

Background: N-type Ca2+ channels (CaV2.2) are clinically validated targets for chronic pain.
Results: Two peptides from CaV2.2 and CaV1.2 perturb binding to a regulatory protein, CRMP2, inhibit calcium influx, and attenuate mechanical hyperalgesia in a rodent model of drug-induced chronic pain.
Conclusion: Ca2+ channel peptides block drug- and nerve injury-induced chronic pain.
Significance: Ca2+ channel peptide therapeutics can be useful in mitigating chronic pain.
Keywords: AIDS, Calcium Channels, Calcium Imaging, Ion Channels, Pain, Co-immunoprecipitation, Microscale Thermophoresis, Transmitter Release, Calcitonin Gene-related Peptide
Abstract
N-type Ca2+ channels (CaV2.2) are a nidus for neurotransmitter release and nociceptive transmission. However, the use of CaV2.2 blockers in pain therapeutics is limited by side effects resulting from inhibition of the physiological functions of CaV2.2 within the CNS. We identified an anti-nociceptive peptide (Brittain, J. M., Duarte, D. B., Wilson, S. M., Zhu, W., Ballard, C., Johnson, P. L., Liu, N., Xiong, W., Ripsch, M. S., Wang, Y., Fehrenbacher, J. C., Fitz, S. D., Khanna, M., Park, C. K., Schmutzler, B. S., Cheon, B. M., Due, M. R., Brustovetsky, T., Ashpole, N. M., Hudmon, A., Meroueh, S. O., Hingtgen, C. M., Brustovetsky, N., Ji, R. R., Hurley, J. H., Jin, X., Shekhar, A., Xu, X. M., Oxford, G. S., Vasko, M. R., White, F. A., and Khanna, R. (2011) Suppression of inflammatory and neuropathic pain by uncoupling CRMP2 from the presynaptic Ca2+ channel complex. Nat. Med. 17, 822–829) derived from the axonal collapsin response mediator protein 2 (CRMP2), a protein known to bind and enhance CaV2.2 activity. Using a peptide tiling array, we identified novel peptides within the first intracellular loop (CaV2.2(388–402), “L1”) and the distal C terminus (CaV1.2(2014–2028) “Ct-dis”) that bound CRMP2. Microscale thermophoresis demonstrated micromolar and nanomolar binding affinities between recombinant CRMP2 and synthetic L1 and Ct-dis peptides, respectively. Co-immunoprecipitation experiments showed that CRMP2 association with CaV2.2 was inhibited by L1 and Ct-dis peptides. L1 and Ct-dis, rendered cell-penetrant by fusion with the protein transduction domain of the human immunodeficiency virus TAT protein, were tested in in vitro and in vivo experiments. Depolarization-induced calcium influx in dorsal root ganglion (DRG) neurons was inhibited by both peptides. Ct-dis, but not L1, peptide inhibited depolarization-stimulated release of the neuropeptide transmitter calcitonin gene-related peptide in mouse DRG neurons. Similar results were obtained in DRGs from mice with a heterozygous mutation of Nf1 linked to neurofibromatosis type 1. Ct-dis peptide, administered intraperitoneally, exhibited antinociception in a zalcitabine (2′-3′-dideoxycytidine) model of AIDS therapy-induced and tibial nerve injury-related peripheral neuropathy. This study suggests that CaV peptides, by perturbing interactions with the neuromodulator CRMP2, contribute to suppression of neuronal hypersensitivity and nociception.
Introduction
The N-type voltage-gated calcium channel (CaV2.2)5 has recently gained immense popularity as one of the key factors in the ascending pain pathway (see reviews by Zamponi et al. (1) and Snutch (2)). As such, regulation of CaV2.2 expression and function is posited to have a major impact on the presentation of multiple pain states. Indeed, inhibition of CaV2.2 by synthetic conopeptides provides analgesic relief in a variety of platforms (3–6). However, given the importance of CaV2.2 integrity in peripheral and central synapses, directly targeting channel function is complicated by a myriad of adverse side effects (7–9). Targeting protein-protein interactions that regulate CaV2.2 may provide analgesic benefits similar to those provided by direct inhibition, while avoiding complications associated with channel block. We recently demonstrated that the interaction between CaV2.2 and collapsin response mediator protein 2 (CRMP2) (10), a positive regulator of channel surface expression, could be disrupted by a 15-aa peptide derived from the C terminus of CRMP2 (TAT CBD3). Interfering with this interaction efficiently reduced pain behaviors associated with a variety of rodent models of chronic neuropathic/inflammatory pain (11–13). Despite achieving similar levels of analgesic relief, TAT CBD3 treatment did not result in the adverse side effects observed with direct channel inhibition. Here we demonstrate similar effects of targeting the reciprocal interface of the interaction using peptides derived from channel domains demonstrated to coordinate CRMP2 binding.
The use of calcium channel peptides as decoys to disrupt binding of regulatory proteins has previously been demonstrated using the II-III cytoplasmic loop (14) and the α interaction domain of CaV2.2 (15). Intracellular injection of a peptide consisting of the II-III loop, containing the synprint interaction site, prevented association of CaV2.2 with the synaptic core complex, reducing synaptic transmission (15). Peptides containing the α interaction domain of CaV2.2 prevented G-protein-mediated inhibition of channel function by disrupting binding of the Gβγ subunit to the channel (14). The success of these studies in altering channel function and neurotransmitter release validates the use of such peptides as both research tools and potential therapeutics. In this study, we demonstrate that 15 amino acid peptides derived from the I-II cytoplasmic loop (L1) and the distal C terminus (Ct-dis) of CaV2.2 and CaV1.2, respectively, effectively disrupt the interaction between CRMP2 and CaV2.2, reducing calcium influx. Importantly, systemic administration of Ct-dis peptide transiently reversed mechanical hypersensitivity associated with HIV retroviral treatment-induced painful peripheral neuropathy and a model of neuropathic pain involving tibial nerve injury.
EXPERIMENTAL PROCEDURES
Materials
TAT control (YGRKKRRQRRRWEAKEMLYFEALVIE; TAT sequence denoted in underlined text), a random sequence with no homology to any known sequence; TAT L1 (YGRKKRRQRRRYLEWIFKAEEVMLAE), and TAT Ct-dis (YGRKKRRQRRRNSSFPSIHCSSSWSEE) were synthesized and HPLC-purified by Antagene Inc. (Sunnyvale, CA). An N-terminal fluorescein isothiocyanate (FITC)-conjugated version of TAT Ct-dis was purchased from Genscript (Piscataway, NJ). All chemicals, unless noted were purchased from Sigma. Lipofectamine 2000 was purchased from Invitrogen. Fura-2/AM was obtained from Teflabs (Austin, TX). Antibodies were purchased as follows: anti-CRMP2 polyclonal antibody, anti-syntaxin monoclonal antibody, mouse anti-FLAG M2 monoclonal antibody (all from Sigma), anti-CaV1.2 monoclonal antibody (University of California Davis Neuromab, Davis, CA), and anti-CaV2.2 polyclonal antibody (Origene Technologies, Inc., Rockville, MD). The CRMP2-His-pET28B cDNA was provided by Dr. Rihe Liu (University of North Carolina), and the CRMP2–3xFLAG construct was provided by Dr. Akihiro Kurimasa (Tottori University, Tottori, Japan).
Synthesis and Blotting of SPOT Membranes
SPOTs blots (16) encompassing the first intracellular loops and distal third of the C termini of N-, P/Q-, R-, and L-type calcium channels were synthesized, using standard 9-fluorenylmethoxycarbonyl (Fmoc) chemistry, in 30 × 20 spot arrays using a Multipep peptide synthesizer adapted for SPOT synthesis (Intavis AG, Cologne, Germany). Membranes were blocked for at least 1 h in Tris-buffered saline containing 0.5% Tween 20 (TBST) with 5% skim milk powder before incubation for 1 h with a purified rat brain synaptosome fraction at room temperature with gentle shaking. Following a series of brief washes in TBST, the blot was probed overnight with a polyclonal CRMP2 antibody at 4 °C. The following day, blots were washed three times for 10 min each time in TBST, incubated in secondary antibody (horseradish peroxidase-conjugated goat anti-rabbit; 1:10,000) for 45 min at room temperature, and washed for 30 min in TBST three more times for 10 min each time before visualizing SPOTs by exposing the membranes to enhanced chemiluminescence reagent.
Construction of Glutathione S-Transferase (GST) Fusion Proteins
Oligonucleotides for the distal third of the C terminus of CaV1.2 (nucleotides 5961–6695 of rat sequence, GenBankTM accession number NM_012517) were purchased from Sigma. This Ct-dis region was amplified from cDNA prepared from post-natal day 1 rat brain and cloned into the BamHI/MfeI-cut pGex-3x-Glu vector. The distal portion of the C termini of CaV2.3 (α1E, GI:14578562) containing nucleotides 6251–6892 was cloned into pGex-3x-Glu vector. The channel fragment was amplified by PCR from rat brain cDNA and inserted using the restriction enzymes BglII/EcoRI. The final product was verified by DNA sequencing and expressed in BL21(DE3)pLysE cells (Bioline, Taunton, MA). Expression of GST-Glu fusion proteins was performed as described previously (10, 11). Construction of the CaV2.2 Ct-dis-pGex-3x-Glu vector has been described previously (10).
Purification of CRMP2-His and CaV1.2 Ct-dis- and CaV2.2 Ct-dis-GST Fusion Proteins
GST fusion proteins were purified from BL21 (DE3) Escherichia coli bacterial lysates as described previously (10, 11), whereas CRMP2-His protein was purified as before (17) with the following modifications (see Fig. 3A). Expression of CRMP2-His was induced with 1 mm isopropyl-β-d-thiogalactopyranoside. For purification, following overnight growth at 16 °C, transformed bacteria were pelleted at 5,000 × g for 20 min at 4 °C and lysed in His lysis buffer (50 mm HEPES, pH 7.4, 150 mm NaCl) using an M-110L Microfluidizer fluid processor (Microfluidics Corp., Newton, MA). The lysate was then clarified by spinning at 30,000 × g for 45 min at 4 °C before being filtered through a 0.45-μm syringe filter. The filtered supernatant was loaded onto a TalonTM metal affinity resin (Clontech, Mountain View, CA) column pre-equilibrated with His wash buffer (50 mm HEPES, pH 7.4, 150 mm NaCl, 10 mm imidazole). The column was washed with 2× column volumes of His wash buffer, and His proteins were eluted with His elution buffer (50 mm HEPES pH 7.4, 150 mm NaCl, 100 mm imidazole). Elution fractions were separated by SDS-PAGE, and fractions containing CRMP2 were combined. The combined elution fractions were then dialyzed against protein storage buffer (10 mm HEPES, pH 7.4, 100 mm NaCl, and 20 mm CaCl2). Purified protein concentration was determined from densitometric scans of protein bands resolved on a SDS-polyacrylamide gel stained by colloidal Coomassie Blue (Pierce) against a bovine serum albumin standard curve. Additionally, protein identity was verified by immunoblotting (data not shown).
FIGURE 3.

CRMP2 binding to L1 and Ct-dis peptides in solution measured by MST. A, grayscale image of Coomassie-stained gel of CRMP2-His purification. CRMP2-His (in the vector pET28B-CRMP2-His) was purified as described under “Experimental Procedures.” In addition to the elution fractions, the flow-through from loading of the lysate along with one of the wash fractions was loaded on the SDS-PAGE. The fractions with the highest concentration of CRMP2-His (fractions 2–5), as determined by a visual comparison with intensity of bands of the BenchMarkTM protein ladder (Invitrogen), were pooled and then dialyzed into an imidazole-free buffer (10 mm HEPES, pH 7.4, 100 mm M NaCl, 20 mm CaCl2). The final dialyzed protein was quantified by SDS-PAGE by comparison with a bovine serum albumin protein standard curve. The final concentration of CRMP2-His was found to be ∼33 μm, and aliquots were flash-frozen on dry ice and stored at −80 °C. That the purified protein was indeed CRMP2 was verified by Western blot analyses with two different CRMP2 antibodies (Sigma and Immunobiological Laboratories (Minneapolis, MN)) and further in a functional assay in which it was found to enhance tubulin polymerization. B, MST assay principle. A standard capillary containing NT647-labeled CRMP2 is locally heated by a IR laser (IR on). CRMP2 diffuses away from the heated spot, causing a local depletion and drop in fluorescence. Peptide binding changes the thermophoretic properties of CRMP2, resulting in a decreased thermodiffusion. C and D, MST time traces of 10–13 different peptide concentrations (ranging from 0 to 5 μm). Thermodiffusion is reduced with increasing peptide concentrations. MST was performed in order to determine the dissociation constant of L1 (E) and Ct-dis (F) to fluorescently labeled CRMP2. 10 μm NT-647-labeled CRMP2 was mixed with increasing peptide concentrations. The normalized fluorescence is plotted for different concentrations of L1 and Ct-dis peptides. A Kd of 3.12 ± 0.31 μm (L1) and 0.64 ± 0.10 μm (Ct-dis) was determined for the interaction to CRMP2. A representative range of data points obtained from at least two measurements is shown.
Microscale Thermophoresis (MST) Binding Analyses
MST, the directed movement of molecules in optically generated microscopic temperature gradients, permits an immobilization-free fluorescence methodology for the analysis of interaction of biomolecules (18, 19). This thermophoretic movement is determined by the entropy of the hydration shell around molecules. The microscopic temperature gradient is generated by an infrared laser. In a typical MST experiment, the concentration of the labeled molecule is kept constant while the concentration of the unlabeled interaction partner is varied. A constant concentration of dye NT647-labeled CRMP2 (labeled protein concentration of 20 μm) was incubated for 10 min at room temperature in the dark with different concentrations of L1 or Ct-dis peptides (up to 5000 nm) in 20 mm Tris, 150 mm NaCl, 0.01 mm EDTA with 0.01% Tween 20. Immediately afterward, 3–5 μl of the samples were loaded into standard glass capillaries (Monolith NT Capillaries, NanoTemper), and the thermophoresis analysis was performed on a NanoTemper Monolith NT.115 instrument (40% LED, 40% IR laser power). The MST curves were fitted with a Hill method using Origin version 8.5 software to obtain Kd values for binding between CRMP2 and peptides.
Catecholamine A-differentiated (CAD) Cell Culture
Transfection of CAD Cells
CAD cells were transfected using polyethyleneimine (PEI) as previously described (22). Briefly, cells were transfected at ∼60% confluence using an experimentally optimized ratio of DNA/PEI. DNA and PEI were mixed and allowed to incubate for 5 min at room temperature prior to the addition to cells.
Generation of CAD Cell, Rat Brain, and Dorsal Root Ganglion (DRG) Lysates, Immunoprecipitation, and Immunoblotting
CAD cells were lysed 2 days following transfection using a modified radioimmune precipitation assay buffer (50 mm Tris-HC1, pH 8.0, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 1 mm EDTA supplemented with fresh protease inhibitors) with gentle agitation at 4 °C for 10 min. Samples were then clarified by centrifugation at 10,000 × g for 20 min. The supernatant was saved and used for subsequent immunoprecipitations and immunoblotting as described previously (10, 11, 23).
To assess if CaV peptides can interfere with the association between CRMP2 and calcium channels, lysate from ∼40–60 ganglia from adult rats, devoid of nerve roots, were lysed in modified radioimmune precipitation assay buffer and subjected to immunoprecipitation with anti-CaV2.2 antibody in the absence or presence of control peptide, TAT L1, and TAT Ct-dis peptides (10 μm each added 30 min prior to the addition of antibody). Post-natal day 2 rat brains were homogenized in radioimmune precipitation assay buffer (without detergents) by sonication and then clarified by centrifugation to remove insoluble material. Immunoblotting was performed as described (11, 24, 25).
Primary DRG Neuronal Cultures
Isolation of sensory neurons from 1–2-month-old wild type C57BL/6J and Nf1+/− C57BL/6J littermates (26) was performed exactly as described previously (11, 27, 28), whereas DRGs from adult rats were prepared as per Brittain et al. (11). All animals were housed and bred and had free access to food and water in the Indiana University Laboratory Animal Research Center and were used in procedures approved by the Animal Use and Care Committee of the Indiana University School of Medicine. All animals were genotyped prior to use in experiments.
Calcium Imaging
DRG neurons were loaded at 37 °C with 2.6 μm Fura-2/AM (Kd = 25 μm, λex 340, 380 nm/λem 512 nm) to follow changes in [Ca2+]c in a standard bath solution containing 139 mm NaCl, 3 mm KCl, 0.8 mm MgCl2, 1.8 mm CaCl2, 10 mm NaHEPES, pH 7.4, 5 mm glucose. Fluorescence imaging was performed with an inverted microscope, Nikon Eclipse TE2000-U, using a Nikon Super Fluor ×20 objective, 0.75 numerical aperture and a Photometrics cooled CCD camera CoolSNAPHQ (Roper Scientific, Tucson, AZ) controlled by MetaFluor version 6.3 software (Molecular Devices, Downingtown, PA). The excitation light was delivered by a Lambda-LS system (Sutter Instruments, Novato, CA). The excitation filters (340 ± 5 and 380 ± 7 nm) were controlled by a Lambda 10-2 optical filter change (Sutter Instruments). Fluorescence was recorded through a 505-nm dichroic mirror at 535 ± 25 nm. To minimize photobleaching and phototoxicity, the images were taken every 15 s during the time course of the experiment using the minimal exposure time that provided acceptable image quality. The changes in [Ca2+]c were monitored by following a ratio of F340/F380, calculated after subtracting the background from both channels.
Stimulated Release of Immunoreactive Calcitonin Gene-related Peptide (iCGRP)
Measurement of stimulus-evoked release and content of iCGRP from isolated sensory neurons was accomplished as described previously (11, 24, 27). After 5–7 days in culture, medium was removed, and the basal or resting release of iCGRP was measured from cells incubated for 10 min in HEPES buffer consisting of 25 mm HEPES, 135 mm NaCl, 3.5 mm KCl, 2.5 mm CaCl2, 1 mm MgCl2, 3.3 mm dextrose, and 0.1% (w/v) bovine serum albumin, pH 7.4, and maintained at 37 °C. The cells were incubated in HEPES buffer containing stimulus (50 mm KCl) for 10 min and then incubated again with HEPES buffer with 3.5 mm KCl to re-establish resting release levels. The amount of iCGRP released in each incubation was measured by a radioimmunoassay. The minimum amount of iCGRP detected by the radioimmunoassay is 5 fmol with a 95% confidence interval (29). After the release protocol, the remaining peptide content in each well was determined by exposing the cells to 2 n acetic acid for 10 min. Aliquots of acid solution were diluted in HEPES, and iCGRP levels were determined by radioimmunoassay. The release of iCGRP during the 10-min incubation period is expressed as a percentage of the total content. A minimum of three different preparations were used for each condition.
2′,3′-Dideoxycytidine (ddC) Model of Painful Peripheral Neuropathy
Mechanical hypersensitivity was established by a single injection (50 mg/kg) of the antiretroviral drug ddC (Sigma) given intraperitoneally in 150–200-g Sprague-Dawley female rats. A single administration of ddC produced a significant bilateral decrease in paw withdrawal threshold to von Frey hair stimulation from postinjection day 3 through 21, the last day of testing.
The von Frey test was performed on six positions spaced across the glabrous side of the hind paw; two distinct locations for the distribution of each nerve branch (saphenous, tibial, and sural) exactly as described previously to determine paw withdrawal threshold (PWT) to tactile stimuli (11, 30). Base-line threshold measurements were performed for 3 successive days prior to intraperitoneal injection of ddC. Stimuli were applied randomly to left and right hind paws to determine the stimulus intensity threshold stiffness required to elicit a paw withdrawal response. The incidence of foot withdrawal was expressed as a percentage of six applications of each filament as a function of force. A Hill equation was fitted to the function (Origin version 6.0, Microcal Software) relating the percentage of indentations eliciting a withdrawal to the force of indentation. From this equation, the threshold force was obtained and defined as the force corresponding to a 50% withdrawal rate. A threshold that exhibits at least a −20 millinewton difference from the base-line threshold of testing in a given animal is representative of neuropathic pain (11, 31).
Tibial Nerve Injury (TNI) Model of Peripheral Neuropathy
This procedure was completed as described previously (32). Under isoflurane (2%) anesthesia, the skin on the lateral surface of the thigh was incised, and a section was made directly through the biceps femoris muscle, exposing the sciatic nerve and its three terminal branches: the sural, common peroneal, and tibial nerves. The surgical procedure comprised an axotomy and ligation of the tibial nerve, leaving the common peroneal and sural nerves intact. The tibial nerve was tightly ligated with 5.0 silk and sectioned distal to the ligation, removing 2–4 mm of the distal nerve stump. An effort was made to avoid contact with or stretching of the intact common peroneal and sural nerves. Muscle and skin were closed in two layers. Sham controls involved exposure of the sciatic nerve and its branches without any lesion.
Assessment of Distribution of FITC-conjugated Ct-dis in Tissue Sections of Naive and ddC-treated Rats
Adult female Sprague-Dawley rats were euthanized with CO2 and transcardially perfused with saline followed by 4% paraformaldehyde. Lumbar ganglia 5 and 6 from behaviorally tested ddC-treated rats (n = 2) were immediately removed and postfixed for 4 h. Additional lumbar DRGs were removed from naive, behaviorally tested rats (n = 2). Sagittal sections of the DRG were serially cut at 14 μm onto SuperFrost Plus microscope slides (Fisher) and counterstained with Hoescht nuclear stain (1:1000; Sigma). At least 6 sections/DRG were obtained for analysis. Tissue was processed such that DRG sections on each slide were at intervals of 80 μm. Images of DRG were taken with an intensified CCD camera (Photometrics CoolSnap HQ2) coupled to a Nikon microscope (Nikon Eclipse Ti) using Nikon Elements software (Nikon Instruments Inc., Melville, NY). Tissue sections were illuminated with a Lambda DG-4 175-watt xenon lamp (Sutter Instruments), and images were captured using the same exposure for all tissue. Raw images are presented without any manipulation.
Statistical Analyses
Differences between means were compared by either paired or unpaired two-tailed Student's t tests. Transmitter release measurements, represented as a percentage of the total content of iCGRP, are expressed as the means ± S.E. All differences in iCGRP release and total content were compared with analyses of variance (ANOVAs) and Dunnett's post hoc analysis or Student's t tests, as indicated. Behavioral threshold values were statistically analyzed for each foot separately, and the significance of differences between the average of at least two preinjection tests and the mean was obtained for each postinjection test. In all tests, base-line data were obtained for the ddC-treated and sham-treated groups before drug or vehicle administration. Within each treatment group, postadministration means were compared with the base-line values by repeated measures ANOVAs followed by post hoc pairwise comparisons (Student-Newman-Keuls method). A p value of <0.05 was used to indicate statistical significance between treatment and non-treatment groups.
RESULTS
Identification of CRMP2 Binding Peptides within CaV
We recently identified CRMP2 as a novel binding partner of CaV2.2 (10). A short peptide derived from CRMP2 (CBD3) could disrupt the CaV2.2-CRMP2 interaction, leading to a physiologically relevant decrease in Ca2+ current and neurotransmitter release and, in turn, suppression of persistent inflammatory and neuropathic hypersensitivity (11). Molecular mapping revealed direct binding between CRMP2 and two intracellular regions on CaV2.2, the first intracellular loop (L1; aa 356–483; Fig. 1A) and the distal third of the C terminus (Ct-dis; aa 2119–2336; see Fig. 2A) (10). Here, we asked whether short peptides from the L1 or Ct-dis regions of the channel could disrupt the interaction and affect Ca2+ signaling, transmitter release, and behavioral hypersensitivity. In order to identify shorter regions within these large L1 (∼128 aa) and Ct-dis (∼217 aa) regions that coordinated binding to CRMP2, we created a peptide array harboring 15-mer peptides, with an overlap of 10 aa, from the L1 and Ct-dis regions of CaV2.2. Because of significant (∼60%) homology of this region to CaV2.1 (P/Q-type), CaV2.3 (R-type), and CaV1.2 (L-type), the L1 regions of these channels were also tiled. Although multiple sequence alignment revealed lesser (<30%) homology between Ct-dis regions of these channels, these regions were also tiled for completion. These spots, along with spots harboring peptides against GST (negative control) were then probed in a far Western manner with a CRMP2 antibody (Fig. 1B). Quantification of the intensity of the CRMP2 immunoreactivity on the spots identified several L1 and Ct-dis peptides from all four Ca2+ channel isoforms (Figs. 1, B and C, and 2C). For L1 peptides, CRMP2 binding clustered around the ninth spot harboring a sequence that was relatively well conserved across the four Ca2+ channel isoforms (Fig. 1, D and E). In contrast, Ct-dis peptides bound CRMP2 at various spots spread throughout the sequence of the four Ca2+ channel isoforms (Fig. 2B). However, despite little homology between the 24th Ct-dis peptide (aa 2014–2028 of L-type; Fig. 2C) and the other three Ca2+ channel isoforms, the corresponding regions in the other three Ca2+ channel isoforms around this peptide displayed relatively high binding to CRMP2.
FIGURE 1.

Identification of first intracellular loop CaV2.2 peptides that bind CRMP2. A, schematic representation of the topological organization of the Ca2+ channel. The first intracellular loop (L1) is highlighted in green. The amino acids (CaV2.2 rat sequence) used for the tiling array are indicated. B, image of CRMP2 bound to an immobilized 15-mer peptide array of the L1 region of CaV2.2. C, matrix representation of normalized fluorescent intensity of CRMP2 binding to immobilized 15-mer L1 peptides of N (CaV2.2)-, P/Q (CaV2.1)-, R (CaV2.3)-, and L (CaV1.2)-type calcium channels. The matrix was generated with Matrix2png software (61). Cyan rectangles represent spots without any peptides. D, amino acid alignment of the L1 peptide (ninth peptide indicated by arrow in C or purple in D) from CaV2.2 that exhibited the highest CRMP2 binding. Asterisks indicate residues that are fully conserved between the channel subtypes. The italicized numbers refer to amino acid residues in CaV2.2 (GenBankTM accession number NM_147141.1, translated from messenger RNA). The amino acid residues and accession numbers for the other sequences are as follows: CaV2.1, aa 392–410 (GenBankTM number NM_012918.2); CaV2.3, aa 381–399 (GenBankTM number NM_019294.2); and CaV1.2, aa 465–483 (GenBankTM number NM_012517.2). E, superposition of the CaV1.2 (purple) and CaV2.2 (green) I-II linker fragments (62). The location of the CaV L1 peptide is indicated in orange. The α interaction domain is denoted by the bracket.
FIGURE 2.

Identification of C terminus CaV peptides that bind CRMP2. A, schematic representation of the topological organization of the Ca2+ channel. The distal third of the C terminus (Ct-dis) is highlighted in brown. The amino acids (CaV1.2 rat sequence) used for the tiling array are indicated. B, matrix representation of normalized fluorescent intensity of CRMP2 binding to immobilized 15-mer L1 peptides of N (CaV2.2)-, P/Q (CaV2.1)-, R(CaV2.3)-, and L (CaV1.2)-type calcium channels. Cyan rectangles represent spots without any peptides. C, amino acid alignment of the Ct-dis peptide (24th peptide indicated by an arrow in B or purple in C) from CaV1.2 that exhibited the highest binding to CRMP2. The italicized numbers refer to amino acid residues in CaV1.2. The amino acid residues (accession numbers provided in Fig. 1) for the other sequences are as follows: CaV2.2, aa 2194–2216; CaV2.1, aa 2248–2267; and CaV2.3, aa 2151–2177.
Biophysical Characterization of CRMP2 Binding to L1 and Ct-dis Peptides
Using in vitro binding assays, we had previously demonstrated that both full-length L1 and the Ct-dis fragments bind CRMP2; Ct-dis bound CRMP2 ∼75-fold tighter than L1 (10). Here, we asked if short 15-mer peptides from these regions, L1(388–402) and Ct-dis(2014–2028), bound to CRMP2 using a new technique called MST (18, 33). Using an infrared laser, precise microscopic temperature gradients are generated within thin glass capillaries filled with a fluorescently labeled protein sample in a buffer, and the atomistic movement of molecules along these temperature gradients is monitored in the presence of increasing concentrations of an unlabeled binding partner (Fig. 3B). Changes in fluorescence intensity in the capillaries are used to calculate binding affinity between biomolecules. MST measurements were made on NT647-labeled CRMP2 in the presence of increasing amounts of L1 or Ct-dis peptides. As the concentration of the peptides increased, they bound to CRMP2 thermodiffusing out of the heated infrared spot, resulting in a decrease in the MST signal and providing a readout of the binding between the CRMP2 and the peptides (Fig. 3, C and D). We fitted the binding curves with the Hill method to obtain Kd values for binding between CRMP2 and peptides. L1 peptide bound to CRMP2 with a Kd of 3.12 ± 0.31 μm, whereas the Ct-dis peptide bound to CRMP2 with a Kd of 0.64 ± 0.10 μm (Fig. 3, E and F). Additional experiments using isothermal titration calorimetry revealed a similar 5–7-fold difference in binding affinities between L1 and Ct-dis binding to CRMP2 (results not shown). These results demonstrate that 15-mer peptides derived from CaV indeed bind CRMP2.
L1 and Ct-dis Peptides Block Interaction between CaV2.2 and CRMP2
Immunoprecipitation from rat dorsal root ganglia with CaV2.2 antibody in the presence of L1 or Ct-dis (10 μm) peptides reduced the amount of CRMP2 but not the prototypical CaV2.2-binding protein, syntaxin (34), that was recovered (Fig. 4). These results suggest that L1 and Ct-dis peptides are sufficient to uncouple the interaction between CaV2.2 and CRMP2.
FIGURE 4.

CaV2.2 L1 and Ct-dis peptides prevent association of CaV2.2 and CRMP2. Immunoprecipitation (IP) with CaV2.2 antibody in the presence of control (ctrl), TAT L1 (L1), or TAT Ct-dis (Ct-dis) peptides reduced the amount of CRMP2 (top) but not syntaxin (bottom) that could be captured from rat DRGs. Peptides (10 μm each) were added for 30 min prior to the addition of the antibodies. A representative blot from three separate experiments is shown. Similar results were obtained in experiments with rat brain lysates.
Although the Ct-dis peptide was able to bind and inhibit the interaction between CaV2.2 and CRMP2 (Figs. 2–4), it is derived from the L-type (i.e. CaV1.2) channel raising the possibility that this Ca2+ channel isoform may bind to CRMP2. To test this possibility, we transfected a neuronal CAD cell line that endogenously expresses L-type channels (Fig. 5A, top left blot) with FLAG-tagged CRMP2 and tested if the proteins could form a complex. Precipitation of CRMP2 (with a FLAG antibody) from lysates of these cells co-precipitated CaV1.2 (Fig. 5A, top right blot). As negative controls, lysates immunoprecipitated with FLAG antibody from cells expressing FLAG-tagged Nipsnap1/2 proteins, which we had previously shown to regulate but not associate with CaV1.2 (22), did not capture CaV1.2.
FIGURE 5.

CRMP2 exists in a biochemical complex with CaV1.2 and CaV2.3. A, CAD cells were transfected with cDNAs encoding Nipsnap-1-, Nipsnap-2-, or CRMP2–3xFLAG. We recently reported a novel role of Nipsnap2 in transcriptional regulation via L-type Ca2+ channels (22). Lysates from these cells were immunoprecipitated (IP w/) with an anti-FLAG antibody. CaV1.2 was detected in CAD lysates expressing CRMP2 (top right blot) but not Nipsnap1 or -2, as reported previously (22). CAD cells express endogenous CaV1.2 (input lanes, top left blot). All FLAG-tagged constructs expressed detectable levels of the fusion protein (input lanes, bottom left blot). B, pull-downs with recombinant CaV1.2- or CaV2.3-Ct-dis-GST-Glu proteins captured CRMP2 protein from post-natal day 2 rat brain lysates (two replicates shown). The input lane was loaded with 3% of the starting material used for the pull-downs, and the adjacent lane contained the protein marker used to determine approximate protein size. Control pull-downs with GST were negative for CRMP2. The blot probed for CRMP2 was subsequently stained for total protein using Amido Black as displayed in the lower blot. Molecular mass markers are indicated in kDa. Representative blots from 2–3 experiments are shown. WB, Western blot.
To examine if the Ct-dis region of CaV1.2 interacts with CRMP2, we cloned and purified bacterially expressed GST fusion CaV1.2 and CaV2.3 Ct-dis and incubated them with post-natal day 2 rat brain lysates. Then in vitro complexes were recovered with glutathione-Sepharose beads, washed extensively, and immunoblotted with CRMP2. Both CaV1.2 and CaV2.3 Ct-dis proteins bound to CRMP2 (Fig. 5B). GST protein alone did not recover CRMP2 (Fig. 5B). CaV2.2 Ct-dis also bound to CRMP2 (data not shown) as reported previously (10). Collectively, these biochemical results demonstrate that Ct-dis peptide is capable of binding CRMP2 and inhibiting the interaction between CaV2.2 and CRMP2.
L1 and Ct-dis Peptides Affect K+-stimulated Ca2+ Influx
Our previous results demonstrated that uncoupling the CaV2.2-CRMP2 interaction with a peptide derived from CRMP2 resulted in reduction of depolarization-induced Ca2+ influx in sensory neurons (11). Here, we asked whether interfering with the CaV2.2-CRMP2 complex with a peptide derived from the opposite interface can achieve the same result. Calcium imaging experiments performed with Fura-2/AM on adult rat DRG neurons showed that stimulation with high K+ (46.5 mm) produced a transient increase of [Ca2+]c in response to plasma membrane depolarization (Fig. 6, A–C). Peak calcium influx was recorded within 20 s of stimulation (Fig. 6, A–C). DRGs incubated for 20 min with L1 or Ct-dis peptides showed a concentration-dependent decrease in K+-stimulated [Ca2+]c influx (Fig. 6D). At 30 μm, peptide-mediated inhibition of [Ca2+]c influx was much more pronounced for Ct-dis than L1, with a control TAT peptide having no effect (Fig. 6D). These results show that L1 and Ct-dis peptides affect Ca2+ influx.
FIGURE 6.

CaV peptides affect K+-stimulated Ca2+ influx in DRG neurons. Shown are differential interference contrast (DIC) (A) and pseudocolored fluorescent images of a field of DRG neurons visualized for Fura-2/AM, before (B) and after stimulation with KCl (Hi K+) (C). Ca2+ imaging was performed on adult rat DRG neurons using the ratiometric Ca2+-sensitive dye Fura-2/AM. Following a 1-min base-line measurement, neurons were stimulated with 46.5 mm KCl for 30–40 s to induce Ca2+ influx. Arrowhead indicates the Ca2+ influx in a sensory neuron. D, bar graph shows the peak fluorescence response (adjusted for background) of DRGs incubated for 20 min with 30 μm TAT alone or 3, 10, or 30 μm TaT L1 or TAT Ct-dis peptides compared with untreated DRGs. Values represent the average ± S.E. (error bars) from three separate imaging experiments, with the number of cells per condition indicated in parentheses. Asterisks indicate statistical significance compared with untreated cells (p < 0.05, one-way ANOVA followed by Dunnett's post hoc test).
Ct-dis but Not L1 Peptide Affects K+-evoked Transmitter Release from Isolated Sensory Neurons
We previously demonstrated that CRMP2 expression levels could affect release of the neuropeptide transmitter CGRP from sensory neurons (24). We further demonstrated that disrupting the CaV2.2-CRMP2 interaction with the CBD3 peptide suppressed evoked CGRP release in sensory neurons in culture as well as spinal cord slices (11), highlighting the importance of this interaction for transmitter release. Here, we asked if peptides derived from the reciprocal interface (i.e. the channel itself) could affect release. The KCl-stimulated release of CGRP was measured from sensory neurons exposed to TAT control, TAT L1, or TAT Ct-dis peptides, at 10 μm, included in the 10 min prior to and throughout the high K+ exposures (total peptide exposure of 30 min). The levels of basal or resting release of iCGRP were not significantly different between the groups: 1.53 ± 0.10% total peptide content/10 min (n = 12 wells) in untreated control neurons, 1.58 ± 0.17% total peptide content/10 min (n = 12 wells) in TAT control peptide-treated neurons, 1.04 ± 0.09 total peptide content/10 min (n = 12 wells) in TAT L1 peptide-treated neurons, and 0.92 ± 0.09% total peptide content/10 min (n = 16 wells) in TAT control peptide-treated neurons (Fig. 7A). A 10-min stimulation with 50 mm KCl evoked a robust increase (∼12–14-fold over basal) in iCGRP release in untreated, TAT control-treated, and TAT L1-treated neurons. In contrast, neurons exposed to TAT Ct-dis peptide had significantly (∼70%) less iCGRP release compared with TAT control (one-way ANOVA; Fig. 7A). The decrease in KCl-stimulated iCGRP release observed in TAT Ct-dis-treated neurons was not caused by a decrease in the total cellular content of iCGRP because there was no significant difference in neuropeptide content in any of the four conditions (Fig. 7A, n = 12–16 wells). Incubation of DRGs with 30 μm TAT Ct-dis or TAT L1 (n = 12 wells each), a concentration at which both peptides attenuated calcium influx (Fig. 6D), did not provide further impairment of iCGRP release (data not shown).
FIGURE 7.

CaV peptides affect K+-stimulated transmitter release in DRG neurons. Adult mouse DRG neurons were maintained in culture for 5–7 days prior to the release experiments. A, bar graph of iCGRP release expressed as mean percentage of total iCGRP content of cells in each well ± S.E. (error bars) (n = 12–16 wells/condition). Neuropeptide release was measured from cells treated with successive incubations of normal HEPES buffer containing 3.5 mm KCl (basal; B), HEPES buffer containing 50 mm KCl (S), and HEPES buffer containing 3.5 mm KCl, again. The peptides were included in the 10 min prior to and throughout the high K+ exposures (total peptide exposure of 30 min). *, statistically significant differences in iCGRP release between TAT Ct-dis and all other groups using an ANOVA with Dunnett's post hoc test (p < 0.05). In all cases, release stimulated by high extracellular K+ was significantly higher than basal release. B, the total content of iCGRP measured at the end of the release experiment. There were no significant differences in iCGRP content between the conditions tested.
We also tested if the CaV peptides could affect the release of CGRP in sensory neurons isolated from Nf+/− mice, which we have previously shown to have increased N-type calcium currents as well as enhanced transmitter release of glutamate (28) and CGRP (27). Neurofibromin, the protein whose deficiency leads to neurofibromatosis type 1, is a binding partner of CRMP2 (35), and it is not known if disrupting the CRMP2-CaV2.2 interaction could affect CGRP release in these neurons. KCl-stimulated release of CGRP was measured from sensory neurons exposed to 10 μm TAT-conjugated peptides, as indicated for wild type neurons. The levels of basal or resting release of iCGRP were not significantly different between the groups (Fig. 8A). A 10-min stimulation with 50 mm KCl evoked a robust increase (∼8–10-fold over basal) in iCGRP release in untreated and TAT control-treated neurons. In contrast, neurons exposed to TAT Ct-dis peptide had significantly (∼70%) less iCGRP release compared with TAT control (one-way ANOVA; Fig. 8A). The CRMP2 peptide CBD3 also inhibited CGRP release by ∼78% compared with TAT control neurons (Fig. 8A). The decrease in KCl-stimulated iCGRP release observed in TAT Ct-dis- or TAT CBD3-treated neurons was not caused by a decrease in the total cellular content of iCGRP because there was no significant difference in neuropeptide content in any of the four conditions (Fig. 7A, n = 12–16 wells). The KCl-stimulated increase in iCGRP occurred largely via N-type Ca2+ channels because 500 nm ω-CTX inhibited iCGRP release by ∼50% compared with untreated neurons (data not shown). Collectively, these results indicate that disrupting CRMP2-CaV2.2 interactions impact release of the neuropeptide transmitter iCGRP in sensory neurons.
FIGURE 8.
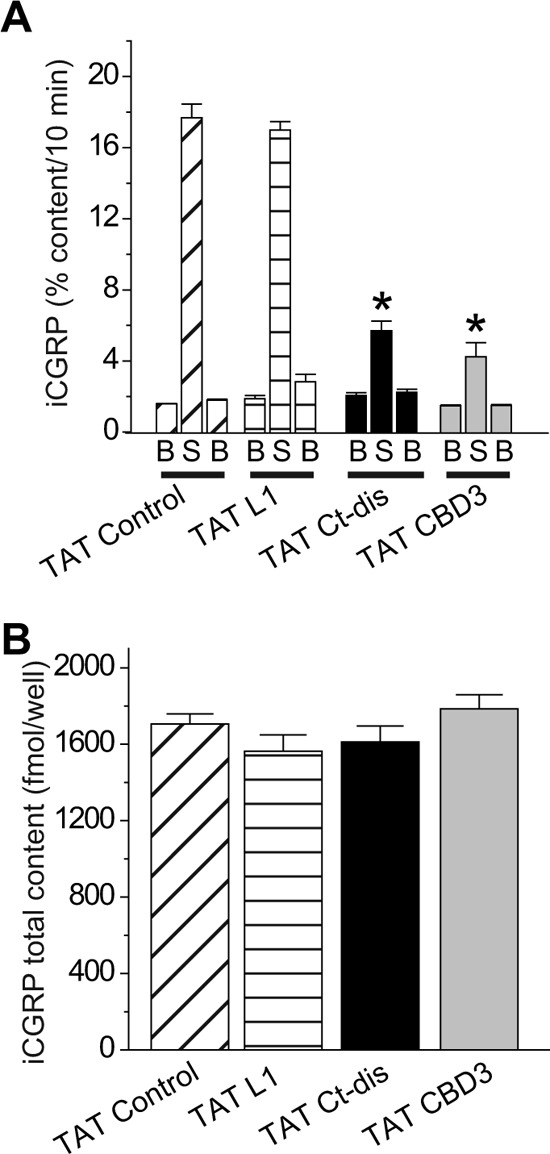
CaV peptides affect K+-stimulated transmitter release in DRG neurons from Nf1+/− mice. Adult Nf1+/− mice DRG neurons were maintained in culture for 5–7 days prior to the release experiments. A, bar graph of iCGRP release expressed as mean percentage of total iCGRP content of cells in each well ± S.E. (error bars) (n = 12 wells/condition). Neuropeptide release was measured from cells exactly as described in the legend to Fig. 7. *, statistically significant differences in iCGRP release between TAT Ct-dis or TAT CBD3 and TAT control using an ANOVA with Dunnett's post hoc test (p < 0.05). B, total content of iCGRP measured at the end of the release experiment. There were no significant differences in iCGRP content between the conditions tested. Error bars, S.E.
Ct-dis Peptide Attenuates AIDS Therapy- and TNI-induced Painful Peripheral Neuropathy
Because the L1 peptide did not affect CGRP (Figs. 7 and 8) or glutamate release (data not shown), we focused on the Ct-dis peptide in this set of experiments. We next examined the effects of the Ct-dis on chronic nociceptive behavior in an animal model of AIDS therapy-induced painful neuropathy (11, 13, 31). Nucleoside reverse transcriptase inhibitors, commonly used for AIDS treatment, produce side effects including painful neuropathies. The ability of peptides to reverse tactile hypersensitivity was evaluated in rats 7 days after a single injection of ddC. TAT Ct-dis produced a time-dependent increase in PWT when administered intraperitoneally (Fig. 9). Almost complete reversal of tactile hypersensitivity was observed at the ∼4-mg/kg dose of TAT Ct-dis 1 h after intraperitoneal injection. Two hours after injection, peptide-induced reversal of hypersensitivity had diminished to prepeptide levels, which may be accounted for by degradation and biodistribution of the peptide. A scramble control peptide (10 mg/kg) or saline controls did not elicit any change in PWT at any time tested following intraperitoneal injection of ddC-injected rats (data not shown). At a higher dose (20 mg/kg), TAT Ct-dis ameliorated pain behavior to a similar level, yet the time scale was slightly altered. Although both doses provided near reversal of pain behaviors at 1 h postinjection, the 20-mg/kg dose was also analgesic at 30 and 90 min postinjection (Fig. 9). The efficacy of TAT Ct-dis was also tested in the TNI model of neuropathic pain. Unlike in the ddC model, injection of 4 mg/kg did not alter tactile hypersensitivity. However, injection of 20 mg/kg was analgesic at 30 and 60 min postinjection and peaked at 90 min, when complete reversal of pain behavior was observed (Fig. 10). Pain behaviors returned to preinjection levels 120 min following treatment.
FIGURE 9.
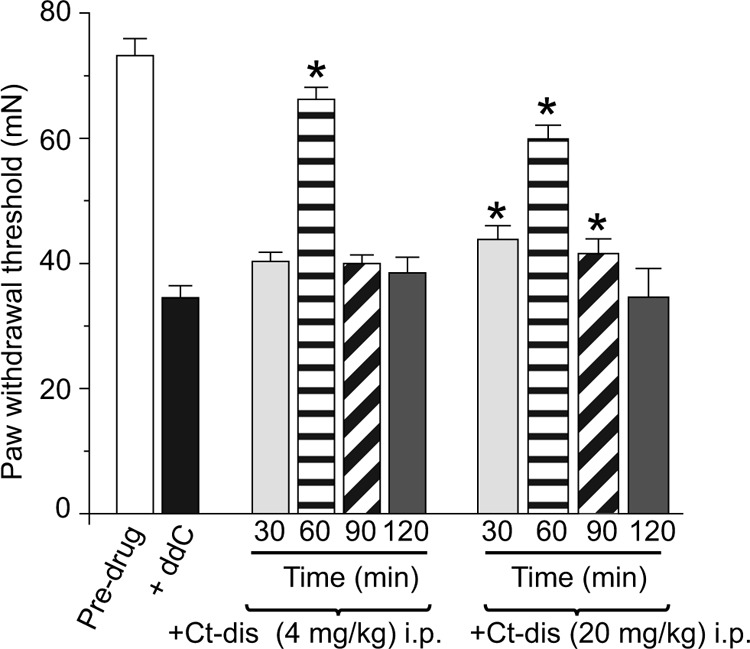
CaV1.2 Ct-dis peptide affects ddC-induced mechanical hypersensitivity. PWTs (in millinewtons; mN) were measured in ddC-treated rodents before and after intraperitoneal (i.p.) administration of the indicated peptides. PWT in rodents subjected to a single injection of ddC was significantly reduced when compared with pre-ddC thresholds (n = 6; black bar). After the induction of neuropathic pain behavior using ddC, TAT Ct-dis (4 or 20 mg/kg body weight) was administered, and bilateral pain behavior was assessed using the von Frey filament test. Behavior was tested at 30, 60, 90, and 120 min. Following administration of TAT Ct-dis, bilateral paw withdrawal thresholds increased to pre-ddC levels (ANOVA with Dunnett's post hoc test; *, p < 0.05). Error bars, S.E.
FIGURE 10.
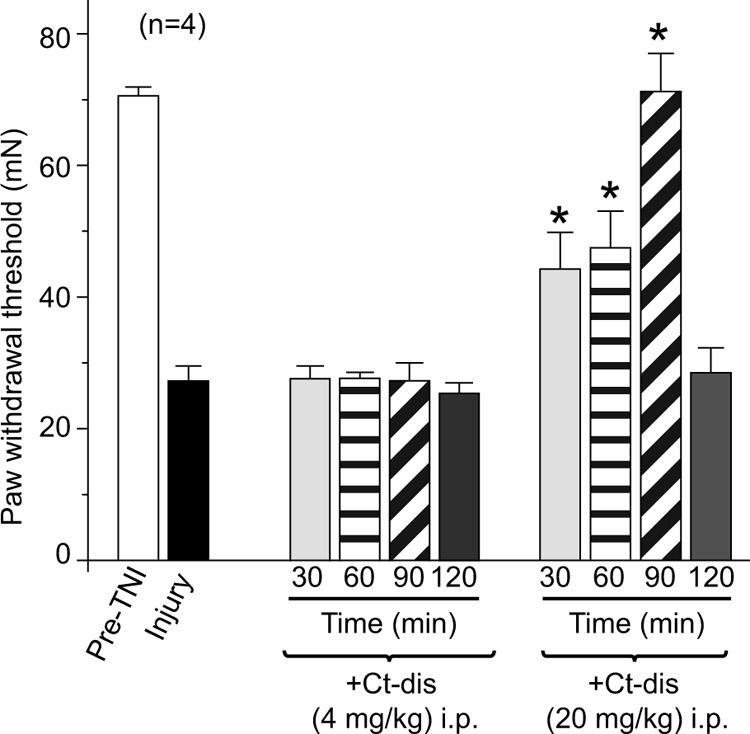
CaV1.2 Ct-dis peptide affects TNI-induced mechanical hypersensitivity. PWTs (in millinewtons; mN) were measured in TNI rodents before and after intraperitoneal administration of the indicated peptides. PWT in TNI rodents was significantly reduced when compared with pre-TNI thresholds (n = 6; black bar). Following intraperitoneal (i.p.) administration of TAT Ct-dis (4 or 20 mg/kg body weight), PWT returned to pre-TNI levels for at least 90 min (ANOVA with Dunnett's post hoc test; *, p < 0.05). Error bars, S.E.
To address the transient effect in the AIDS drug-induced painful peripheral neuropathy, FITC-labeled TAT Ct-dis was injected (20 mg/kg) into naïve and ddC-injured animals. Uptake into the DRG was observed 30 and 60 min postinjection in both naïve and ddC animals (Fig. 11). Similar to what was previously observed with other TAT peptides, uptake appeared relatively restricted to the soma of the DRG (11, 23) because neither nerve fibers in the peripheral nervous system nor other organs, such as the liver, exhibit any uptake (data not shown). Although the extent of DRG labeling was similar at 30 and 60 min, labeling intensity appeared higher in DRG derived from ddC-injured rodents at both time points.
FIGURE 11.

Distribution of FITC-conjugated TAT Ct-dis peptides in DRGs from naive and ddC-treated rats following intraperitoneal injection. Shown are sections of lumbar DRGs from naive (i-vi) or ddC-treated (vii-xii) rodents, the latter having developed mechanical hypersensitivity, 30 and 60 min following injection with FITC-labeled TAT Ct-dis (20 mg/kg intraperitoneally). Nuclei of all cells were stained with Hoechst (blue). Scale bar represents 20 μm.
DISCUSSION
Our findings demonstrate that synthetic peptides derived from the voltage-gated Ca2+ channel can be employed as decoys to interrupt binding of regulatory proteins. These channel regions are known to coordinate interactions between the channel and many other regulatory proteins. The I-II cytoplasmic loop contains interaction sites for CaVβ proteins (36) as well as Gβγ subunits (37), whereas the C terminus also contains interaction sites for CaVβ and Gβγ subunits (38) as well as the calcium/calmodulin-dependent serine protein kinase (39, 40). Importantly, the 15-aa peptides we report here do not overlap with the binding sites for calmodulin or the Gβγ subunits (41, 42). However, there is partial overlap of the L1 peptide with the C-terminal portion of the α interaction domain, which is responsible for binding CaVβ. Here, molecular mapping revealed two distinct regions with direct binding to CRMP2. Two short (15-amino acid) peptides from these regions that exhibited the highest degree of CRMP2-binding were selected. Although both peptides bound CRMP2 with fair affinity, Ct-dis exhibited tighter binding compared with L1. Both Ct-dis and L1 sufficiently interrupted the interaction between CRMP2 and CaV2.2. Uncoupling of the CaV2.2-CRMP2 complex by these peptides resulted in an overall decrease in calcium influx.
Interestingly, TAT Ct-dis reduced release of the neuropeptide CGRP from isolated sensory neurons, whereas TAT L1 did not. The ∼5-fold lower binding affinity of TAT L1 versus TAT Ct-dis to CRMP2 coupled with the ostensibly higher reduction on calcium influx may explain the lack of effect of TAT L1 on release. Thus, a smaller reduction in Ca2+ influx effected by TAT L1 may not translate into a meaningful change in release despite the non-linear relationship between calcium influx and neurotransmitter release (43). This is also consistent with a recent study by Bucci et al. (14), who demonstrated an ∼37% inhibition in excitatory postsynaptic potential amplitude recorded in synaptically coupled superior cervical ganglion neurons using a relatively high (1 mm) concentration of a slightly overlapping (6 of 15 aa) α interaction domain peptide from L1 of CaV2.2. Because it was effective at reducing CGRP release, TAT Ct-dis was chosen for assessment of analgesic properties in an animal model of AIDS therapy-induced peripheral neuropathy. Systemic administration of TAT Ct-dis led to a transient reversal of pain behavior. Unfortunately, the mechanisms for clearance, elimination, and metabolism of TAT-conjugated peptides are not well understood (44). Although these processes may depend largely on the protein cargo (45), total clearance of the TAT motif has been observed 2 h after a bolus tail vein injection (46). Such rapidity of metabolism and clearance may account for the transience of the behavioral effect of TAT Ct-dis. Additionally, intracellular degradation may play an important role. Bioactivity of a TAT-conjugated peptide of similar length (17 aa) was demonstrated to peak around 60 min and rapidly declined shortly thereafter (47). This transience is also consistent with what we observed here and previously (11–13) in animal models of peripheral neuropathy with similar sized peptides.
Injection of a FITC-labeled TAT Ct-dis led to uptake within the DRG of both injured and naïve animals. Although labeling appeared similar at both 30 and 60 min postinjection, uptake into the DRG was increased in injured animals compared with naive DRGs. It is possible that the antiretroviral drug, ddC, may lead to greater permeability of the vascular bed of the DRG due to changes in endothelial cell fenestrations (48), allowing increased penetration of the TAT-conjugated peptide. Alternatively, increased transduction of TAT peptides has been demonstrated following application of a low voltage pulse (47). Because ddC-induced peripheral neuropathy leads to hyperexcitability within the DRG (49), increased activity may allow for increased uptake of TAT Ct-dis.
The efficacy of targeting VGCC regulation in alleviating pain behavior validates the importance of calcium channels in pain signaling. Highly expressed on nociceptive fibers and within the dorsal horn of the spinal cord, CaV2.2 plays a fundamental role in relaying pain signals from the periphery. Alternative splice variants found on small diameter nociceptive neurons are associated with increased thermal and mechanical hyperalgesia (50, 51). CaV2.2 is believed to be responsible for increased neurotransmitter release commonly associated with chronic and neuropathic pain conditions (1, 2, 52, 53). Consistent with the role of CaV2.2 in pain signaling, genetic deletion, as well as pharmacologic block of CaV2.2, impairs nociceptive processing (3, 54). Additionally, central blockade of CaV2.2 is effective in treating some cases of chronic pain that have been intractable to other interventions (8).
Although the N-type channel appears to predominate in neuropathic pain conditions, the L-type channel also appears to play a role in acute and inflammatory pain signaling (55). Because CRMP2 interacts with CaV1.2 as well as CaV2.2, TAT Ct-dis is probably interrupting binding between CRMP2 and both N- and L-type channels. The ability to simultaneously target both interactions may aid in its efficacy. Pharmacologic block of L-type channels reduces acute response to noxious peripheral stimuli (56). Administration of the L-type blocker, nifedipine, into the dorsal horn of the spinal cord prevented secondary thermal and mechanical hyperalgesia following injection of capsaicin into the hind paw (57). The L-type channel also appears to play a role in inflammatory-mediated pain signaling. Spinal block of L-type channels attenuates the enhanced response of primary afferents to noxious and innocuous stimuli during inflammation-induced central sensitization (58). Consequently, intrathecal administration of nifedipine reduces late-phase nociceptive behavior following formalin administration (3). Interestingly, a recent report that attributes the maintenance of chronic neuropathic pain (spinal nerve ligation model) to CaV1.2 (59). Favereaux et al. (60) also reported bidirectional regulation of CaV1.2 by a single microRNA, thus implicating CaV1.2 as a novel possible therapeutic target in neuropathic chronic pain. The complementary roles of these two calcium channels in both peripheral and spinal processing of pain signaling indicate opportunities for therapeutic intervention through control of their regulation. Although the impact of CRMP2 on L-type channels is unknown, the potential ability of CRMP2 to regulate both channels positions it as a prime target for modulation of pain signaling. Subsequently, by binding and sequestering CRMP2, TAT Ct-dis may simultaneously affect N-type and L-type channel function.
Previously, we determined that a peptide derived from CRMP2 could reverse pain behavior associated with chronic inflammatory and neuropathic pain conditions via interruption of the CRMP2-CaV2.2 complex (11–13). Here, we demonstrate that the reciprocal approach of targeting this interaction also has analgesic potential. A channel peptide responsible for CRMP2 binding also reversed pain behavior associated with peripheral neuropathy. There is an advantage to the ability to target the same interaction through multiple strategies. Each approach may present different levels of efficacy and potency, which become important in the search for larger therapeutic windows. Also, separate approaches may allow for individual tailoring of treatment strategies to avoid adverse effects seen as the result of one approach but not the other. Targeting protein interactions that regulate voltage-gated calcium channels as an alternative to direct channel block has multiple advantages. Similar levels of efficacy can be achieved by targeting channel regulation compared with channel function. However, many of the adverse side effects associated with direct channel block may potentially be averted by targeting channel regulation. Here, we demonstrate that protein-protein interactions provide the additional benefit of targeting one interaction from at least two separate avenues.
Acknowledgments
We thank Dr. Ana Lazic (NanoTemper Technologies Inc.) for use of the Monolith NT.115 instrument for analysis of peptide-CRMP2 interactions and Dr. May Khanna (Indiana University School of Medicine (IUSM)) for discussions on the crystal structures of the CaV I-II linkers. An antibody to CGRP was generously provided by Dr. Michael R. Vasko (IUSM) and originally produced by Michael J. Iadarola (National Institutes of Health). The peptide blot was synthesized at the Stark Neurosciences Research Institute core facility.
This work was supported, in whole or in part, by National Institutes of Health (NIH), NCRR, Project Development Team Grant RR025761 in support of the Indiana Clinical and Translational Sciences Institute (to R. K); NIH, NINDS, Grant NS049136-06 (to F. A. W.); and NIH, NIDA, Grant DA026040-04 (to F. A. W.). This work was also supported by a Clinical and Translational Sciences Award from the Indiana State Department of Health Spinal Cord and Brain Injury Fund (A70-9-079138 to R. K.), American Heart Association National Scientist Development Grant SDG5280023 (to R. K.), Department of Defense Congressionally Directed Military Medical Research and Development Program Neurofibromatosis New Investigator Award NF1000099 (to R. K.), a Ralph W. and Grace M. Showalter Foundation grant (to R. K.), Indiana University Biomedical Committee Research Support Funds Grant 2286501 (to R. K), a Research Inventions and Scientific Commercialization grant (to R. K.) from the Indiana Clinical and Translational Sciences Institute, and the Elwert Award in Medicine (to R. K.).
- CaV2.2
- N-type voltage-gated Ca2+ channel
- CaV2.1
- P/Q-type voltage-gated Ca2+ channel
- CaV1.2
- L-type voltage-gated Ca2+ channel
- CaV2.3
- R-type voltage-gated Ca2+ channel
- CGRP
- calcitonin gene-related peptide
- iCGRP
- immunoreactive CGRP
- CRMP2
- collapsin response mediator protein 2
- Ct-dis
- distal end of the C terminus
- ddC
- 2′,3′-dideoxycytidine
- DRG
- dorsal root ganglion
- L1
- first intracellular loop
- MST
- microscale thermophoresis
- PWT
- paw withdrawal threshold
- TAT
- transactivator of transcription
- TNI
- tibial nerve injury
- aa
- amino acid(s)
- CAD
- catecholamine A-differentiated.
REFERENCES
- 1. Zamponi G. W., Lewis R. J., Todorovic S. M., Arneric S. P., Snutch T. P. (2009) Role of voltage-gated calcium channels in ascending pain pathways. Brain Res. Rev. 60, 84–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Snutch T. P. (2005) Targeting chronic and neuropathic pain. The N-type calcium channel comes of age. NeuroRx. 2, 662–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Malmberg A. B., Yaksh T. L. (1994) Voltage-sensitive calcium channels in spinal nociceptive processing. Blockade of N- and P-type channels inhibits formalin-induced nociception. J. Neurosci. 14, 4882–4890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bowersox S. S., Gadbois T., Singh T., Pettus M., Wang Y. X., Luther R. R. (1996) Selective N-type neuronal voltage-sensitive calcium channel blocker, SNX-111, produces spinal antinociception in rat models of acute, persistent, and neuropathic pain. J. Pharmacol. Exp. Ther. 279, 1243–1249 [PubMed] [Google Scholar]
- 5. White D. M., Cousins M. J. (1998) Effect of subcutaneous administration of calcium channel blockers on nerve injury-induced hyperalgesia. Brain Res. 801, 50–58 [DOI] [PubMed] [Google Scholar]
- 6. Souza A. H., Ferreira J., Cordeiro Mdo N., Vieira L. B., De Castro C. J., Trevisan G., Reis H., Souza I. A., Richardson M., Prado M. A., Prado V. F., Gomez M. V. (2008) Analgesic effect in rodents of native and recombinant Phα1β toxin, a high voltage-activated calcium channel blocker isolated from armed spider venom. Pain 140, 115–126 [DOI] [PubMed] [Google Scholar]
- 7. McGivern J. G. (2007) Ziconotide. A review of its pharmacology and use in the treatment of pain. Neuropsychiatr. Dis. Treat. 3, 69–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schmidtko A., Lötsch J., Freynhagen R., Geisslinger G. (2010) Ziconotide for treatment of severe chronic pain. Lancet 375, 1569–1577 [DOI] [PubMed] [Google Scholar]
- 9. Bowersox S. S., Singh T., Nadasdi L., Zukowska-Grojec Z., Valentino K., Hoffman B. B. (1992) Cardiovascular effects of ω-conopeptides in conscious rats. Mechanisms of action. J Cardiovasc. Pharmacol. 20, 756–764 [PubMed] [Google Scholar]
- 10. Brittain J. M., Piekarz A. D., Wang Y., Kondo T., Cummins T. R., Khanna R. (2009) An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J. Biol. Chem. 284, 31375–31390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brittain J. M., Duarte D. B., Wilson S. M., Zhu W., Ballard C., Johnson P. L., Liu N., Xiong W., Ripsch M. S., Wang Y., Fehrenbacher J. C., Fitz S. D., Khanna M., Park C. K., Schmutzler B. S., Cheon B. M., Due M. R., Brustovetsky T., Ashpole N. M., Hudmon A., Meroueh S. O., Hingtgen C. M., Brustovetsky N., Ji R. R., Hurley J. H., Jin X., Shekhar A., Xu X. M., Oxford G. S., Vasko M. R., White F. A., Khanna R. (2011) Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca2+ channel complex. Nat. Med. 17, 822–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wilson S. M., Brittain J. M., Piekarz A. D., Ballard C. J., Ripsch M. S., Cummins T. R., Hurley J. H., Khanna M., Hammes N. M., Samuels B. C., White F. A., Khanna R. (2011) Further insights into the antinociceptive potential of a peptide disrupting the N-type calcium channel-CRMP-2 signaling complex. Channels 5, 449–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ripsch M. S., Ballard C. J., Khanna M., Hurley J. H., White F. A., Khanna R. (2012) A Peptide Uncoupling CRMP-2 from the Presynaptic Ca2+ Channel Complex Demonstrates Efficacy in Animal Models of Migraine and AIDS Therapy-induced Neuropathy. Transl. Neurosci. 3, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bucci G., Mochida S., Stephens G. J. (2011) Inhibition of synaptic transmission and G protein modulation by synthetic CaV2.2 Ca2+ channel peptides. J. Physiol. 589, 3085–3101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mochida S., Sheng Z. H., Baker C., Kobayashi H., Catterall W. A. (1996) Inhibition of neurotransmission by peptides containing the synaptic protein interaction site of N-type Ca2+ channels. Neuron 17, 781–788 [DOI] [PubMed] [Google Scholar]
- 16. Frank R. (2002) The SPOT synthesis technique. Synthetic peptide arrays on membrane supports. Principles and applications. J. Immunol. Methods 267, 13–26 [DOI] [PubMed] [Google Scholar]
- 17. Chan A. W., Khanna R., Li Q., Stanley E. F. (2007) Munc18. A presynaptic transmitter release site N type (CaV2.2) calcium channel interacting protein. Channels 1, 11–20 [PubMed] [Google Scholar]
- 18. Wienken C. J., Baaske P., Rothbauer U., Braun D., Duhr S. (2010) Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 1, 100. [DOI] [PubMed] [Google Scholar]
- 19. van den Bogaart G., Meyenberg K., Diederichsen U., Jahn R. (2012) Phosphatidylinositol 4,5-bisphosphate increases Ca2+ affinity of synaptotagmin-1 by 40-fold. J. Biol. Chem. 287, 16447–16453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang Y., Khanna R. (2011) Voltage-gated calcium channels are not affected by the novel anti-epileptic drug lacosamide. Transl. Neurosci. 2, 13–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Y., Brittain J. M., Jarecki B. W., Park K. D., Wilson S. M., Wang B., Hale R., Meroueh S. O., Cummins T. R., Khanna R. (2010) In silico docking and electrophysiological characterization of lacosamide binding sites on collapsin response mediator protein-2 identifies a pocket important in modulating sodium channel slow inactivation. J. Biol. Chem. 285, 25296–25307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brittain J. M., Wang Y., Wilson S. M., Khanna R. (2012) Regulation of CREB signaling through L-type Ca2+ channels by Nipsnap-2. Channels 6, 94–102 [DOI] [PubMed] [Google Scholar]
- 23. Brittain J. M., Chen L., Wilson S. M., Brustovetsky T., Gao X., Ashpole N. M., Molosh A. I., You H., Hudmon A., Shekhar A., White F. A., Zamponi G. W., Brustovetsky N., Chen J., Khanna R. (2011) Neuroprotection against traumatic brain injury by a peptide derived from the collapsin response mediator protein 2 (CRMP2). J. Biol. Chem. 286, 37778–37792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chi X. X., Schmutzler B. S., Brittain J. M., Wang Y., Hingtgen C. M., Nicol G. D., Khanna R. (2009) Regulation of N-type voltage-gated calcium channels (Cav2.2) and transmitter release by collapsin response mediator protein-2 (CRMP-2) in sensory neurons. J. Cell Sci. 122, 4351–4362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Khanna R., Chang M. C., Joiner W. J., Kaczmarek L. K., Schlichter L. C. (1999) hSK4/hIK1, a calmodulin-binding KCa channel in human T lymphocytes. Roles in proliferation and volume regulation. J. Biol. Chem. 274, 14838–14849 [DOI] [PubMed] [Google Scholar]
- 26. Jacks T., Shih T. S., Schmitt E. M., Bronson R. T., Bernards A., Weinberg R. A. (1994) Tumor predisposition in mice heterozygous for a targeted mutation in Nf1. Nat. Genet. 7, 353–361 [DOI] [PubMed] [Google Scholar]
- 27. Hingtgen C. M., Roy S. L., Clapp D. W. (2006) Stimulus-evoked release of neuropeptides is enhanced in sensory neurons from mice with a heterozygous mutation of the Nf1 gene. Neuroscience 137, 637–645 [DOI] [PubMed] [Google Scholar]
- 28. Wang Y., Brittain J. M., Wilson S. M., Hingtgen C. M., Khanna R. (2010) Altered Calcium Currents and Axonal Growth in Nf1 Haploinsufficient Mice. Transl. Neurosci. 1, 106–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen J. J., Barber L. A., Dymshitz J., Vasko M. R. (1996) Peptidase inhibitors improve recovery of substance P and calcitonin gene-related peptide release from rat spinal cord slices. Peptides 17, 31–37 [DOI] [PubMed] [Google Scholar]
- 30. Bhangoo S. K., Ren D., Miller R. J., Chan D. M., Ripsch M. S., Weiss C., McGinnis C., White F. A. (2007) CXCR4 chemokine receptor signaling mediates pain hypersensitivity in association with antiretroviral toxic neuropathy. Brain Behav. Immun. 21, 581–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Joseph E. K., Chen X., Khasar S. G., Levine J. D. (2004) Novel mechanism of enhanced nociception in a model of AIDS therapy-induced painful peripheral neuropathy in the rat. Pain 107, 147–158 [DOI] [PubMed] [Google Scholar]
- 32. Wang Y., Wilson S. M., Brittain J. M., Ripsch M. S., Salomé C., Park K. D., White F. A., Khanna R., Kohn H. (2011) Merging structural motifs of functionalized amino acids and α-aminoamides results in novel anticonvulsant compounds with significant effects on slow and fast inactivation of voltage-gated sodium channels and in the treatment of neuropathic pain. ACS Chem. Neurosci. 2, 317–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zillner K., Jerabek-Willemsen M., Duhr S., Braun D., Längst G., Baaske P. (2012) Microscale thermophoresis as a sensitive method to quantify protein. Nucleic acid interactions in solution. Methods Mol. Biol. 815, 241–252 [DOI] [PubMed] [Google Scholar]
- 34. Sheng Z. H., Rettig J., Takahashi M., Catterall W. A. (1994) Identification of a syntaxin-binding site on N-type calcium channels. Neuron 13, 1303–1313 [DOI] [PubMed] [Google Scholar]
- 35. Patrakitkomjorn S., Kobayashi D., Morikawa T., Wilson M. M., Tsubota N., Irie A., Ozawa T., Aoki M., Arimura N., Kaibuchi K., Saya H., Araki N. (2008) Neurofibromatosis type 1 (NF1) tumor suppressor, neurofibromin, regulates the neuronal differentiation of PC12 cells via its associating protein, CRMP-2. J. Biol. Chem. 283, 9399–9413 [DOI] [PubMed] [Google Scholar]
- 36. Pragnell M., De Waard M., Mori Y., Tanabe T., Snutch T. P., Campbell K. P. (1994) Calcium channel β-subunit binds to a conserved motif in the I-II cytoplasmic linker of the α1-subunit. Nature 368, 67–70 [DOI] [PubMed] [Google Scholar]
- 37. De Waard M., Liu H., Walker D., Scott V. E., Gurnett C. A., Campbell K. P. (1997) Direct binding of G-protein βγ complex to voltage-dependent calcium channels. Nature 385, 446–450 [DOI] [PubMed] [Google Scholar]
- 38. Li B., Zhong H., Scheuer T., Catterall W. A. (2004) Functional role of a C-terminal Gβγ-binding domain of Cav2.2 channels. Mol. Pharmacol. 66, 761–769 [DOI] [PubMed] [Google Scholar]
- 39. Maximov A., Südhof T. C., Bezprozvanny I. (1999) Association of neuronal calcium channels with modular adaptor proteins. J. Biol. Chem. 274, 24453–24456 [DOI] [PubMed] [Google Scholar]
- 40. Spafford J. D., Munno D. W., Van Nierop P., Feng Z. P., Jarvis S. E., Gallin W. J., Smit A. B., Zamponi G. W., Syed N. I. (2003) Calcium channel structural determinants of synaptic transmission between identified invertebrate neurons. J. Biol. Chem. 278, 4258–4267 [DOI] [PubMed] [Google Scholar]
- 41. Van Petegem F., Clark K. A., Chatelain F. C., Minor D. L., Jr. (2004) Structure of a complex between a voltage-gated calcium channel β-subunit and an α-subunit domain. Nature 429, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tedford H. W., Kisilevsky A. E., Vieira L. B., Varela D., Chen L., Zamponi G. W. (2010) Scanning mutagenesis of the I-II loop of the Cav2.2 calcium channel identifies residues arginine 376 and valine 416 as molecular determinants of voltage-dependent G protein inhibition. Mol. Brain. 3, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dodge F. A., Jr., Rahamimoff R. (1967) On the relationship between calcium concentration and the amplitude of the end-plate potential. J. Physiol. 189, 90P–92P [PubMed] [Google Scholar]
- 44. Chen L., Harrison S. D. (2007) Cell-penetrating peptides in drug development. Enabling intracellular targets. Biochem. Soc. Trans. 35, 821–825 [DOI] [PubMed] [Google Scholar]
- 45. Chauhan A., Tikoo A., Kapur A. K., Singh M. (2007) The taming of the cell-penetrating domain of the HIV Tat. Myths and realities. J. Control Release 117, 148–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Polyakov V., Sharma V., Dahlheimer J. L., Pica C. M., Luker G. D., Piwnica-Worms D. (2000) Novel Tat peptide chelates for direct transduction of technetium 99m and rhenium into human cells for imaging and radiotherapy. Bioconjug. Chem. 11, 762–771 [DOI] [PubMed] [Google Scholar]
- 47. Manceur A. P., Driscoll B. D., Sun W., Audet J. (2009) Selective enhancement of the uptake and bioactivity of a TAT-conjugated peptide inhibitor of glycogen synthase kinase-3. Mol. Ther. 17, 500–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jimenez-Andrade J. M., Herrera M. B., Ghilardi J. R., Vardanyan M., Melemedjian O. K., Mantyh P. W. (2008) Vascularization of the dorsal root ganglia and peripheral nerve of the mouse. Implications for chemical-induced peripheral sensory neuropathies. Mol. Pain. 4, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Piekarz A. D., Due M. R., Khanna M., Wang B., Ripsch M. S., Wang R., Meroueh S. O., Vasko M. R., White F. A., Khanna R. (2012) CRMP-2 peptide-mediated decrease of high and low voltage-activated calcium channels, attenuation of nociceptor excitability, and anti-nociception in a model of AIDS therapy-induced painful peripheral neuropathy. Mol. Pain. 8, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bell T. J., Thaler C., Castiglioni A. J., Helton T. D., Lipscombe D. (2004) Cell-specific alternative splicing increases calcium channel current density in the pain pathway. Neuron 41, 127–138 [DOI] [PubMed] [Google Scholar]
- 51. Altier C., Dale C. S., Kisilevsky A. E., Chapman K., Castiglioni A. J., Matthews E. A., Evans R. M., Dickenson A. H., Lipscombe D., Vergnolle N., Zamponi G. W. (2007) Differential role of N-type calcium channel splice isoforms in pain. J. Neurosci. 27, 6363–6373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Winquist R. J., Pan J. Q., Gribkoff V. K. (2005) Use-dependent blockade of Cav2.2 voltage-gated calcium channels for neuropathic pain. Biochem. Pharmacol. 70, 489–499 [DOI] [PubMed] [Google Scholar]
- 53. Cizkova D., Marsala J., Lukacova N., Marsala M., Jergova S., Orendacova J., Yaksh T. L. (2002) Localization of N-type Ca2+ channels in the rat spinal cord following chronic constrictive nerve injury. Exp. Brain Res. 147, 456–463 [DOI] [PubMed] [Google Scholar]
- 54. Saegusa H., Kurihara T., Zong S., Kazuno A., Matsuda Y., Nonaka T., Han W., Toriyama H., Tanabe T. (2001) Suppression of inflammatory and neuropathic pain symptoms in mice lacking the N-type Ca2+ channel. EMBO J. 20, 2349–2356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vanegas H., Schaible H. (2000) Effects of antagonists to high threshold calcium channels upon spinal mechanisms of pain, hyperalgesia, and allodynia. Pain 85, 9–18 [DOI] [PubMed] [Google Scholar]
- 56. Neugebauer V., Vanegas H., Nebe J., Rümenapp P., Schaible H. G. (1996) Effects of N- and L-type calcium channel antagonists on the responses of nociceptive spinal cord neurons to mechanical stimulation of the normal and the inflamed knee joint. J. Neurophysiol. 76, 3740–3749 [DOI] [PubMed] [Google Scholar]
- 57. Sluka K. A. (1997) Blockade of calcium channels can prevent the onset of secondary hyperalgesia and allodynia induced by intradermal injection of capsaicin in rats. Pain 71, 157–164 [DOI] [PubMed] [Google Scholar]
- 58. Neugebauer V., Rümenapp P., Schaible H. G. (1996) Calcitonin gene-related peptide is involved in the spinal processing of mechanosensory input from the rat's knee joint and in the generation and maintenance of hyperexcitability of dorsal horn neurons during development of acute inflammation. Neuroscience 71, 1095–1109 [DOI] [PubMed] [Google Scholar]
- 59. Fossat P., Dobremez E., Bouali-Benazzouz R., Favereaux A., Bertrand S. S., Kilk K., Léger C., Cazalets J. R., Langel U., Landry M., Nagy F. (2010) Knockdown of L calcium channel subtypes. Differential effects in neuropathic pain. J. Neurosci. 30, 1073–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Favereaux A., Thoumine O., Bouali-Benazzouz R., Roques V., Papon M. A., Salam S. A., Drutel G., Léger C., Calas A., Nagy F., Landry M. (2011) Bidirectional integrative regulation of Cav1.2 calcium channel by microRNA miR-103. Role in pain. EMBO J. 30, 3830–3841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pavlidis P., Noble W. S. (2003) Matrix2png. A utility for visualizing matrix data. Bioinformatics 19, 295–296 [DOI] [PubMed] [Google Scholar]
- 62. Almagor L., Chomsky-Hecht O., Ben-Mocha A., Hendin-Barak D., Dascal N., Hirsch J. A. (2012) The role of a voltage-dependent Ca2+ channel intracellular linker. A structure-function analysis. J. Neurosci. 32, 7602–7613 [DOI] [PMC free article] [PubMed] [Google Scholar]