

Background: The role of HDAC1 in the regulation of neuronal survival is unresolved.
Results: In cooperation with HDRP, HDAC1 promotes neuronal survival, but when it interacts with HDAC3, HDAC1 promotes neuronal death.
Conclusion: HDAC1 can protect neurons or promote neuronal death depending on whether it interacts with HDRP or HDAC3.
Significance: Our results provide insight into the role of HDAC1 in the regulation of neuronal survival.
Keywords: Akt PKB, Glycogen Synthase Kinase 3, Histone Deacetylase, Neurodegeneration, Neurons, HDAC1, HDAC3, HDRP, Neuronal Survival
Abstract
Both neuroprotective and neurotoxic roles have previously been described for histone deacetylase-1 (HDAC1). Here we report that HDAC1 expression is elevated in vulnerable brain regions of two mouse models of neurodegeneration, the R6/2 model of Huntington disease and the Ca2+/calmodulin-dependent protein kinase (CaMK)/p25 double-transgenic model of tauopathic degeneration, suggesting a role in promoting neuronal death. Indeed, elevating HDAC1 expression by ectopic expression promotes the death of otherwise healthy cerebellar granule neurons and cortical neurons in culture. The neurotoxic effect of HDAC1 requires interaction and cooperation with HDAC3, which has previously been shown to selectively induce the death of neurons. HDAC1-HDAC3 interaction is greatly elevated under conditions of neurodegeneration both in vitro and in vivo. Furthermore, the knockdown of HDAC3 suppresses HDAC1-induced neurotoxicity, and the knockdown of HDAC1 suppresses HDAC3 neurotoxicity. As described previously for HDAC3, the neurotoxic effect of HDAC1 is inhibited by treatment with IGF-1, the expression of Akt, or the inhibition of glycogen synthase kinase 3β (GSK3β). In addition to HDAC3, HDAC1 has been shown to interact with histone deacetylase-related protein (HDRP), a truncated form of HDAC9, whose expression is down-regulated during neuronal death. In contrast to HDAC3, the interaction between HDRP and HDAC1 protects neurons from death, an effect involving acquisition of the deacetylase activity of HDAC1 by HDRP. We find that elevated HDRP inhibits HDAC1-HDAC3 interaction and prevents the neurotoxic effect of either of these two proteins. Together, our results suggest that HDAC1 is a molecular switch between neuronal survival and death. Its interaction with HDRP promotes neuronal survival, whereas interaction with HDAC3 results in neuronal death.
Introduction
Histone deacetylases (HDACs)2 are a family of proteins originally identified on the basis of their ability to deacetylate lysine residues on histones, thus resulting in the compaction of chromatin into a transcriptionally repressed state (1, 2). It has since been found that HDACs also deacetylate a variety of other nuclear, cytoplasmic, and mitochondrial proteins, thereby regulating diverse cellular events. Vertebrates express 18 HDACs that are organized into four classes based on their similarity to yeast proteins: the Class I RPD3-like proteins (HDAC1, HDAC2, HDAC3, and HDAC8), the Class II HDA1-like proteins (HDAC4, HDAC5, HDAC6, HDAC7, HDAC9, HDRP, and HDAC10), the Class III SIR2-like proteins (SIRT1–7), and HDAC11, which shares sequence homology with both Class I and Class II HDACs and is the sole member of the Class IV subfamily. Although Class I, II, and IV HDACs are zinc-dependent enzymes collectively referred to as classical HDACs, Class III HDACs require NAD+ for their activity and are referred to as sirtuins (1, 2).
There is a growing consensus that HDACs play a role in the regulation of neuronal survival (3–5). Much of this is based on the protective effects afforded by chemical inhibitors of HDACs in a variety of experimental models of neurodegenerative diseases. Surprisingly, the analysis of individual HDAC proteins has revealed that several of them including HDAC4, HDAC6, HDAC7, HDRP (a truncated form of HDAC9), and Sirt1 protect neurons rather than promote degeneration (6–13). Although the identities of the targets of the HDAC inhibitors in the context of neurodegeneration have yet to be conclusively identified, we have found that HDAC3 displays a high level of toxicity in neurons (14), suggesting that its inhibition might account, at least in part, for the extensively described neuroprotective effects of HDAC inhibitors.
Another HDAC protein that regulates neuronal viability is HDAC1. We previously reported that although lacking a catalytic domain, the neuroprotective effect of HDRP requires deacetylase activity, which is acquired through its interaction with HDAC1 (7). This suggests a neuroprotective role for HDAC1. This conclusion was supported by studies from another laboratory that found that HDAC1 protected neurons in cell culture and mouse models of Alzheimer disease and ischemic stroke (10). Perplexingly, other groups have reported that HDAC1 promotes axonal and neuronal degeneration (15, 16). Hence whether HDAC1 protects against neurodegeneration or is involved in promoting it remains equivocal. In this study, we show that HDAC1 can be both neuroprotective and neurotoxic depending on whether it interacts with the Class IIA HDAC, HDRP, or with HDAC3, a member of the Class I family. The neurotoxic effect of HDAC1 is dependent on GSK3β activity and can be inhibited by IGF-1 treatment or activation of PI3K-Akt signaling. We also find that HDAC1 expression is elevated selectively in vulnerable brain regions in mouse models of HD and tauopathy.
EXPERIMENTAL PROCEDURES
Materials
All cell culture media and reagents were purchased from Invitrogen, and all chemicals were from Sigma-Aldrich, unless stated otherwise. Expression plasmids for HDAC1-FLAG, HDAC1-GFP, HDAC1-Mut-GFP, HDAC3-FLAG, constitutively active Akt (CA-Akt), and dominant-negative GSK3β (DN-GSK3β) were purchased from Addgene (Cambridge, MA). FLAG antibody was from Sigma-Aldrich, HDAC1 antibody used for Western blotting was from Sigma-Aldrich (catalog number WH0003065), and the antibody used for immunoprecipitation was from Santa Cruz Biotechnology (Santa Cruz, CA; catalog number sc6298). HDAC3 antibody used for Western blotting was from Santa Cruz Biotechnology (catalog number sc-11417), and that used for immunoprecipitation was from Sigma-Aldrich (catalog number H3034). HDAC1 shRNA plasmids (TRCN0000039400 and TRCN0000039402), HDAC3 shRNA plasmids (TRCN0000039391 and TRCN0000039392), and a control plasmid (pLKO1) were purchased from Sigma-Aldrich. The ability of the HDAC3 shRNAs to suppress expression was previously documented (14).
Culturing, Treatment, and Transfection of Neurons
Cerebellar granule neurons (CGNs) were cultured from 7- or 8-day-old Wistar rats and plated as described previously (17). To prevent non-neuronal cells from replicating, cytosine arabinofuranoside (10 μm) was added to the culture medium 18–22 h after plating. These cultures were treated with high potassium medium (HK; serum-free basal medium Eagle with 25 mm KCl) or low potassium medium (LK; serum-free basal medium Eagle) 7 days after plating unless the cells were used for transfections. Cortical neurons were cultured from Wistar rats at embryonic day 17 or 18 as described previously (6, 18, 19). The neurons were treated with homocysteic acid (HCA) 1 or 2 days after plating by directly adding HCA (to a final concentration of 1 mm) to the culture medium. Neuronal viability was quantified by staining cell nuclei with 4′,6′-diamidino-2-phenylindole hydrochloride (DAPI). Cells with condensed or fragmented nuclei were scored as apoptotic.
Transfection and treatment of neurons were performed as described previously (14, 20, 21). Briefly, unless stated otherwise, transfections were performed on day 5 after plating for CGNs. Eight hours after transfection, medium was switched to either HK or LK for 24 h when viability of transfected neurons was quantified. For the experiments in which IGF-1 or inhibitors were used, treatment was done at the time of transfection and again when the medium was changed to HK or LK. The treatments were performed at the following concentrations: IGF-1 at 50 ng/ml, SP600125 at 10 μm, roscovitine at 50 μm, SB415286 at 30 μm, and SB216763 at 5 μm. The ability of the inhibitors to inhibit their targets at the doses used was confirmed in control experiments.
Expression Studies
RNA was extracted from cultured neurons using TRIzol (Invitrogen), and cDNA was prepared using the SuperScript II first strand synthesis system for RT-PCR (Invitrogen). PCR was performed with GoTaq Green master mix (Promega). Western blot analysis was performed as described previously. Briefly, cell lysate was prepared using 1× cell lysis buffer (Cell Signaling Technology) and subjected to SDS-PAGE after protein normalization and heat denaturation.
Immunoprecipitation and in Vitro Kinase Assay
GFP, FLAG-tagged HDAC3, or FLAG-tagged HDAC1 was overexpressed in HEK293 cells, and cell lysates were collected after 24 h. The lysates were incubated with 5 μg of either FLAG or GFP antibody and 25 μl of protein A/G PLUS-agarose beads (Santa Cruz Biotechnology) overnight. Immunoprecipitates were collected after centrifugation and washed twice with cell lysis buffer and once with kinase buffer. The immunoprecipitated proteins were incubated with or without active GSK3β and with 5 μl of [γ-32P]ATP mixture for 30 min. The reaction was stopped by adding 25 μl of 3× SDS and boiled at 95 °C for 5 min. The samples were run on an SDS-polyacrylamide gel and subjected to autoradiography.
Site-directed Mutagenesis
HDAC3 deacetylase activity mutant H134Q containing the mutations H134Q and H135A was constructed using the QuikChange site-directed mutagenesis kit from Stratagene (La Jolla, CA) using the manufacturer's protocol. The primers used for the constructs were as follows: 5′-CTG GTG GTC TGC AGG CTG CCA AGA AGT TTG-3′ and 5′-CAA ACT TCT TGG CAG CCT GCA GAC CAC CAG-3′.
shRNA-mediated Knockdown
For HDAC1 knockdown studies, the shRNA or control constructs were transfected into CGNs on day 5 after plating along with GFP plasmid in the ratio of 6.5:1. The shRNA was allowed to express for 48 h, after which HK or LK treatment was done and viability was assessed 24 h later. The cells were co-stained with HDAC1 antibody to ensure that the protein was knocked down in the cells co-expressing GFP. When shRNA constructs were co-transfected with other tagged proteins, (HDAC1 shRNA with HDAC3-FLAG, or HDAC3 shRNA with HDAC1-FLAG), the shRNA constructs and the tagged proteins were transfected at a ratio of 2:1, and the proteins were visualized using the appropriate tag antibody.
Generation of Hdac3−/− Conditional Knock-out Mice and Cultures
Mice homozygous for loxP sites in the Hdac3 locus (Hdac3neo-loxP) were a kind gift from the Olson laboratory at the University of Texas Southwestern Medical Center (22). These mice were bred to Nes-Cre transgenic mice purchased from The Jackson Laboratory (Bar Harbor, ME), which express the Cre recombinase throughout the central nervous system (CNS), allowing for the generation of CNS-specific Hdac3+/− mice. These Hdac3+/− mice were then interbred, and embryos were extracted at day 17 or 18. Each embryo was genotyped, and cortical neuronal cultures were prepared from individual Hdac3−/− and Hdac3+/+ embryos. The cultures were transfected with either GFP or HDAC1-FLAG on day 7 after plating, and viability was assessed 20 h later.
The R6/2 Transgenic Mouse Model of HD
Female mice hemizygous for the ovarian transplant of the truncated mutant huntingtin (Htt) transgene were bred with wild-type male mice. Both mice were on a C57BL/6J background and purchased from The Jackson Laboratory. The offspring were genotyped 10–12 days after birth. At 6, 9, and 13 weeks after birth, mice with the transgene (R6/2) or their WT littermates of the same gender were sacrificed, the brains were dissected, and the striatum was separated. Lysates were made from the brain tissue and used for Western blotting or immunoprecipitation assays.
The CaMK-p25 Double-transgenic Mouse Model
A bitransgenic mouse line expressing p25-GFP selectively in the forebrain was generated as described previously (23, 24). Briefly, mice containing the p25-GFP transgene under the direction of the tetOp were crossed with mice containing the tetracycline tTA gene coupled to the forebrain-specific CaMK2a promoter. Both transgenic lines were purchased from The Jackson Laboratory and produced on mixed C57BL/6 backgrounds. Breeding dams and offspring were kept on doxycycline-containing food pellets (Bio-Serv, Frenchtown, NJ) to suppress transcription of the p25-GFP transgene. After genotyping, double transgenic mice expressing both the p25-GFP transgene and tTA were either taken off the doxycycline-containing food (Tg-ON) or kept on the food (Tg-OFF) for 8 weeks, after which the mice were sacrificed and lysates were prepared from the cortex, hippocampus, and cerebellum. As reported by Le-Huei Tsai and co-workers (23, 24), who developed this model, we find that the p25-GFP transgene is expressed highly in the cortex and hippocampus and that the neurodegeneration in these brain regions is obvious by 6 weeks of transgene induction.
Statistical Analysis
All statistical analyses were done using GraphPad Prism software. Student's t test was performed for all quantification experiments. All quantification experiments were performed at least three times and were done in duplicates each time. For cell viability quantification, at least 150 cells were counted for each coverslip. * denotes a p value < 0.05.
RESULTS
HDAC1 Promotes Neuronal Death
Both neuroprotective and neurotoxic roles have been described for HDAC1 (7, 10, 15, 16). As a step toward gaining a better understanding of the role of HDAC1 in the regulation of neuronal survival and death, we examined its expression in the R6/2 transgenic mouse model of Huntington disease. R6/2 mice are transgenic for the first exon of the human huntingtin gene carrying about 120 CAG repeats. These mice exhibit a progressive neurological phenotype that mimics many features of human HD including selective striatal neuropathology, intracellular aggregates, reduced motor performance, and shortened lifespan (25). As shown in Fig. 1A, HDAC1 expression is elevated in the striatum of R6/2 mice, but not in extra-striatal tissues, which are relatively unaffected by neuropathology. It is known that neuropathology and behavioral deficits in R6/2 mice are not obvious at 6 weeks, but clearly discernible at 10 weeks of age (25). As shown in Fig. 1A, HDAC1 expression in R6/2 mice is similar to wild-type littermates at 6 weeks but is increased at 9 weeks. Expression is increased further at 13 weeks when neuropathology is known to be robust (Fig. 1A). In comparison, the induction of HDAC1 expression in extra-striatal tissue of R6/2 mice when compared with their wild-type littermates is modest, if any. We also examined HDAC1 expression in the inducible CaMK-p25/CDK5 double-transgenic mouse model of tauopathic degeneration (23, 24). In these mice, degeneration occurs selectively starting at about 5 weeks after doxycycline-mediated induction of p25 through the CaMK promoter. As described previously (23, 24), expression of the p25-GFP transgene occurs specifically in the cortex and hippocampus, areas of the brain that display neurodegeneration. The expression of HDAC1 is increased specifically in these areas of the brain (Fig. 1B). These results suggest that elevated HDAC1 expression contributes to neurodegeneration.
FIGURE 1.

Induction of HDAC1 expression in vulnerable brain region in mouse models of neurodegenerative disease. A, Western blot analysis of lysates from the striatum (St.) or extra-striatal tissue (EST) of R6/2 mice at 6, 9, and 13 weeks were analyzed for expression of HDAC1 or tubulin as loading control. B, in the CaMK/p25 transgenic line, p25-GFP expression is driven through a doxycycline-inducible CaMK promoter. Left panel, expression of p25-GFP transgene is observed only when its expression is induced and in the cortex and hippocampus where the CaMK promoter is active. Right panel, HDAC1 expression is elevated in cortex and hippocampus, but not in cerebellum. Lysates for both panels were from mice in which transgene expression was induced for 8 weeks at which time neurodegeneration is obvious in the cortex and hippocampus (our observations and Ref. 23). Tg-ON, transgenic mice taken off the doxycycline-containing food; Tg-OFF, transgenic mice kept on the food. C, lysates from CGN cultures treated with either HK or LK for 6, 9, 12, or 15 h were subjected to Western blotting and probed for HDAC1. The blot was reprobed with tubulin to show equal loading. The lack of alterations in HDAC1 expression was confirmed in multiple experiments. D, cultured cortical neurons were either left untreated or treated with HCA for 3, 6, 9, 12, or 15 h, and the lysates were subjected to Western blotting and probed for HDAC1. The blot was reprobed with tubulin to show equal loading. The absence of major alterations in HDAC1 expression was confirmed in additional experiments.
To investigate the effect of elevated HDAC1 expression on neuronal viability, we used cultured CGNs. These neurons undergo apoptosis when switched from depolarizing HK medium to nondepolarizing LK medium (17). In contrast to the in vivo models of neurodegeneration, HDAC1 expression was not increased following LK treatment (Fig. 1C). However, when the level of HDAC1 is enhanced by ectopic expression, over 30% of the neurons die within 24 h (Fig. 2A). No further increase in cell death is observed in LK medium overexpressing HDAC1, suggesting that death resulting from elevated HDAC1 involves the same molecular mechanism as LK-induced death. We extended our studies to embryonic cortical neurons induced to die by treatment with HCA, which induces oxidative stress. As observed with CGNs, there was no increase in HDAC1 expression in cortical neurons after treatment with HCA (Fig. 1D). When HDAC1 is overexpressed in cortical neurons, viability is reduced by about 40% (Fig. 2B). HDAC1-expressing neurons treated with HCA displayed only a marginal increase in toxicity over cultures treated with HCA alone.
FIGURE 2.

Elevated HDAC1 is toxic to neurons. A, CGNs were transfected with either GFP or HDAC1 and then switched to HK or LK medium 8 h later. Viability of transfected neurons was assessed after 24 h. Viability is normalized to cultures transfected with GFP and treated with HK. B, cortical neurons were transfected with GFP or HDAC1 for 6 h and were either left untreated (Unt) or treated with HCA. Viability of transfected neurons was assessed after 14 h and is normalized to GFP-transfected untreated cultures. Error bars indicate S.D.
HDAC1-induced Neurotoxicity Is GSK3-dependent and Is Inhibited by PI3K-Akt Signaling
To understand the mechanism by which HDAC1 neurotoxicity is mediated, we treated CGNs overexpressing HDAC1 with IGF-1, which is the physiological survival factor for these neurons in vivo. Treatment with IGF-1 blocks HDAC1-induced neurotoxicity (Fig. 3A). IGF-1 is an activator of the PI3K-Akt signaling pathway (26, 27). To examine whether activation of this signaling pathway was involved in the suppression of HDAC1 neurotoxicity by IGF-1, we expressed a constitutively active form of Akt (CA-Akt). As shown in Fig. 3A, co-expression of active Akt also blocks the neurotoxic effect of HDAC1. A well established downstream substrate of Akt is GSK3β, a kinase known to be involved in promoting cell death in a variety of experimental models of neurodegeneration (28, 29). Phosphorylation of GSK3β by Akt inhibits its kinase activity and its ability to promote neuronal death (30, 31). To examine whether HDAC1 neurotoxicity is dependent on GSK3β, we blocked its activity with two structurally distinct pharmacological inhibitors, SB216763 and SB415826. Both GSK3β inhibitors protect CGNs against HDAC1 neurotoxicity (Fig. 3B). Similarly, inhibiting GSK3β with a dominant-negative form of the enzyme also blocks HDAC1-induced neurotoxicity (Fig. 3A). Two other kinases known to be involved in promoting neuronal death are c-Jun N-terminal kinase (JNK) and cyclin-dependent kinases (CDKs) (32, 33). In contrast to GSK3β, the pharmacological inhibition of JNK and CDKs using SB600125 and roscovitine, respectively, had no protective effect against HDAC1-induced neuronal death (Fig. 3B).
FIGURE 3.
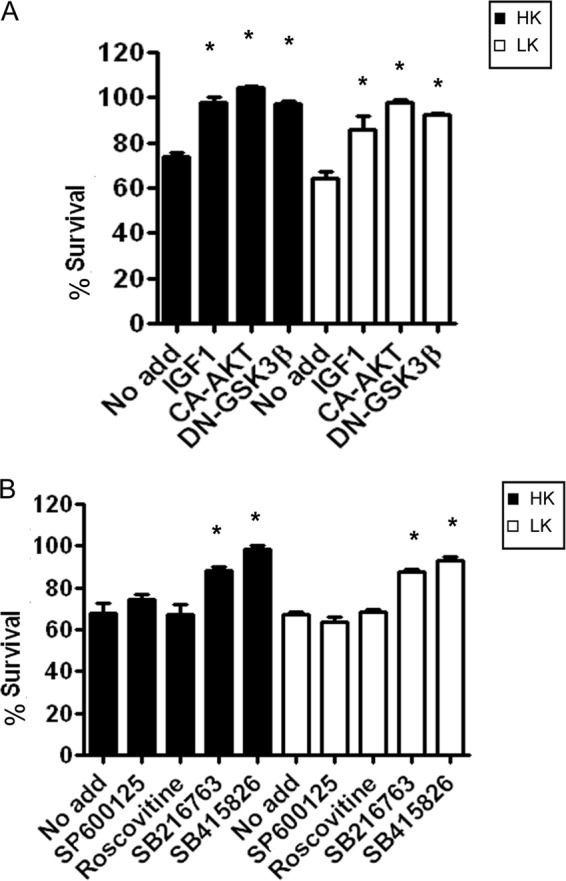
HDAC1 toxicity can be prevented by inhibiting GSK3β or by activating the PI3K-AKT pathway. A, CGNs were transfected with HDAC1 and treated with HK or LK medium 8 h later. The cultures were either left with no additives (No add) or treated with IGF-1, or co-transfected with CA-Akt or DN-GSK3β. Viability was assessed 24 h later and normalized to cultures transfected with GFP and treated with HK. B, CGNs were transfected with HDAC1 and treated with HK or LK medium 8 h later. The cultures were either left with no additives or supplemented with JNK inhibitor (SP600125), CDK inhibitor (roscovitine), or two different GSK3β inhibitors (SB216763 and SB415826). Viability was assessed 24 h later and normalized to cultures transfected with GFP and treated with HK. Error bars indicate S.D.
HDAC1 and HDAC3 Cooperate to Promote Neuronal Death
We previously demonstrated that forced expression of HDAC3 also promotes the death of neurons (14). Coincidentally, HDAC3-mediated neurotoxicity is also inhibited by IGF-1, by active Akt expression, and by treatment with GSK3β inhibitors or expression of a dominant-negative construct of this kinase (14). This raised the possibility that HDAC1 and HDAC3 cooperate to promote neuronal death. As a step toward examining this possibility, we investigated whether the two proteins interact. As shown in Fig. 4A, interaction between HDAC1 and HDAC3 is observed when the two proteins are co-expressed in HEK293 cells. Interaction of endogenous HDAC1 with HDAC3 is also seen in neurons (Fig. 4B). Consistent with the possibility that the proteins cooperate to promote death, HDAC1-HDAC3 interaction is substantially higher in neurons primed to die when compared with healthy neurons (Fig. 4B).
FIGURE 4.
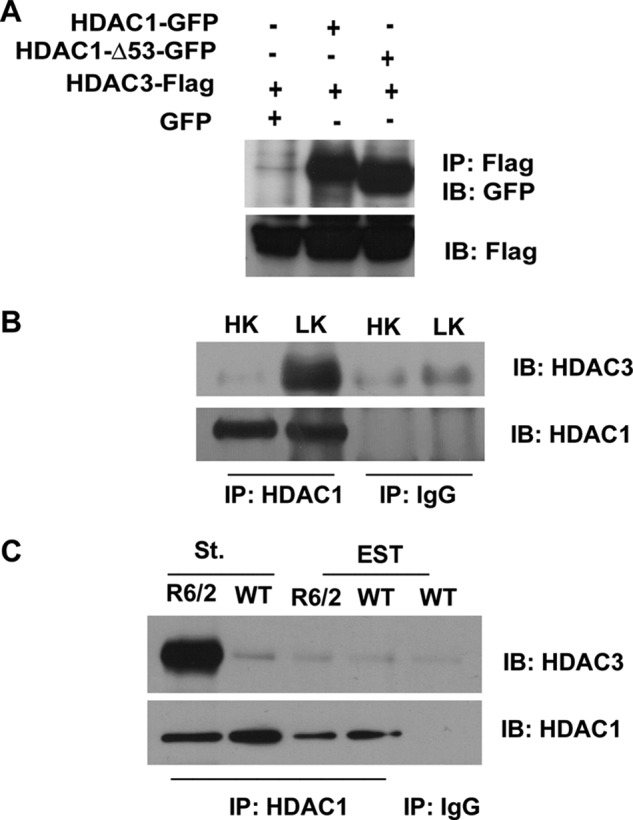
Full-length HDAC1 and HDAC1-Δ53 interact with HDAC3. A, HEK293 cells were co-transfected with FLAG-tagged HDAC3 and GFP, HDAC1, or HDAC1-Δ53 for 24 h, after which lysates were collected and immunoprecipitation (IP) was performed using FLAG antibody. Lysates were subjected to Western blot (IB) analysis and probed with GFP antibody. The same membrane was then reprobed with FLAG antibody to show the immunoprecipitated HDAC3 protein. B, CGNs were treated with either HK or LK for 6 h. Lysates were collected, and immunoprecipitation was performed using either HDAC1 or IgG antibody as negative control. The immunoprecipitate was subjected to Western blot analysis and probed with HDAC3 antibody. The membrane was reprobed with HDAC1 antibody to show pulldown of HDAC1. C, lysates from the striatum (St.) and extra-striatal tissue (EST) of R6/2 mice or their wild-type (WT) littermates were used to immunoprecipitate HDAC1 and were then run on a Western gel and probed with HDAC3 antibody. The membrane was reprobed with HDAC1 to show pulldown of the endogenous protein.
To examine whether HDAC1-HDAC3 interaction also occurred in vivo, we used the R6/2 mouse model. As shown in Fig. 4C, robust interaction between HDAC1 and HDAC3 is seen in the striatum of R6/2 mice, but not in wild-type littermates. In contrast to the striatum, interaction is barely detectable in lysates from extra-striatal tissue (Fig. 4C). This finding supports the conclusion that HDAC1-HDAC3 interaction promotes neuronal death.
To ascertain more directly whether HDAC1 and HDAC3 function together to promote neuronal death, we co-expressed the two proteins in CGNs (Fig. 5A). The extent of toxicity is higher when both proteins are co-expressed than when only one of them is overexpressed (and hence the other one is in limiting amounts with regard to interaction). We also examined the effect of knocking down the expression of one of these proteins on the ability of the other protein to promote neuronal death. The knockdown of HDAC1 using two different shRNA constructs reduced the toxicity of HDAC3 (Fig. 5B). Likewise, the suppression of HDAC3 using two different shRNAs reduced the toxicity of HDAC1 (Fig. 5C). The efficacy of the two HDAC3 shRNAs was previously demonstrated (14). This result was verified using cortical neurons cultured from Hdac3 conditional knock-out mice in which the Hdac3 gene was ablated in the CNS by crossing Hdac3-flox mice with transgenic mice expressing Cre under the control of the nestin promoter. In contrast to its effect in wild-type neurons, HDAC3-deficient neurons are resistant to HDAC1-induced toxicity (Fig. 5D). Taken together, these results demonstrate that HDAC1 and HDAC3 require each other to promote neuronal death.
FIGURE 5.

HDAC1 and HDAC3 cooperate to produce toxicity. A, CGNs transfected with GFP, HDAC1, HDAC3, or co-transfected with HDAC1 and HDAC3 were treated with HK or LK medium for 24 h. The viability of cells was assessed after immunocytochemistry and normalized to GFP-transfected cultures in HK. The panel on the right shows knockdown of HDAC1 by the two shRNAs in CGNs co-transfected with GFP and either control (Ctrl) or one of two HDAC1 shRNAs (H1-sh-1 or H1-sh-2). Cells expressing shRNA are identified based on GFP staining and knockdown of endogenous HDAC1evaluated using an HDAC1 antibody. B, CGNs were co-transfected with HDAC3 and either control (Ctrl) or one of two HDAC1 shRNAs (HDAC1-sh1 or HDAC1-sh2) and then treated with HK or LK medium for 24 h, after which cells were fixed, subjected to immunocytochemistry, and assessed for viability. Viability was normalized to GFP-transfected cultures in HK. C, CGNs were co-transfected with HDAC1 and either control (Ctrl) or one of two HDAC3 shRNAs (HDAC3-sh1 or HDAC3-sh2) for 48 h and then treated with HK or LK medium for 24 h after which cells were fixed, subjected to immunocytochemistry, and assessed for viability. D, upper panel, cortical neurons cultured from Nes-Cre Hdac3−/− mice or from their wild-type littermates were transfected with either GFP or HDAC1, and viability was assessed after 20 h. Lower panel, whole brain lysates from Hdac3−/− conditional KO mice (−/−) and wild-type littermates (+/+) were analyzed by Western blot using an HDAC3 antibody. The membrane was reprobed with tubulin. Error bars in panels A–D indicate S.D. E, lysates from HEK293 cells transfected with GFP or FLAG-tagged HDAC1 or HDAC3 were immunoprecipitated using either GFP or FLAG antibodies. The immunoprecipitated proteins were used in an in vitro kinase assay with or without active GSK3β as indicated. Autoradiograph shows a phosphorylated band in the HDAC3 lane, but no band is discernible for HDAC1. WCL, whole cell lysate; IB, immunoblot.
Because HDAC3 is phosphorylated directly by GSK3β (14), we examined whether HDAC1 is also a GSK3β substrate. Interestingly and in contrast to HDAC3, phosphorylation of HDAC1 could not be detected (Fig. 5E). This suggests that the inhibition of HDAC1 toxicity by GSK3β inhibitors is an indirect effect of inhibition of HDAC3 and that HDAC1 needs HDAC3 for its toxic effect.
Deacetylase Activity of Either HDAC1 or HDAC3 is Necessary for Neurotoxicity of the HDAC1-HDAC3 Complex
To test whether the catalytic activity of HDAC1 and HDAC3 is necessary for toxicity, we expressed HDAC1-Δ53, a deletion mutant lacking the N-terminal 53 residues of HDAC1 and HDAC3-H134Q, in which His-134 and -135 are mutated to glutamine and alanine, respectively. Both of these mutants have previously been reported to lack catalytic activity and can function by a dominant-negative mechanism to inhibit activity of the endogenous HDAC1 or HDAC3 proteins (34, 35). We independently confirmed that HDAC1-Δ53 is without activity and that HDAC3-H134Q has reduced activity when compared with wild-type HDAC3 (supplemental Fig. 1). Surprisingly, HDAC1-Δ53 was as toxic as wild-type HDAC1 when expressed in HK- or LK-treated CGNs (Fig. 6A). Similarly, HDAC3-H134Q was just as neurotoxic as wild-type HDAC3 (Fig. 6B). Although this observation suggests that HDAC1 and HDAC3 act through a deacetylase-independent mechanism, it was possible that the HDAC1 and HDAC3 acquire deacetylase activity through interaction with endogenous HDAC3 and HDAC1, respectively. Consistent with this possibility, immunoprecipitation of ectopically expressed HDAC1-Δ53 pulls down endogenous HDAC3 (Fig. 6C), and likewise, endogenous HDAC1 co-precipitates with HDAC3-H134Q (Fig. 6D). When deacetylase-deficient HDAC1 and HDAC3 mutants are expressed together, there is no toxicity in HK (Fig. 6E). Although toxicity is seen in LK, it is significantly reduced when compared with HDAC3-H134Q alone (Fig. 6B). Taken together, these results suggest that deacetylase activity is required for the toxicity of HDAC1 and HDAC3, but that this activity can be contributed to the HDAC1-HDAC3 complex by either of the two HDAC proteins.
FIGURE 6.

Deacetylase activity of either HDAC1 or HDAC3 Is sufficient for neurotoxicity. A, CGNs transfected with GFP, HDAC1, or HDAC1-Δ53 were treated with HK or LK medium after 8 h. Viability was assessed 24 h later. B, CGNs transfected with GFP, HDAC3, or HDAC3-H134Q were treated with HK or LK medium 8 h later for 24 h, and viability was assessed. C, HEK293 cells were transfected with either GFP-tagged HDAC1 or HDAC1-Δ53. The proteins were immunoprecipitated (IP) using either GFP or IgG antibody and analyzed by Western blotting (IB) with HDAC3 antibody. Both HDAC1 and HDAC1-Δ53 pull down endogenous HDAC3. The lower panel shows the input probed with GFP antibody showing overexpressed HDAC1 proteins. D, lysates from HEK293 cells transfected with FLAG-tagged HDAC3 or HDAC3-H134Q were immunoprecipitated with either FLAG or IgG antibody and analyzed by Western blotting with HDAC1 antibody. Both HDAC3 and HDAC3-H134Q pull down HDAC1. The lower panel shows the input probed with FLAG antibody showing overexpressed HDAC3 proteins. E, CGNs were transfected with GFP, HDAC1-Δ53, or HDAC3-H134Q or co-transfected with HDAC1-Δ53 and HDAC3-H134Q and treated with HK or LK medium after 8 h for 24 h, after which viability was quantified. Error bars in panels A, B, and E indicate S.D.
HDAC1 Is a Molecular Switch Promoting Both Neuronal Survival and Neuronal Death
The expression of HDRP, a truncated form of HDAC9 generated by alternative splicing and completely lacking the HDAC catalytic domain, is high in healthy neurons but is sharply down-regulated in neurons primed to die (7). Forced suppression of HDRP expression induces death in otherwise healthy CGNs, whereas HDRP overexpression protects neurons from LK-induced death. Although overexpressed HDRP is predominantly cytoplasmic in HK, it translocates to the nucleus following LK treatment where neuroprotection is mediated (7). We previously described that HDRP-mediated neuroprotection depends on deacetylase activity, which is acquired through interaction with HDAC1 (7). As described above, HDAC1 also contributes to the neurotoxic effect of HDAC3. This suggests that HDAC1 can contribute to both the survival and the death of neurons depending on whether it interacts with HDRP or HDAC3, respectively.
We tested the possibility that HDRP protects neurons by disrupting the neurotoxic interaction between HDAC1 and HDAC3 through the sequestration of HDAC1. In accordance with this model, the association between HDAC1 and HDAC3 is reduced when HDRP is expressed (Fig. 7A). Furthermore, HDRP interacts with HDAC1, but no interaction is observed with HDAC3 (Fig. 7B). Consistent with the requirement for interaction in HDAC1-HDAC3 toxicity, neuronal death induced by HDAC3 expression is reduced when HDRP is overexpressed (Fig. 7C). Because HDAC3 does not interact directly with HDRP, our results suggest that the reduction of toxicity by HDRP results from its sequestration of HDAC1.
FIGURE 7.

HDRP competes with HDAC3 for binding to HDAC1 and can reduce HDAC1 and HDAC3 toxicity. A, HEK293 cells were transfected with either GFP or HDRP-FLAG for 36 h. Endogenous HDAC3 was immunoprecipitated, and the immunoprecipitate (IP) was subjected to Western blotting and probed with HDAC1 antibody. The membrane was reprobed with HDAC3 antibody to show equal pulldown. The lower two panels show input lanes with overexpressed HDRP and equal endogenous HDAC1 expression. WCL, whole cell lysate. B, HEK293 cells were co-transfected with HDRP-FLAG and GFP, HDAC3-FLAG, HDAC1-Δ53-GFP, or HDAC1-GFP for 36 h. Immunoprecipitation was carried out using either GFP or HDAC3 antibody, and the immunoprecipitates were subjected to Western blot analysis and probed with FLAG antibody. The membrane was reprobed with GFP to show pulldown of the HDAC1 proteins. The bottom panel shows pulldown of FLAG-tagged HDAC3. C, CGNs were transfected with HDAC1, HDAC3, HDAC1-Δ53, or HDRP or co-transfected HDRP with HDAC1, HDAC3, or HDAC1-Δ53. The cultures were switched to HK or LK medium 8 h later, and viability was assessed 24 h later. Error bars indicate S.D.
Interaction between HDRP and HDAC1 occurs within the N-terminal 53 amino acids of HDAC1 as HDAC1-Δ53 does not co-precipitate with HDRP (Fig. 7B). Moreover, although the toxicity resulting from overexpression of HDAC1 can be inhibited by HDRP overexpression, the toxicity of HDAC1-Δ53 is not inhibited (Fig. 7C). These results strengthen the idea that HDRP inhibits HDAC1 neurotoxicity by interacting with it.
DISCUSSION
The role of HDAC1 in the regulation of neuronal survival has been unclear. Although we and other laboratories have previously shown that HDAC1 protects neurons from death (7, 10), others have shown that HDAC1 promotes axonal and neuronal degeneration (15, 16). In this study, we find that HDAC1 can have a dual role in the regulation of neuronal viability; it can promote both neuronal survival and neuronal death. Although neuroprotection by HDAC1 is mediated through association with survival-promoting HDACs such as HDRP (36), neuronal death involves its interaction with HDAC3.
It remains to be clarified where interaction between HDAC1 and HDAC3 occurs within the cells. We have found that HDAC1 is predominantly nuclear, whereas HDAC3 localizes to both the nucleus and the cytoplasm in neurons.3 Although this suggests that interaction occurs in the nucleus, one study has reported that in degenerating neurons, HDAC1 translocates from the nucleus to the cytoplasm and that cytoplasmic localization of HDAC1 is necessary for neuronal and axonal degeneration (16). Although our studies have failed to observe translocation of HDAC1, we cannot rule out the possibility that a small amount of HDAC1 does move out of the nucleus, where it interacts with cytoplasmic HDAC3.
Assuming that HDAC1 and HDAC3 interaction occurs within the nucleus, how is it prevented in healthy neurons? HDAC3 is phosphorylated by GSK3β, a modification that is necessary for its neurotoxic effect (14). It is possible that GSK3β phosphorylation of HDAC3 promotes its interaction with HDAC1. The finding that inhibition of GSK3β blocks neurotoxicity by HDAC1 expression, although it does not directly phosphorylate HDAC1, is consistent with this possibility. Our results suggest that via direct interaction, HDAC1 and HDAC3 cooperate to promote neuronal death. Indeed, a knockdown of either HDAC1 or HDAC3 is sufficient to prevent neuronal death by either of these proteins. Additionally, manipulations that inhibit HDAC3 neurotoxicity, such as IGF-1 treatment or expression of active Akt, also inhibit the ability of HDAC1 to induce neuronal death.
We have previously shown that overexpression of HDRP, a Class IIA HDAC and an alternatively spliced form of HDAC9, protects neurons from neuronal death (7). The neuroprotective effect of ectopically expressed HDRP involves its translocation from the cytoplasm to the nucleus (7). Our results suggest that within the nucleus, HDRP interacts with HDAC1 and in doing so denies access to HDAC3 (7). The finding that overexpressed HDRP blocks interaction between HDAC1 and HDAC3 without interacting with HDAC3 is consistent with this idea. Besides inhibiting association with HDAC3, HDRP acquires deacetylase activity through its interaction with HDAC1. We have previously shown that this activity is used by HDRP to repress the c-jun promoter (7). Thus, although promoting neuronal death when associated with HDAC3, HDAC1 is neuroprotective when it partners with HDRP.
A recent study described impressive protection by chemical compounds that inhibited both HDAC1 and HDAC3 in tissue culture and in fly as well as mouse models of HD (37). This is consistent with our results showing cooperation between HDAC1 and HDAC3 in promoting neurodegeneration. Based on our observations, it is likely that such HDAC1-HDAC3 inhibitors will also be protective in models of neurodegenerative diseases other than HD. In addition to pharmacological inhibitors of catalytic activity, peptides or other agents that inhibit HDAC1-HDAC3 interaction could be useful in preventing neurodegeneration.
In summary, we propose that HDAC1 acts as a molecular switch providing the balance between neuroprotection and neurotoxicity. Whether HDAC1 promotes neuronal survival or death depends on whether it interacts with proteins such as HDRP or HDAC3. Our findings reconcile the opposing roles previously ascribed to HDAC1 in the regulation of neuronal death.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants NS40408 and NS058462 (to S. R. D.).
This article was selected as a Paper of the Week.

This article contains supplemental Fig. 1.
F. H. Bardai, V. Price, M. Zaayman, L. Wang, and S. R. D'Mello, unpublished observations.
- HDAC
- histone deacetylase
- HD
- Huntington disease
- CaMK
- calmodulin-dependent protein kinase
- HDRP
- histone deacetylase-related protein
- GSK3β
- glycogen synthase kinase 3β
- HK
- high potassium medium
- LK
- low potassium medium
- HCA
- homocysteic acid
- CGN
- cerebellar granule neuron
- CDK
- cyclin-dependent kinase.
REFERENCES
- 1. Yang X. J., Seto E. (2008) The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 9, 206–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Haberland M., Montgomery R. L., Olson E. N. (2009) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 10, 32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. D'Mello S. R. (2009) Histone deacetylases as targets for the treatment of human neurodegenerative diseases. Drug News Perspect. 22, 513–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kazantsev A. G., Thompson L. M. (2008) Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat. Rev. Drug Discov. 7, 854–868 [DOI] [PubMed] [Google Scholar]
- 5. Sleiman S. F., Basso M., Mahishi L., Kozikowski A. P., Donohoe M. E., Langley B., Ratan R. R. (2009) Putting the “HAT” back on survival signaling: the promises and challenges of HDAC inhibition in the treatment of neurological conditions. Expert Opin. Investig. Drugs 18, 573–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Majdzadeh N., Wang L., Morrison B. E., Bassel-Duby R., Olson E. N., D'Mello S. R. (2008) HDAC4 inhibits cell cycle progression and protects neurons from cell death. Dev. Neurobiol. 68, 1076–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Morrison B. E., Majdzadeh N., Zhang X., Lyles A., Bassel-Duby R., Olson E. N., D'Mello S. R. (2006) Neuroprotection by histone deacetylase-related protein. Mol. Cell Biol. 26, 3550–3564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pandey U. B., Nie Z., Batlevi Y., McCray B. A., Ritson G. P., Nedelsky N. B., Schwartz S. L., DiProspero N. A., Knight M. A., Schuldiner O., Padmanabhan R., Hild M., Berry D. L., Garza D., Hubbert C. C., Yao T. P., Baehrecke E. H., Taylor J. P. (2007) HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447, 859–863 [DOI] [PubMed] [Google Scholar]
- 9. Pfister J. A., Ma C., Morrison B. E., D'Mello S. R. (2008) Opposing effects of sirtuins on neuronal survival: SIRT1-mediated neuroprotection is independent of its deacetylase activity. PLoS ONE 3, e4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim D., Frank C. L., Dobbin M. M., Tsunemoto R. K., Tu W., Peng P. L., Guan J. S., Lee B. H., Moy L. Y., Giusti P., Broodie N., Mazitschek R., Delalle I., Haggarty S. J., Neve R. L., Lu Y., Tsai L. H. (2008) Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron 60, 803–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim D., Nguyen M. D., Dobbin M. M., Fischer A., Sananbenesi F., Rodgers J. T., Delalle I., Baur J. A., Sui G., Armour S. M., Puigserver P., Sinclair D. A., Tsai L. H. (2007) SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer disease and amyotrophic lateral sclerosis. EMBO J. 26, 3169–3179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Qin W., Yang T., Ho L., Zhao Z., Wang J., Chen L., Zhao W., Thiyagarajan M., MacGrogan D., Rodgers J. T., Puigserver P., Sadoshima J., Deng H., Pedrini S., Gandy S., Sauve A. A., Pasinetti G. M. (2006) Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J. Biol. Chem. 281, 21745–21754 [DOI] [PubMed] [Google Scholar]
- 13. Chen J., Zhou Y., Mueller-Steiner S., Chen L. F., Kwon H., Yi S., Mucke L., Gan L. (2005) SIRT1 protects against microglia-dependent amyloid-β toxicity through inhibiting NF-κB signaling. J. Biol. Chem. 280, 40364–40374 [DOI] [PubMed] [Google Scholar]
- 14. Bardai F. H., D'Mello S. R. (2011) Selective toxicity by HDAC3 in neurons: regulation by Akt and GSK3β. J. Neurosci. 31, 1746–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jeong H., Then F., Melia T. J., Jr., Mazzulli J. R., Cui L., Savas J. N., Voisine C., Paganetti P., Tanese N., Hart A. C., Yamamoto A., Krainc D. (2009) Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell 137, 60–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim J. Y., Shen S., Dietz K., He Y., Howell O., Reynolds R., Casaccia P. (2010) HDAC1 nuclear export induced by pathological conditions is essential for the onset of axonal damage. Nat. Neurosci. 13, 180–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. D'Mello S. R., Galli C., Ciotti T., Calissano P. (1993) Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc. Natl. Acad. Sci. U.S.A. 90, 10989–10993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang L., Ankati H., Akubathini S. K., Balderamos M., Storey C. A., Patel A. V., Price V., Kretzschmar D., Biehl E. R., D'Mello S. R. (2010) Identification of novel 1,4-benzoxazine compounds that are protective in tissue culture and in vivo models of neurodegeneration. J. Neurosci. Res. 88, 1970–1984 [DOI] [PubMed] [Google Scholar]
- 19. Chen H. M., Wang L., D'Mello S. R. (2008) A chemical compound commonly used to inhibit PKR, {8-(imidazol-4-ylmethylene)-6H-azolidino[5,4-g] benzothiazol-7-one}, protects neurons by inhibiting cyclin-dependent kinase. Eur. J. Neurosci. 28, 2003–2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ghosh S., Feany M. B. (2004) Comparison of pathways controlling toxicity in the eye and brain in Drosophila models of human neurodegenerative diseases. Hum. Mol. Genet. 13, 2011–2018 [DOI] [PubMed] [Google Scholar]
- 21. Ma C., D'Mello S. R. (2011) Neuroprotection by histone deacetylase-7 (HDAC7) occurs by inhibition of c-jun expression through a deacetylase-independent mechanism. J. Biol. Chem. 286, 4819–4828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Montgomery R. L., Potthoff M. J., Haberland M., Qi X., Matsuzaki S., Humphries K. M., Richardson J. A., Bassel-Duby R., Olson E. N. (2008) Maintenance of cardiac energy metabolism by histone deacetylase-3 in mice. J. Clin. Invest. 118, 3588–3597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cruz J. C., Tseng H. C., Goldman J. A., Shih H., Tsai L. H. (2003) Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron 40, 471–483 [DOI] [PubMed] [Google Scholar]
- 24. Fischer A., Sananbenesi F., Pang P. T., Lu B., Tsai L. H. (2005) Opposing roles of transient and prolonged expression of p25 in synaptic plasticity and hippocampus-dependent memory. Neuron 48, 825–838 [DOI] [PubMed] [Google Scholar]
- 25. Li J. Y., Popovic N., Brundin P. (2005) The use of the R6 transgenic mouse models of Huntington disease in attempts to develop novel therapeutic strategies. NeuroRx 2, 447–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. D'Mello S. R., Borodezt K., Soltoff S. P. (1997) Insulin-like growth factor and potassium depolarization maintain neuronal survival by distinct pathways: possible involvement of PI 3-kinase in IGF-1 signaling. J. Neurosci. 17, 1548–1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dudek H., Datta S. R., Franke T. F., Birnbaum M. J., Yao R., Cooper G. M., Segal R. A., Kaplan D. R., Greenberg M. E. (1997) Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275, 661–665 [DOI] [PubMed] [Google Scholar]
- 28. Hernández F., Nido J. D., Avila J., Villanueva N. (2009) GSK3 inhibitors and disease. Mini Rev. Med. Chem. 9, 1024–1029 [DOI] [PubMed] [Google Scholar]
- 29. Bhat R. V., Budd Haeberlein S. L., Avila J. (2004) Glycogen synthase kinase 3: a drug target for CNS therapies. J. Neurochem. 89, 1313–1317 [DOI] [PubMed] [Google Scholar]
- 30. Shaw M., Cohen P., Alessi D. R. (1997) Further evidence that the inhibition of glycogen synthase kinase-3β by IGF-1 is mediated by PDK1/PKB-induced phosphorylation of Ser-9 and not by dephosphorylation of Tyr-216. FEBS Lett. 416, 307–311 [DOI] [PubMed] [Google Scholar]
- 31. Cross D. A., Alessi D. R., Cohen P., Andjelkovich M., Hemmings B. A. (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 378, 785–789 [DOI] [PubMed] [Google Scholar]
- 32. Borsello T., Forloni G. (2007) JNK signaling: a possible target to prevent neurodegeneration. Curr. Pharm. Des. 13, 1875–1886 [DOI] [PubMed] [Google Scholar]
- 33. Greene L. A., Liu D. X., Troy C. M., Biswas S. C. (2007) Cell cycle molecules define a pathway required for neuron death in development and disease. Biochim. Biophys. Acta 1772, 392–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grégoire S., Xiao L., Nie J., Zhang X., Xu M., Li J., Wong J., Seto E., Yang X. J. (2007) Histone deacetylase-3 interacts with and deacetylates myocyte enhancer factor 2. Mol. Cell Biol. 27, 1280–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tou L., Liu Q., Shivdasani R. A. (2004) Regulation of mammalian epithelial differentiation and intestine development by Class I histone deacetylases. Mol. Cell Biol. 24, 3132–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Morrison B. E., Majdzadeh N., D'Mello S. R. (2007) Histone deacetylases: focus on the nervous system. Cell. Mol. Life Sci. 64, 2258–2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jia H., Pallos J., Jacques V., Lau A., Tang B., Cooper A., Syed A., Purcell J., Chen Y., Sharma S., Sangrey G. R., Darnell S. B., Plasterer H., Sadri-Vakili G., Gottesfeld J. M., Thompson L. M., Rusche J. R., Marsh J. L., Thomas E. A. (2012) Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington disease. Neurobiol. Dis. 46, 351–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.