 protein is a soluble mitochondrial protein of 166 amino acids, with the mature protein being 137 amino acids in length. Using the S. cerevisiae ortholog Sdh5, it was found that SdhAF2 is required for covalent FAD insertion into SdhA. Recently, a bacterial ortholog of SdhAF2/Sdh5 has also been found to be necessary for flavinylation of complex II in Serratia (80). This ortholog, termed SdhE, is homologous to a gene/protein (YgfY) previously of unknown function in E. coli (80). Sequence analyses suggest that SdhAF2/Sdh5/SdhE belongs to a very large family conserved throughout Eubacteria and Eukarya (supplemental Fig. S6). The bacterial proteins are about half the size (∼80–90 amino acids) of the eukaryotic counterparts (∼160–170 amino acids). The mechanism of how SdhAF2 is involved in incorporation of the covalent FAD linkage in SdhA remains unclear. The available studies suggest that it has a chaperone-like function in binding and stabilizing the SdhA subunit (46, 80), but there is conflicting evidence as to whether Sdh5/SdhE binds FAD directly (80),4 potentially passing this cofactor to SdhA.
Although the role of Sdh5/SdhE was unknown until recently, structures of homologs from E. coli (YgfY), Neisseria meningitidis (NMA1147), Vibrio cholera (VC_2471), and S. cerevisiae4 were determined as part of structural genomics projects (Protein Data Bank code 2JR5) (81, 82)4 and show a fold based upon a five-helix bundle (Fig. 4). The yeast Sdh5 NMR4 fragment corresponds to the sequence region that is common to both prokaryotes and eukaryotes and aligns with E. coli SdhE with a root mean square deviation of 1.95 Å over 79 Cα atoms (Fig. 4). Importantly, sequence and structural comparisons have identified a signature motif for this fold, RGxxE, located between helices 1 and 2 of the five-helix bundle. Analysis of the structure of E. coli SdhE suggested that this sequence is important for folding and stability because the N-terminal arginine forms hydrogen-bonding contacts with the backbone between helices 3 and 4 of the five-helix bundle (81). Inspection of the NMR structure of Sdh54 and the prokaryotic homologs VC_2471 (Protein Data Bank code 2JR5) and NMA1147 (82) suggests that this stabilizing contact may be conserved throughout the family, although it is only explicitly demonstrated in the coordinates of VC_2471 (Protein Data Bank code 2JR5). An alternative proposal is that this signature sequence forms part of the SdhA-binding site.4 The RGxxE motif starts at position 77 of the human SdhAF2 sequence, and Gly-78, which is mutated in the affected patient, is the second amino acid.
FIGURE 4.

Structure of Sdh5/SdhE. The structure of the yeast Sdh5 fragment (Protein Data Bank code 2LM4)4 is colored with the N terminus in blue and the C terminus in red and is shown overlaid with the structure of E. coli SdhE (Protein Data Bank code 1X6I (81)) colored in light gray. The location of the G78R mutation is highlighted as a red sphere.
Remaining Questions
The correlation between the clinical phenotype and biochemistry of complex II mutations raises a number of questions as to how the enzyme acts in the intact organism. The two clinical presentations discussed here, those of neurodegeneration and early death (generally, alteration of SdhA) versus tumor formation (generally, alteration of other subunits and assembly factors of complex II), have different symptoms and severity. Although the biochemical consequences of some of these mutations can be predicted in many cases, it is not readily apparent why the two distinct clinical manifestations are associated with the mutations that cause them. It can be suggested that mutation of SdhA influences succinate oxidation, an important chemical reaction within the Krebs cycle. If the loss of this activity is sufficiently severe, this can result in neurodegeneration and death, i.e. the same clinical presentation as the mutation of other respiratory chain enzymes. Conversely, mutation of the SdhB, -C, or -D subunit of complex II, which alters either the electron transfer conduit to or the integrity of the quinone reduction site, may perturb mitochondrial metabolism so that increased levels of succinate alter signaling pathways, or increased reactive oxygen species could contribute to tumors. The assembly factors for complex II are an exciting recent development and deserve further study. It is noteworthy that two mutations mapping near the surface of SdhA and loss of the covalent FAD assembly factor SdhAF2 both lead to paragangliomas. This is the first link between mutations in SdhA and paragangliomas usually associated with SdhB, -C, and -D mutations. Evidently, further study is needed to fully understand the biochemical and clinical consequences of deficits in complex II function.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants GM61606 and GM79419. This work was also supported by Department of Veterans Affairs Biomedical Laboratory Research and Development.

This article contains supplemental Figs. S1–S6, Tables 1–4, and additional references.
A. Eletsky, M.-Y. Jeong, H. Kim, H.-W. Lee, R. Xiao, D. J. Pagliarini, J. H. Prestegard, D. R. Winge, G. T. Montelione, and T. Szyperski, submitted for publication (Solution NMR structure of yeast succinate dehydrogenase flavinator protein Sdh5 reveals a putative Sdh1-binding site).
- SQR
- succinate:quinone oxidoreductase
- QFR
- quinol:fumarate reductase
- SdhAF1
- succinate dehydrogenase assembly factor 1.
REFERENCES
- 1. Beinert H. (2002) Spectroscopy of succinate dehydrogenases, a historical perspective. Biochim. Biophys. Acta 1553, 7–22 [DOI] [PubMed] [Google Scholar]
- 2. Bourgeron T., Rustin P., Chretien D., Birch-Machin M., Bourgeois M., Viegas-Péquignot E., Munnich A., Rötig A. (1995) Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat. Genet. 11, 144–149 [DOI] [PubMed] [Google Scholar]
- 3. Bardella C., Pollard P. J., Tomlinson I. (2011) SDH mutations in cancer. Biochim. Biophys. Acta 1807, 1432–1443 [DOI] [PubMed] [Google Scholar]
- 4. Maklashina E., Hellwig P., Rothery R. A., Kotlyar V., Sher Y., Weiner J. H., Cecchini G. (2006) Differences in protonation of ubiquinone and menaquinone in fumarate reductase from Escherichia coli. J. Biol. Chem. 281, 26655–26664 [DOI] [PubMed] [Google Scholar]
- 5. Guest J. R. (1981) Partial replacement of succinate dehydrogenase function by phage- and plasmid-specified fumarate reductase in Escherichia coli. J. Gen. Microbiol. 122, 171–179 [DOI] [PubMed] [Google Scholar]
- 6. Maklashina E., Berthold D. A., Cecchini G. (1998) Anaerobic expression of Escherichia coli succinate dehydrogenase: functional replacement of fumarate reductase in the respiratory chain during anaerobic growth. J. Bacteriol. 180, 5989–5996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Iverson T. M., Luna-Chavez C., Cecchini G., Rees D. C. (1999) Structure of the Escherichia coli fumarate reductase respiratory complex. Science 284, 1961–1966 [DOI] [PubMed] [Google Scholar]
- 8. Lancaster C. R., Kröger A., Auer M., Michel H. (1999) Structure of fumarate reductase from Wolinella succinogenes at 2.2 Å resolution. Nature 402, 377–385 [DOI] [PubMed] [Google Scholar]
- 9. Yankovskaya V., Horsefield R., Törnroth S., Luna-Chavez C., Miyoshi H., Léger C., Byrne B., Cecchini G., Iwata S. (2003) Architecture of succinate dehydrogenase and reactive oxygen species generation. Science 299, 700–704 [DOI] [PubMed] [Google Scholar]
- 10. Huang L. S., Sun G., Cobessi D., Wang A. C., Shen J. T., Tung E. Y., Anderson V. E., Berry E. A. (2006) 3-Nitropropionic acid is a suicide inhibitor of mitochondrial respiration that, upon oxidation by complex II, forms a covalent adduct with a catalytic base arginine in the active site of the enzyme. J. Biol. Chem. 281, 5965–5972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sun F., Huo X., Zhai Y., Wang A., Xu J., Su D., Bartlam M., Rao Z. (2005) Crystal structure of mitochondrial respiratory membrane protein complex II. Cell 121, 1043–1057 [DOI] [PubMed] [Google Scholar]
- 12. Shimizu H., Osanai A., Sakamoto K., Inaoka D. K., Shiba T., Harada S., Kita K. (2012) Crystal structure of mitochondrial quinol:fumarate reductase from the parasitic nematode Ascaris suum. J. Biochem. 151, 589–592 [DOI] [PubMed] [Google Scholar]
- 13. Bayley J. P., Devilee P., Taschner P. E. (2005) The SDH mutation database: an online resource for succinate dehydrogenase sequence variants involved in pheochromocytoma, paraganglioma, and mitochondrial complex II deficiency. BMC Med. Genet. 6, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wong L. J. (2012) Mitochondrial syndromes with leukoencephalopathies. Semin. Neurol. 32, 55–61 [DOI] [PubMed] [Google Scholar]
- 15. Horváth R., Abicht A., Holinski-Feder E., Laner A., Gempel K., Prokisch H., Lochmüller H., Klopstock T., Jaksch M. (2006) Leigh syndrome caused by mutations in the flavoprotein (Fp) subunit of succinate dehydrogenase (SDHA). J. Neurol. Neurosurg. Psychiatry 77, 74–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Parfait B., Chretien D., Rötig A., Marsac C., Munnich A., Rustin P. (2000) Compound heterozygous mutations in the flavoprotein gene of the respiratory chain complex II in a patient with Leigh syndrome. Hum. Genet. 106, 236–243 [DOI] [PubMed] [Google Scholar]
- 17. Tomasiak T. M., Maklashina E., Cecchini G., Iverson T. M. (2008) A threonine on the active site loop controls transition state formation in Escherichia coli respiratory complex II. J. Biol. Chem. 283, 15460–15468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ackrell B. A., Kearney E. B., Mayr M. (1974) Role of oxaloacetate in the regulation of mammalian succinate dehydrogenase. J. Biol. Chem. 249, 2021–2027 [PubMed] [Google Scholar]
- 19. Ackrell B. A., Cochran B., Cecchini G. (1989) Interactions of oxaloacetate with Escherichia coli fumarate reductase. Arch. Biochem. Biophys. 268, 26–34 [DOI] [PubMed] [Google Scholar]
- 20. Birch-Machin M. A., Taylor R. W., Cochran B., Ackrell B. A., Turnbull D. M. (2000) Late-onset optic atrophy, ataxia, and myopathy associated with a mutation of a complex II gene. Ann. Neurol. 48, 330–335 [PubMed] [Google Scholar]
- 21. Blaut M., Whittaker K., Valdovinos A., Ackrell B. A., Gunsalus R. P., Cecchini G. (1989) Fumarate reductase mutants of Escherichia coli that lack covalently bound flavin. J. Biol. Chem. 264, 13599–13604 [PubMed] [Google Scholar]
- 22. Levitas A., Muhammad E., Harel G., Saada A., Caspi V. C., Manor E., Beck J. C., Sheffield V., Parvari R. (2010) Familial neonatal isolated cardiomyopathy caused by a mutation in the flavoprotein subunit of succinate dehydrogenase. Eur. J. Hum. Genet. 18, 1160–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pagnamenta A. T., Hargreaves I. P., Duncan A. J., Taanman J. W., Heales S. J., Land J. M., Bitner-Glindzicz M., Leonard J. V., Rahman S. (2006) Phenotypic variability of mitochondrial disease caused by a nuclear mutation in complex II. Mol. Genet. Metab. 89, 214–221 [DOI] [PubMed] [Google Scholar]
- 24. Van Coster R., Seneca S., Smet J., Van Hecke R., Gerlo E., Devreese B., Van Beeumen J., Leroy J. G., De Meirleir L., Lissens W. (2003) Homozygous Gly555Glu mutation in the nuclear-encoded 70-kDa flavoprotein gene causes instability of the respiratory chain complex II. Am. J. Med. Genet. Part A 120A, 13–18 [DOI] [PubMed] [Google Scholar]
- 25. Brouillet E., Condé F., Beal M. F., Hantraye P. (1999) Replicating Huntington disease phenotype in experimental animals. Prog. Neurobiol. 59, 427–468 [DOI] [PubMed] [Google Scholar]
- 26. Túnez I., Tasset I., Pérez-De La Cruz V., Santamaría A. (2010) 3-Nitropropionic acid as a tool to study the mechanisms involved in Huntington disease: past, present, and future. Molecules 15, 878–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Horsefield R., Yankovskaya V., Sexton G., Whittingham W., Shiomi K., Omura S., Byrne B., Cecchini G., Iwata S. (2006) Structural and computational analysis of the quinone-binding site of complex II (succinate:ubiquinone oxidoreductase). A mechanism of electron transfer and proton conduction during ubiquinone reduction. J. Biol. Chem. 281, 7309–7316 [DOI] [PubMed] [Google Scholar]
- 28. Ruprecht J., Yankovskaya V., Maklashina E., Iwata S., Cecchini G. (2009) Structure of Escherichia coli succinate:quinone oxidoreductase with an occupied and empty quinone-binding site. J. Biol. Chem. 284, 29836–29846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ullrich K., Flott-Rahmel B., Schluff P., Musshoff U., Das A., Lücke T., Steinfeld R., Christensen E., Jakobs C., Ludolph A., Neu A., Röper R. (1999) Glutaric aciduria type I: pathomechanisms of neurodegeneration. J. Inherit. Metab. Dis. 22, 392–403 [DOI] [PubMed] [Google Scholar]
- 30. Alston T. A., Mela L., Bright H. J. (1977) 3-Nitropropionate, the toxic substance of Indigofera, is a suicide inactivator of succinate dehydrogenase. Proc. Natl. Acad. Sci. U.S.A. 74, 3767–3771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang L. S., Shen J. T., Wang A. C., Berry E. A. (2006) Crystallographic studies of the binding of ligands to the dicarboxylate site of complex II and the identity of the ligand in the “oxaloacetate-inhibited” state. Biochim. Biophys. Acta 1757, 1073–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tomasiak T. M., Archuleta T. L., Andréll J., Luna-Chávez C., Davis T. A., Sarwar M., Ham A. J., McDonald W. H., Yankovskaya V., Stern H. A., Johnston J. N., Maklashina E., Cecchini G., Iverson T. M. (2011) Geometric restraint drives on- and off-pathway catalysis by the Escherichia coli menaquinol:fumarate reductase. J. Biol. Chem. 286, 3047–3056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ghezzi D., Goffrini P., Uziel G., Horvath R., Klopstock T., Lochmüller H., D'Adamo P., Gasparini P., Strom T. M., Prokisch H., Invernizzi F., Ferrero I., Zeviani M. (2009) SDHAF1, encoding a LYR complex II-specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nat. Genet. 41, 654–656 [DOI] [PubMed] [Google Scholar]
- 34. Dibrov E., Fu S., Lemire B. D. (1998) The Saccharomyces cerevisiae TCM62 gene encodes a chaperone necessary for the assembly of the mitochondrial succinate dehydrogenase (complex II) J. Biol. Chem. 273, 32042–32048 [DOI] [PubMed] [Google Scholar]
- 35. Tzagoloff A., Jang J., Glerum D. M., Wu M. (1996) FLX1 codes for a carrier protein involved in maintaining a proper balance of flavin nucleotides in yeast mitochondria. J. Biol. Chem. 271, 7392–7397 [DOI] [PubMed] [Google Scholar]
- 36. Giancaspero T. A., Wait R., Boles E., Barile M. (2008) Succinate dehydrogenase flavoprotein subunit expression in Saccharomyces cerevisiae–involvement of the mitochondrial FAD transporter Flx1p. FEBS J. 275, 1103–1117 [DOI] [PubMed] [Google Scholar]
- 37. Warburg O., Posener K., Negelein E. (1924) The metabolism of tumors. Biochem. Z. 152, 319–344 [Google Scholar]
- 38. Bayley J. P., Devilee P. (2010) Warburg tumors and the mechanisms of mitochondrial tumor suppressor genes. Barking up the right tree? Curr. Opin. Genet. Dev. 20, 324–329 [DOI] [PubMed] [Google Scholar]
- 39. Baysal B. E., Ferrell R. E., Willett-Brozick J. E., Lawrence E. C., Myssiorek D., Bosch A., van der Mey A., Taschner P. E., Rubinstein W. S., Myers E. N., Richard C. W., 3rd, Cornelisse C. J., Devilee P., Devlin B. (2000) Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287, 848–851 [DOI] [PubMed] [Google Scholar]
- 40. Frezza C., Pollard P. J., Gottlieb E. (2011) Inborn and acquired metabolic defects in cancer. J. Mol. Med. 89, 213–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. King A., Selak M. A., Gottlieb E. (2006) Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene 25, 4675–4682 [DOI] [PubMed] [Google Scholar]
- 42. Selak M. A., Armour S. M., MacKenzie E. D., Boulahbel H., Watson D. G., Mansfield K. D., Pan Y., Simon M. C., Thompson C. B., Gottlieb E. (2005) Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 7, 77–85 [DOI] [PubMed] [Google Scholar]
- 43. Koivunen P., Hirsilä M., Remes A. M., Hassinen I. E., Kivirikko K. I., Myllyharju J. (2007) Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates. Possible links between cell metabolism and stabilization of HIF. J. Biol. Chem. 282, 4524–4532 [DOI] [PubMed] [Google Scholar]
- 44. Smith E. H., Janknecht R., Maher L. J., 3rd (2007) Succinate inhibition of α-ketoglutarate-dependent enzymes in a yeast model of paraganglioma. Hum. Mol. Genet. 16, 3136–3148 [DOI] [PubMed] [Google Scholar]
- 45. Cervera A. M., Bayley J. P., Devilee P., McCreath K. J. (2009) Inhibition of succinate dehydrogenase dysregulates histone modification in mammalian cells. Mol. Cancer 8, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hao H. X., Khalimonchuk O., Schraders M., Dephoure N., Bayley J. P., Kunst H., Devilee P., Cremers C. W., Schiffman J. D., Bentz B. G., Gygi S. P., Winge D. R., Kremer H., Rutter J. (2009) SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 325, 1139–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kowal A. T., Werth M. T., Manodori A., Cecchini G., Schröder I., Gunsalus R. P., Johnson M. K. (1995) Effect of cysteine to serine mutations on the properties of the [4Fe-4S] center in Escherichia coli fumarate reductase. Biochemistry 34, 12284–12293 [DOI] [PubMed] [Google Scholar]
- 48. Manodori A., Cecchini G., Schröder I., Gunsalus R. P., Werth M. T., Johnson M. K. (1992) [3Fe-4S] to [4Fe-4S] cluster conversion in Escherichia coli fumarate reductase by site-directed mutagenesis. Biochemistry 31, 2703–2712 [DOI] [PubMed] [Google Scholar]
- 49. Werth M. T., Cecchini G., Manodori A., Ackrell B. A., Schröder I., Gunsalus R. P., Johnson M. K. (1990) Site-directed mutagenesis of conserved cysteine residues in Escherichia coli fumarate reductase: modification of the spectroscopic and electrochemical properties of the [2Fe-2S] cluster. Proc. Natl. Acad. Sci. U.S.A. 87, 8965–8969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Benn D. E., Gimenez-Roqueplo A. P., Reilly J. R., Bertherat J., Burgess J., Byth K., Croxson M., Dahia P. L., Elston M., Gimm O., Henley D., Herman P., Murday V., Niccoli-Sire P., Pasieka J. L., Rohmer V., Tucker K., Jeunemaitre X., Marsh D. J., Plouin P. F., Robinson B. G. (2006) Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J. Clin. Endocrinol. Metab. 91, 827–836 [DOI] [PubMed] [Google Scholar]
- 51. Lima J., Feijão T., Ferreira da Silva A., Pereira-Castro I., Fernandez-Ballester G., Máximo V., Herrero A., Serrano L., Sobrinho-Simões M., Garcia-Rostan G. (2007) High frequency of germ-line succinate dehydrogenase mutations in sporadic cervical paragangliomas in northern Spain: mitochondrial succinate dehydrogenase structure-function relationships and clinical-pathological correlations. J. Clin. Endocrinol. Metab. 92, 4853–4864 [DOI] [PubMed] [Google Scholar]
- 52. Neumann H. P., Bausch B., McWhinney S. R., Bender B. U., Gimm O., Franke G., Schipper J., Klisch J., Altehoefer C., Zerres K., Januszewicz A., Eng C., Smith W. M., Munk R., Manz T., Glaesker S., Apel T. W., Treier M., Reineke M., Walz M. K., Hoang-Vu C., Brauckhoff M., Klein-Franke A., Klose P., Schmidt H., Maier-Woelfle M., Peçzkowska M., Szmigielski C., Eng C., and Freiburg-Warsaw-Columbus Pheochromocytoma Study Group (2002) Germ-line mutations in nonsyndromic pheochromocytoma. N. Engl. J. Med. 346, 1459–1466 [DOI] [PubMed] [Google Scholar]
- 53. Neumann H. P., Pawlu C., Peczkowska M., Bausch B., McWhinney S. R., Muresan M., Buchta M., Franke G., Klisch J., Bley T. A., Hoegerle S., Boedeker C. C., Opocher G., Schipper J., Januszewicz A., Eng C. (2004) Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 292, 943–951 [DOI] [PubMed] [Google Scholar]
- 54. Sevilla M. A., Hermsen M. A., Weiss M. M., Grimbergen A., Balbín M., Llorente J. L., Rodrigo J. P., Suárez C. (2009) Chromosomal changes in sporadic and familial head and neck paragangliomas. Otolaryngol. Head Neck Surg. 140, 724–729 [DOI] [PubMed] [Google Scholar]
- 55. Srirangalingam U., Walker L., Khoo B., MacDonald F., Gardner D., Wilkin T. J., Skelly R. H., George E., Spooner D., Monson J. P., Grossman A. B., Akker S. A., Pollard P. J., Plowman N., Avril N., Berney D. M., Burrin J. M., Reznek R. H., Kumar V. K., Maher E. R., Chew S. L. (2008) Clinical manifestations of familial paraganglioma and pheochromocytomas in succinate dehydrogenase B (SDHB) gene mutation carriers. Clin. Endocrinol. 69, 587–596 [DOI] [PubMed] [Google Scholar]
- 56. Brouwers F. M., Eisenhofer G., Tao J. J., Kant J. A., Adams K. T., Linehan W. M., Pacak K. (2006) High frequency of SDHB germ-line mutations in patients with malignant catecholamine-producing paragangliomas: implications for genetic testing. J. Clin. Endocrinol. Metab. 91, 4505–4509 [DOI] [PubMed] [Google Scholar]
- 57. Burnichon N., Rohmer V., Amar L., Herman P., Leboulleux S., Darrouzet V., Niccoli P., Gaillard D., Chabrier G., Chabolle F., Coupier I., Thieblot P., Lecomte P., Bertherat J., Wion-Barbot N., Murat A., Venisse A., Plouin P. F., Jeunemaitre X., Gimenez-Roqueplo A. P. (2009) The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J. Clin. Endocrinol. Metab. 94, 2817–2827 [DOI] [PubMed] [Google Scholar]
- 58. Korpershoek E., Petri B. J., van Nederveen F. H., Dinjens W. N., Verhofstad A. A., de Herder W. W., Schmid S., Perren A., Komminoth P., de Krijger R. R. (2007) Candidate gene mutation analysis in bilateral adrenal pheochromocytoma and sympathetic paraganglioma. Endocr. Relat. Cancer 14, 453–462 [DOI] [PubMed] [Google Scholar]
- 59. Cecchini G. (2003) Function and structure of complex II of the respiratory chain. Annu. Rev. Biochem. 72, 77–109 [DOI] [PubMed] [Google Scholar]
- 60. Cecchini G., Sices H., Schröder I., Gunsalus R. P. (1995) Aerobic inactivation of fumarate reductase from Escherichia coli by mutation of the [3Fe-4S]/quinone-binding domain. J. Bacteriol. 177, 4587–4592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Guo J., Lemire B. D. (2003) The ubiquinone-binding site of the Saccharomyces cerevisiae succinate:ubiquinone oxidoreductase is a source of superoxide. J. Biol. Chem. 278, 47629–47635 [DOI] [PubMed] [Google Scholar]
- 62. Huang J., Lemire B. D. (2009) Mutations in the C. elegans succinate dehydrogenase iron-sulfur subunit promote superoxide generation and premature aging. J. Mol. Biol. 387, 559–569 [DOI] [PubMed] [Google Scholar]
- 63. Ishii N., Fujii M., Hartman P. S., Tsuda M., Yasuda K., Senoo-Matsuda N., Yanase S., Ayusawa D., Suzuki K. (1998) A mutation in succinate dehydrogenase cytochrome b causes oxidative stress and ageing in nematodes. Nature 394, 694–697 [DOI] [PubMed] [Google Scholar]
- 64. Tran Q. M., Rothery R. A., Maklashina E., Cecchini G., Weiner J. H. (2006) The quinone-binding site in Escherichia coli succinate dehydrogenase is required for electron transfer to the heme b. J. Biol. Chem. 281, 32310–32317 [DOI] [PubMed] [Google Scholar]
- 65. Szeto S. S., Reinke S. N., Sykes B. D., Lemire B. D. (2007) Ubiquinone-binding site mutations in the Saccharomyces cerevisiae succinate dehydrogenase generate superoxide and lead to the accumulation of succinate. J. Biol. Chem. 282, 27518–27526 [DOI] [PubMed] [Google Scholar]
- 66. Bacca A., Sellari Franceschini S., Carrara D., Bernini M., Zampa V., Taddei S., Miccoli P., Congregati C., Simi P., Ferrari M., Bernini G. (2012) Sporadic or familial head neck paragangliomas enrolled in a single center: clinical presentation and genotype/phenotype correlations. Head Neck 10.1002/hed.22910 [DOI] [PubMed] [Google Scholar]
- 67. Piccini V., Rapizzi E., Bacca A., Di Trapani G., Pulli R., Giachè V., Zampetti B., Lucci-Cordisco E., Canu L., Corsini E., Faggiano A., Deiana L., Carrara D., Tantardini V., Mariotti S., Ambrosio M. R., Zatelli M. C., Parenti G., Colao A., Pratesi C., Bernini G., Ercolino T., Mannelli M. (2012) Head and neck paragangliomas: genetic spectrum and clinical variability in 79 consecutive patients. Endocr. Relat. Cancer 19, 149–155 [DOI] [PubMed] [Google Scholar]
- 68. Poeppel T. D., Yuece A., Boy C., Metz K. A., Kaminsky E., Neumann H. P., Rosenbaum S. J., Mann K., Moeller L. C. (2011) Novel SDHD gene mutation (H102R) in a patient with metastatic cervical paraganglioma effectively treated by peptide receptor radionuclide therapy. J. Clin. Oncol. 29, e812–e815 [DOI] [PubMed] [Google Scholar]
- 69. Antonello M., Piazza M., Menegolo M., Opocher G., Deriu G. P., Grego F. (2008) Role of the genetic study in the management of carotid body tumor in paraganglioma syndrome. Eur. J. Vasc. Endovasc. Surg. 36, 517–519 [DOI] [PubMed] [Google Scholar]
- 70. Fish J. H., Klein-Weigel P., Biebl M., Janecke A., Tauscher T., Fraedrich G. (2007) Systematic screening and treatment evaluation of hereditary neck paragangliomas. Head Neck 29, 864–873 [DOI] [PubMed] [Google Scholar]
- 71. Milunsky J. M., Maher T. A., Michels V. V., Milunsky A. (2001) Novel mutations and the emergence of a common mutation in the SDHD gene causing familial paraganglioma. Am. J. Med. Genet. 100, 311–314 [DOI] [PubMed] [Google Scholar]
- 72. Schiavi F., Savvoukidis T., Trabalzini F., Grego F., Piazza M., Amistà P., Demattè S., Del Piano A., Cecchini M. E., Erlic Z., De Lazzari P., Mantero F., Opocher G. (2006) Paraganglioma syndrome: SDHB, SDHC, and SDHD mutations in head and neck paragangliomas. Ann. N.Y. Acad. Sci. 1073, 190–197 [DOI] [PubMed] [Google Scholar]
- 73. Oyedotun K. S., Sit C. S., Lemire B. D. (2007) The Saccharomyces cerevisiae succinate dehydrogenase does not require heme for ubiquinone reduction. Biochim. Biophys. Acta 1767, 1436–1445 [DOI] [PubMed] [Google Scholar]
- 74. Silkin Y., Oyedotun K. S., Lemire B. D. (2007) The role of Sdh4p Tyr-89 in ubiquinone reduction by the Saccharomyces cerevisiae succinate dehydrogenase. Biochim. Biophys. Acta 1767, 143–150 [DOI] [PubMed] [Google Scholar]
- 75. Tran Q. M., Rothery R. A., Maklashina E., Cecchini G., Weiner J. H. (2007) Escherichia coli succinate dehydrogenase variant lacking the heme b. Proc. Natl. Acad. Sci. U.S.A. 104, 18007–18012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. De Preter K., Vandesompele J., Hoebeeck J., Vandenbroecke C., Smet J., Nuyts A., Laureys G., Combaret V., Van Roy N., Roels F., Van Coster R., Praet M., De Paepe A., Speleman F. (2004) No evidence for involvement of SDHD in neuroblastoma pathogenesis. BMC Cancer 4, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Burnichon N., Brière J. J., Libé R., Vescovo L., Rivière J., Tissier F., Jouanno E., Jeunemaitre X., Bénit P., Tzagoloff A., Rustin P., Bertherat J., Favier J., Gimenez-Roqueplo A. P. (2010) SDHA is a tumor suppressor gene causing paraganglioma. Hum. Mol. Genet. 19, 3011–3020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Korpershoek E., Favier J., Gaal J., Burnichon N., van Gessel B., Oudijk L., Badoual C., Gadessaud N., Venisse A., Bayley J. P., van Dooren M. F., de Herder W. W., Tissier F., Plouin P. F., van Nederveen F. H., Dinjens W. N., Gimenez-Roqueplo A. P., de Krijger R. R. (2011) SDHA immunohistochemistry detects germ-line SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J. Clin. Endocrinol. Metab. 96, E1472–E1476 [DOI] [PubMed] [Google Scholar]
- 79. Pantaleo M. A., Astolfi A., Indio V., Moore R., Thiessen N., Heinrich M. C., Gnocchi C., Santini D., Catena F., Formica S., Martelli P. L., Casadio R., Pession A., Biasco G. (2011) SDHA loss-of-function mutations in KIT-PDGFRA wild-type gastrointestinal stromal tumors identified by massively parallel sequencing. J. Natl. Cancer Inst. 103, 983–987 [DOI] [PubMed] [Google Scholar]
- 80. McNeil M. B., Clulow J. S., Wilf N. M., Salmond G. P., Fineran P. C. (2012) SdhE is a conserved protein required for flavinylation of succinate dehydrogenase in bacteria. J. Biol. Chem. 287, 18418–18428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lim K., Doseeva V., Demirkan E. S., Pullalarevu S., Krajewski W., Galkin A., Howard A., Herzberg O. (2005) Crystal structure of the YgfY from Escherichia coli, a protein that may be involved in transcriptional regulation. Proteins 58, 759–763 [DOI] [PubMed] [Google Scholar]
- 82. Liu G., Sukumaran D. K., Xu D., Chiang Y., Acton T., Goldsmith-Fischman S., Honig B., Montelione G. T., Szyperski T. (2004) NMR structure of the hypothetical protein NMA1147 from Neisseria meningitidis reveals a distinct 5-helix bundle. Proteins 55, 756–758 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.