

Abstract
Two DCM mutants (E40K and E54K) of tropomyosin (Tm) were examined using the thin-filament extraction/reconstitution technique. The effects of the Ca2+, ATP, phosphate (Pi), and ADP concentrations on isometric tension and its transients were studied at 25°C, and the results were compared to those for the WT protein. Our results indicate that both E40K and E54K have a significantly lower T HC (high Ca2+ tension at pCa 4.66) (E40K: 1.21±0.06 T a, ±SEM, N = 34; E54K: 1.24±0.07 T a, N = 28), a significantly lower T LC (low- Ca2+ tension at pCa 7.0) (E40K: 0.07±0.02 T a, N = 34; E54K: 0.06±0.02 T a, N = 28), and a significantly lower T act (Ca2+ activatable tension) (T act = T HC–TLC, E40K: 1.15±0.08 T a, N = 34; E54K: 1.18±0.06 T a, N = 28) than WT (T HC = 1.53±0.07 T a, T LC = 0.12±0.01 T a, T act = 1.40±0.07 T a, N = 25). All tensions were normalized to T a ( = 13.9±0.8 kPa, N = 57), the tension of actin-filament reconstituted cardiac fibers (myocardium) under the standard activating conditions. The Ca2+ sensitivity (pCa50) of E40K (5.23±0.02, N = 34) and E54K (5.24±0.03, N = 28) was similar to that of the WT protein (5.26±0.03, N = 25). The cooperativity increased significantly in E54K (3.73±0.25, N = 28) compared to WT (2.80±0.17, N = 25). Seven kinetic constants were deduced using sinusoidal analysis at pCa 4.66. These results enabled us to calculate the cross-bridge distribution in the strongly attached states, and thereby deduce the force/cross-bridge. The results indicate that the force/cross-bridge is ∼15% less in E54K than WT, but remains similar to that of the WT protein in the case of E40K. We conclude that over-inhibition of the actomyosin interaction by E40K and E54K Tm mutants leads to a decreased force-generating ability at systole, which is the main mechanism underlying the early pathogenesis of DCM.
Introduction
Dilated cardiomyopathy (DCM) is a myocardial disorder that leads to heart failure and sudden cardiac death [1]. In United States, DCM affects 30–40 people in 100,000 [2] and accounts for ∼10,000 deaths (∼10% of those affected) annually [3]. The main clinical manifestation of DCM is dilation of the left ventricle (LV) and systolic dysfunction. In vitro studies have suggested that the systolic dysfunction observed in DCM may be directly related to a decrease in the ATP hydrolysis rate [4].
Although the causes of DCM are diverse and thus general conclusions are often inappropriate, it is now clear that at least 25–30% of the DCM is caused by inherited genetic defects. Hence it is also called familial dilated cardiomyopathy (DCM) [5], [6]. Mutations in more than 20 genes, including several that encode sarcomeric proteins, have been implicated in DCM [7]. Unlike familial hypertrophic cardiomyopathy (FHC, HCM), where mutations in genes encoding sarcomeric proteins play a major role in disease pathogenesis, in the case of DCM the number of such mutations is small and they have not been studied extensively. So far, 12 α-tropomyosin (Tm) mutants are known to cause HCM, whereas only three Tm mutants (E40K, E54K and D230N) are known to cause DCM [8], [9].
Notably, different mutations within a single gene encoding a sarcomeric protein (actin, myosin, troponin, MyBP-C, etc.) can lead to one of two distinct diseases, HCM or DCM [10]. It would be interesting to learn why mutations within a single gene result in two different phenotypes. Previous studies have indicated that the pCa50 (Ca sensitivity, or [Ca2+] when tension reaches half of the maximum tension and defined by Eq. 1 in Methods) alteration caused by a particular mutation might be critical to emergence of a specific disease state: in vitro ATPase and motility assays have shown that HCM mutants increase pCa50, whereas DCM mutants decrease pCa50 [11]. However, in isometric tension studies, investigators also reported increased pCa50 in DCM myocardium [12], [13]. McConnell et al. have reported that in transgenic mice the pathogenesis, and thus disease phenotype, depend on the expression level of the mutant protein [14].
Since HCM generally results in diastolic dysfunction [15], and DCM generally results in systolic dysfunction [3], it has been hypothesized that a difference in myocardial contractility is the key determinant of a distinct disease phenotype. Also, the Potter’s group has shown that in DCM-associated Tn mutant mice, the decrease in the capacity for force generation is likely caused by reduced actomyosin binding [16]. Recently, we have reported that an HCM-associated Tm mutant caused elevated tension at low Ca2+ (pCa 8), from which we inferred that the molecular mechanism responsible is impaired relaxation [17]. Consequently, a tension-based study of the DCM-related mutants would be beneficial in investigating the difference between DCM and HCM pathogenesis.
Tm is a coiled-coil molecule stabilized by a heptad repeat: E40 (at the e position) of one chain interacts with R35 (g) of another, and E54 (e) of one chain interacts with K49 (g) of another, both through an electrostatic force [18], [19]. Also, it should be noted that E40 and E54 are located within the actin-binding motif of the Tm N-terminal region [20]. Mutants carrying an amino acid of the opposite charge may be destabilized, resulting in a change in the affinity of Tm for actin. Furthermore, E40K is located in period 1b, which interacts with actin in the presence of Ca2+, whereas E54K is located in period 2a, which interacts with actin in the absence of Ca2+ [21]. Previous studies have shown that the Ca2+ sensitivity of myocardium containing either of these mutants is decreased [22], and this is thought to be critical to DCM pathogenesis [11], [23]. At the same time, the maximum ATPase activity of E40K was shown to be lower than that of the WT protein [23]. A solution study performed in the absence of Tn showed that E40K reduced the cross-bridge turnover rate (decreased K T) and weakened the affinity of Tm for actin in the presence of myosin [23]. E54K showed only a weakened Tm-actin affinity in the absence of Tn and myosin (other parameters stayed unchanged from WT), leading to the hypothesis that E40K and E54K cause the DCM phenotype through different mechanisms [23].
The effect of a DCM-related Tm mutant on cross-bridge kinetics has never been investigated. Furthermore, most earlier studies did not include information on stiffness in the myocardial system under systolic and diastolic conditions. Here we report the effects of two DCM-related Tm mutants on isometric force, stiffness, and cross-bridge kinetics, as assessed using the thin-filament removal/reconstitution technique combined with sinusoidal analysis [24], [25], [26], and compared the results to those for WT Tm. Our results demonstrate that: 1) the systolic dysfunction in DCM is caused by decreased myocardial contractility, as indicated by the significant decrease in Ca2+-activatable tension in both mutants; 2) the pCa50 of the reconstituted myocardium does not change for either mutant in a solution mimicking the physiological composition; and 3) diastolic dysfunction in Tm mutation related DCM is caused by over-inhibition of the myocardium (based on a significant decrease in the number of cycling cross-bridges and reduced isometric tension at pCa 7.0).
Materials and Methods
Experimental Materials and the Thin-Filament Extraction/Reconstitution Technique
All animal studies conducted at the University of Iowa were in accordance with institutional guidelines and protocols approved by the Animal Care and Use Committee (ACUC). The University of Iowa has an Animal Welfare Assurance on file with the Office of Laboratory Animal Welfare (OLAW), National Institutes of Health. Research projects performed at Kawai’s laboratory using animals were approved by institutional ACUC.
Bovine cardiac muscles were dissected from trabeculae that were freely suspended in the right ventricle, split into small bundles, chemically skinned, and stored at −20°C [24]. These bundles can be stored for up to 2–3 months without altering their activity. On the day an experiment was carried out, these were further split into thin bundles (length ∼2 mm, diameter 90–110 µm). The latter were attached to the experimental apparatus using nail polish, and further skinned in 1% Triton X100 for 20 min in the relaxing solution (Table S1 in Ref. [17]). Preparations were then stretched to a sarcomere length of ∼2.1 µm, and the diameter and the length were measured. These values were used to calculate isometric tension, and the elastic and viscous moduli.
The preparations were then used for extraction, reconstitution, and subsequent mechanical measurements of the thin filament. The technique for extracting and reconstituting the thin filament was originally developed in the Ishiwata laboratory [26] and refined in the Kawai laboratory [25], and was performed as described [17]. Typical slow pen traces of tension time courses, light and electron micrographs at each stage of extraction and reconstitution, and associated SDS-PAGE were published previously [24], [25], [26], [27], [28]. In brief, the thin-filament (contains actin, tropomyosin, and troponin) was first extracted from skinned muscle fibers using gelsolin, a plasma protein which severs actin and thin filaments. The actin filament was then reconstituted in the fibers, in which active tension was measured and termed as T a. Then the regulatory proteins, Tm and Tn, were reconstituted in the fibers (this was when mutant Tm was used) to complete the reconstitution process.
Proteins
G-actin was purified from rabbit fast-twitch skeletal muscle as described [29], and bovine cardiac Tn was purified from bovine hearts as described [30]. Both proteins were purified in the Kawai laboratory. Human WT and mutant α-Tms were expressed as recombinant proteins in E. coli and purified in the Dr. James D. Potter labortory. 100% pure mutant Tm was used in the reconstitution. These Tms have two extra amino acids, Ala-Ser, at the N-terminus, and these functionally substitute for acetylation [31]. Previous studies showed that this N-terminal extension is necessary for normal Tm function but has little effect on either protein stability [22] or Ca2+ sensitivity [32].
Experimental Solutions
For the ATP, Pi and ADP studies, exactly the same solutions were used as previously reported (Table S1 in [17]). Two sets of solutions were used for the pCa studies. One set has been used for many years by us, is called “high ionic strength (IS) solutions”, and contains: 6 mM total of K2H2EGTA and K2CaEGTA, 6.1 mM Na2H2ATP, 6.6 mM MgAc2 (Ac = acetate), 8 mM K1.5H1.5Pi, 54 mM KAc, 3 mM NaAc, 10 mM NaN3, 10 mM MOPS, 15 mM creatine phosphate (Na2CP) and 320 units/ml creatine kinase (CK). The IS of this solution is 200 mM, [MgATP2-] is 5 mM, free [Mg2+] 1 mM, [Na+] 55 mM, and the pH is adjusted to 7.00 using KOH [17]. The other set was used by Chang and Potter in their study on E40K and E54K, is called “low-IS solution”, and contains: 7 mM total of K2H2EGTA and K2CaEGTA, 4.2 mM MgAc2, 40 mM KAc, 2.5 mM Na2MgATP, no added Pi, 20 mM MOPS, 20 mM Na2CP, and 15 U/ml CK. The IS of this solution is 150 mM, [MgATP2-] is 2.2 mM, free [Mg2+] 1.3 mM, [Na+] 45 mM, and the pH is adjusted to 7.00 using KOH [22].
pCa-Tension Study
The pCa-tension study was performed as described [32], in the range of pCa 7.0–4.0. The tension and stiffness at pCa 7.0 were called low Ca2+ tension (T LC) and low Ca2+ stiffness (Y LC). Here the stiffness is defined as Y ∞ [33], but the ∞ symbol is dropped for simplicity. The tension and stiffness at pCa 4.0 were called high Ca2+ tension (T HC) and high Ca2+ stiffness (Y HC). The tension baseline was defined as that existing in the “super relaxing” solution (Table S1 in [17]) at 0°C, in the presence of 6 mM EGTA and 40 mM BDM. There is no significant tension or cross-bridge cycling under these conditions. Subsequent tension measurements were performed as the tension was incrementally increased from the baseline level. The pCa-tension relationship was studied in both the high-IS and low-IS pCa solutions. The results were fitted to the Hill equation:
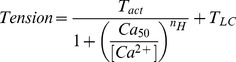 |
(1) |
where pCa = –log10[Ca2+], T act is the Ca2+ activatable tension, and Ca50 is the apparent Ca2+ dissociation constant, which represents the Ca2+ concentration at half tension. pCa50 ( = –log10Ca50) represents Ca2+-sensitivity, and n H (the Hill factor) represents the cooperativity. T HC = T act+T LC is the tension at high [Ca2+]. pCa-tension curves were individually fitted to Eq. 1, and the fitted parameters were averaged. All tension values were normalized to T a of the actin-filament reconstituted myocardium without Tm or Tn, in the standard activating solution (5S8P, Table S2 in [17]). T a averaged to 13.9±0.8 KPa (N = 57). All experiments were carried out at 25°C.
Sinusoidal Analysis
The elementary steps of the cross-bridge cycle based on six states (Fig. 1) were characterized by sinusoidal analysis performed as described [33], [34] in sets of solutions with 200 mM IS. Sinusoidal length changes of small amplitude (0.125% L0) were applied to the reconstituted myocardium at 18 different frequencies (f) in the range of 0.13 Hz to 100 Hz. The resulting tension transients were analyzed and the complex modulus Y(f) was calculated. Y(f) is the ratio of the stress change to the strain change represented in the frequency domain. Y(f) was fitted to Eq. 2, which incorporates 2 exponential processes [33]:
Figure 1. Elementary steps of the cross-bridge cycle.

The uppercase letters K indicate the association or equilibrium constants, and the lowercase letters k the rate constants of the elementary steps. Collectively these are referred to as the “kinetic constants”. A = actin, M = Myosin, D = MgADP, S = MgATP, and P = Pi = Phosphate.
Process B Process C
| (2) |
where 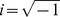 ; 2πb and 2πc (b<c) are the apparent rate constants of exponential processes B and C, respectively; B and C are their respective magnitudes (amplitudes), and H is a constant. Y
∞ = H−B+C is the modulus extrapolated to the infinite frequency; all of these are real numbers. B, C, H, and Y
∞ have the same units as Y(f) and isometric tension, and thus are normalized to T
a. In cardiac muscle fibers, process A was not observed at ≤25°C [25], [34], [35]. The Y(f) thus measured includes the effect of series compliance, which may [36] or may not [37] affect the apparent rate constants. However, the series compliance is not a concern in this report because it does not differ between the mutant and WT proteins.
; 2πb and 2πc (b<c) are the apparent rate constants of exponential processes B and C, respectively; B and C are their respective magnitudes (amplitudes), and H is a constant. Y
∞ = H−B+C is the modulus extrapolated to the infinite frequency; all of these are real numbers. B, C, H, and Y
∞ have the same units as Y(f) and isometric tension, and thus are normalized to T
a. In cardiac muscle fibers, process A was not observed at ≤25°C [25], [34], [35]. The Y(f) thus measured includes the effect of series compliance, which may [36] or may not [37] affect the apparent rate constants. However, the series compliance is not a concern in this report because it does not differ between the mutant and WT proteins.
The apparent rate constants 2πb and 2πc were studied as functions of the MgATP (S), Pi (P) and MgADP (D) concentrations. The data were fitted to the following equations, which were derived from the cross-bridge scheme in Fig. 1, assuming that steps 0, 1 and 5 are in fast equilibrium, and that step 6 is the slowest forward step of the cross-bridge cycle [38].
| (3) |
| (4) |
where
| (5) |
K 1 is the ATP association constant, K 0 is the ADP association constant, k 2 is the forward rate constant of cross-bridge detachment step 2, and k −2 is its reversal step. k 4 is the forward rate constant of the force generation (isomerization) step 4, and k −4 is its reversal step. K 5 is the Pi association constant. σ in Eq. 5 was calculated from K 1 and K 2 obtained from the MgATP study and S = 5 mM, the condition of the Pi study. The reconstituted myocardium was studied at 8 different [MgATP] (0.05, 0.1, 0.2, 0.5, 1, 2, 5, 10 mM) with a fixed [Pi] (8 mM) and pCa 4.66, and the data were fitted to Eq. 3 to deduce the rate and association constants of steps 1 and 2. D = 0 was assumed in this study because of the presence of CP and CK. The effect of phosphate (Pi) was studied at 6 different [Pi] (0, 2, 4, 8, 16, 32 mM) with a fixed [MgATP] (5 mM) and pCa 4.66, and the data were fitted to Eq. 4 to deduce the rate and association constants of steps 4 and 5. The effect of MgADP was studied at 4 different [MgADP] (0, 1, 2, 3 mM), with fixed [Pi] (8 mM) and [MgATP] (2 mM), at pCa 4.66, and the data were fitted to Eq. 3 to deduce K 0.
Results
pCa-tension and pCa-stiffness Studies
To determine the effects of Tm mutants E40K and E54K on Ca2+-sensitivity and cooperativity, tension and stiffness of the thin filament-reconstituted myocardium were studied as functions of [Ca2+]. The pCa-tension plots comparing mutant and WT Tms are displayed in Fig. 2. Their parameters, as fitted to Eq. 1, are plotted in Fig. 3. As seen in these figures, both mutants affected the pCa-tension relationship. They also both showed significantly decreased T HC (WT: 1.59±0.09 T a, N = 25; E40K: 1.28±0.07 T a, N = 34; E54K: 1.29±0.08 T a, N = 28), T act (WT: 1.54±0.08 T a, N = 25; E40K: 1.24±0.06 T a, N = 34; E54K: 1.20±0.07 T a, N = 28) and T LC (WT: 0.13±0.01 T a, N = 25; E40K: 0.08±0.01 T a, N = 34; E54K: 0.06±0.01 T a, N = 28) compared to WT (P<0.005), indicating that contractility was decreased during systole (T HC) and over-inhibited during diastole (T LC). The trend was the same for stiffness (Fig. 3B). The pCa50 of the E40K (5.20±0.03, N = 34, p = 0.25) or E54K (5.23±0.03, N = 28, p = 0.36)-reconstituted myocardium was not significantly different from that of WT (5.26±0.03, N = 25), demonstrating that Ca2+-sensitivity was unaltered (Fig. 3C). The cooperativity (n H) of E40K was similar to that of WT, whereas that of E54K was significantly greater than that of WT (Fig. 3D) (WT 2.80±0.17, E40K 3.01±0.25, E54K 3.64±0.26; see also Table S1 in [17]).
Figure 2. pCa-tension plots.

(A, B) pCa-tension plots comparing mutant Tms and WT-Tm in high-IS solution. N = 25 for WT Tm; N = 34 for E40K; N = 28 for E54K. (C, D) pCa-tension plots comparing mutant Tms and WT-Tm in low-IS solution [22]. N = 8 for WT Tm; N = 10 for E40K; N = 9 for E54K. The means and their SEMs are shown. Curved lines are calculated from Eq. 1, based on best-fit parameters. Tension is normalized to that of actin-filament reconstituted fibers (Ta = 13.9±0.8 kPa, N = 57).
Figure 3. Summary of Tension (A and E), Stiffness (B and F), pCa50 (C and G), and Cooperativity (D and H).

(A–D) Data obtained in high-IS solutions. (E–H) Data obtained in low-IS solutions [22]. (A, E) Low calcium tension (T LC) represents the tension at pCa 7. High calcium tension (T HC) represents the tension at pCa 4.66. Active tension (T act) is the tension developed on Ca2+ activation (T act = T HC–T LC). (B, F) Results of the stiffness study. Tension and stiffness were normalized to Ta. (C, G) pCa50 (Ca2+ sensitivity). (D, H) Cooperativity. *: p<0.05.
pCa-tension Study using the Chang and Potter pCa Solution
Independent studies have indicated that both E40K and E54K cause a decrease in Ca2+ sensitivity, based on an ATPase assay, skinned fiber force measurement, and an in vitro motility assay [22], [39], [40]. Although the experimental conditions varied among these studies, the pCa solutions used for the ATPase assay and tension measurement analysis shared two common features: 150 mM ionic strength (IS) and no added Pi [22], [40]. These conditions are significantly different from those of our study: we used 200 mM IS and 8 mM Pi (high-IS solution, see Methods). To investigate the possible effects of these differences on the pCa50, we studied the pCa-tension curve of the reconstituted myocardium using the solution developed by Chang and Potter [22] (low IS solution, Methods). The result was also fitted to the Hill equation (Eq. 1).
In the low-IS solution, T HC was generally 2–3x that of the high-IS solution (Fig. 3E vs. 3A). In general, the trend was not the same as that in the high-IS solution (Fig. 3E–H vs. 3A–D). E40K and E54K caused a significant decrease in pCa50 (E40K: 5.91±0.02, N = 10; E54K: 5.88±0.03, N = 9) compared to WT (6.14±0.05, N = 8) (Fig. 3G). E40K and E54K caused a significant decrease in T LC (E40K: 0.08±0.02 T a, N = 10; E54K: 0.27±0.05 T a, N = 9) compared to WT (0.63±0.09 T a, N = 8). However, the data for T HC were more scattered and the average values did not differ significantly among the mutant- and WT-reconstituted preparations (WT: 3.46±0.47 T a, N = 8; E40K: 4.08±0.2 T a, N = 10; E54K: 3.13±0.42 T a, N = 9). T act was significantly increased in E40K (4.00±0.19 T a, N = 10), but T act of E54K (2.87±0.45 T a, N = 9) remained similar to that in WT (2.83±0.45 T a, N = 8). The cooperativity remained similar between E54K (1.31±0.16, N = 9) and WT (1.28±0.18, N = 8), but increased significantly in E40K (1.99±0.21, N = 10) (Fig. 3H).
Sinusoidal Analysis and Cross-bridge Kinetics
Sinusoidal analysis was performed as described [25], [33] to assess cross-bridge kinetics and to characterize the elementary steps of the cross-bridge cycle. Nyquist plots comparing data for mutant (open symbols and dashed lines) and WT (filled symbols and solid lines) Tms are shown in Fig. 4, together with curves fitted to Eq. 2: panels A and B show Nyquist plots at pCa 4.66 under the standard activating condition (5S8P, Table S2 in [17]). Also shown are baseline records (+, ×) under the super-relaxing condition (Table S2 in [17]). Panels C and D show the data obtained at pCa 7. The Nyquist plot for the WT fibers demonstrates that an appreciable fraction of cross-bridges actively cycled and performed energy transduction, even at pCa 7 (black squares in Fig. 4C and D), as evidenced by the presence of processes B and C (represented by two contiguous semicircles), and as reported earlier for the myocardium [41]. However, in the cases of E40K and E54K, the magnitude of process C is significantly diminished, to ¼∼½, and process B is very small (B is close to 0) (red squares in Fig. 4C and D), indicating that energy transduction is significantly reduced; this result is consistent with the decreased T LC (Fig. 3A) of these two mutants. The diameters of the semicircles are proportional to the number of actively cycling cross-bridges [38]. The data in Fig. 4C and D demonstrate that, in the case of WT, only ∼13% of the cross-bridges at pCa 4.66 are active at pCa 7.0. In both E40K and E54K, the diameter of the Nyquist plots was significantly smaller than that for WT, demonstrating that only 5–7% of cross-bridges cycle actively at pCa 7. We conclude, therefore, that the regulatory proteins over-inhibit the actomyosin interaction, and that this results in a decrease in T LC and decreases in the magnitudes of B and C in both mutants at pCa 7.
Figure 4. Nyquist plots of the mutants (red) compared to WT (black).

(A–B) Two mutants are compared to WT at pCa 4.66; (C–D) Two mutants are compared to WT at pCa 7.0. Also included in A and B are the baselines taken in the relaxing solution at 0°C (× for the mutant, and + for WT). The data in A–D were obtained in high-IS solutions. (E–F) Two mutants are compared to WT at pCa 4.66 in low-IS solution [22]. Range of frequency used: 0.13–100 Hz. The frequency increases in the clock-wise direction. Note the difference in scale for both axes in panels A–B vs. panels C–D, and panels A–B vs. panels E–F.
The Nyquist plots for E40K and E54K in low-IS solution (pCa 4.66) are shown in Fig. 4E and F. It is clear that process B is absent (B = 0) when the low-IS solution is used; this is primarily due to the absence of Pi in the activating solution [38].
Two apparent rate constants, 2πb and 2πc, were measured as functions of [MgATP] (Fig. 5A and B) and [Pi] (Fig. 5C and D) at pCa 4.66. E40K caused a significant increase in 2πc (Fig. 5A) and a small increase in 2πb (Fig. 5C) compared to WT. E54K also caused increases in these rate constants (Fig. 5B and D), but these were not significant. The rate and association constants (together called “kinetic constants”) of the elementary steps were deduced by fitting the ligand concentration dependence of the apparent rate constants to Eqs. 3 and 4 of [38]. The kinetic constants of the mutants and WT are compared in Fig. 6. All kinetic constants were changed in the mutants, but only the following changes were significant (p<0.05) compared to WT. E40K exhibited an ∼70% decrease in K 0, an ∼610% increase in K 1, an ∼80% increase in K 2, an ∼70% decrease in K 4, an ∼50% decrease in K 5, and an ∼70% increase in k 2, and an ∼190% increase in k -4. E54K exhibited an ∼60% increase in K 0, an ∼260% increase in K 1, an ∼29% decrease in k 2, an ∼28% decrease in k -2, and an ∼50% increase in k -4. Interestingly, k 4 (the rate constant of the force generating step) did not change in either mutant.
Figure 5. Apparent rate constants.

The apparent rate constants for myocardium reconstituted with mutant Tms (red) and WT Tm (black). Symbols represent the mean±SEM. Continuous curves were generated by fitting the data to Eq. 3 (A and B) or Eq. 4 (C and D). (A–B): 2πc (in s−1) is plotted against [MgATP] (mM). (C–D): 2πb (in s−1) is plotted against [Pi] (mM).
Figure 6. Kinetic constants.

The kinetic constants of the cross-bridge cycle (Scheme 1) in reconstituted cardiac fibers are compared among mutant and WT Tms. (A) Rate constants. (B) Equilibrium constants. Note that K 0 was divided by 20 and K 5 was multiplied by 5. *: p<0.05.
Cross-bridge Distribution and Force per Cross-bridge
There are two possible mechanisms through which the observed decrease in isometric tension during Ca2+ activation (T HC, Fig. 3A) can be explained: either the number of force-generating cross-bridges, or the force generated by each cross-bridge is decreased. To find out which one of these mechanisms is at work, we calculated the distribution of cross-bridges in each state under the standard activating condition [42]; the result is shown in Fig. 7. Both mutants showed a significant decrease in the AM state (E40K: ∼74%; E54K: ∼60%, p<0.05) and an increase in the Det state (E40K: ∼88%; E54K: ∼30%, p<0.05) compared to WT. The Det state includes weakly attached states (AMS and AMDP) and truly detached states (MS and MDP) (Fig. 1). E40K showed an ∼40% decrease in AM*DP, and E54K showed an ∼53% increase in AM*S compared to WT (p<0.05). Att indicates the sum of all strongly attached (force generating) cross-bridges: Att = AMD+AM+AM*S+AM*DP+AM*D = 1–Det. Both mutants showed a significantly decreased Att state (E40K: ∼17%; E54K: ∼6%) compared to WT, indicating that the number of force-generating cross-bridges is decreased. Force per cross-bridge was obtained as T HC/Xatt, which remained similar to that of WT in E40K, but decreased by ∼13±6% in E54K (with error propagation) compared to WT.
Figure 7. Cross-bridge distributions.

From the equilibrium constants, the cross-bridge distribution over six states under the standard activating condition (5S8P, Table S2 in [17]) was calculated for two mutant and the WT Tm forms. Att indicates the sum of all strongly attached (force generating) cross-bridges: Att = AMD+AM+AM*S+AM*DP+AM*D.
The values of tension (T a = 13.9 kPa) produced in actin-filament reconstituted fibers in the current study are less than those reported in previous studies (eg: [43], [44], 39–75 kPa). This is because regulatory proteins, Tm and Tn, were absent when T a was measured. Furthermore, 8 mM Pi and 200 mM ionic strength were used in our standard activating solution and the temperature at which the experiments was carried out was 25°C. In many previous experiments, no Pi was added, an ionic strength of 150 mM was often used, and some experiments were carried out at higher temperatures such as 37°C. In addition, these experiments were carried out in the presence of regulatory proteins. An addition of the regulatory proteins increases tension to 1.5×[45], [46]. The deletion of 8 mM Pi increases tension to 2×[17], [25], [47], and a decrease of ionic strength from 200 mM to 150 mM increases tension to 1.2×[48], [49]. See also Fig. 3A vs. 3E. An increase in temperature from 25°C to 37°C increases tension to 1.2–1.5 [46], [50], [51], [52]. When we consider the effects of regulatory proteins and Pi alone, the net effect is 3×( = 1.5×2), therefore 3T a = 42 kPa, which compares reasonably well with other reports [43], [44]. The solution used in this study is more physiological condition, because physiological [Pi] in contracting cardiomyocytes was reported to be ∼6 mM [53], and the ionic strength ∼215 mM [54].
Discussion
The clinical manifestations of DCM are highly diverse and caused by both genetic and non-genetic factors [55]. Mutations within 33 genes are known to cause DCM, and 23 of these (including all sarcomeric proteins and 5 of the Z-line proteins) are involved in the energy transduction processes [56]. Therefore, it is reasonable to postulate that the molecular pathogenesis of familial DCM is associated with deficits in myocardium contractility. Recent studies support the idea that sarcomeres are a key component of contraction and its regulation, rather than a simple assembly of force generators [57], [58], [59], [60]. The functional and regulatory properties of cardiac sarcomeres have been shown to be responsible for many of the mechanical properties of the heart [61], [62], [63], [64]. Tm is critical for maintaining normal function of the heart, as it is an essential element of the excitation-contraction coupling process and the thin-filament regulatory mechanism. In this report, we investigated early consequences of gene alterations of Tm, which eventually lead to DCM.
Compared to transgenic studies, our method has two advantages. 1) Transgenic studies are subject to significant secondary effects such as cardiac remodeling [65], myocyte disarray [66], interstitial fibrosis [67], altered post translational modifications including phosphorylation and dephosphorylation [68], and a loss of cardiac tissue [69], making it difficult to sort out the primary cause of pathogenesis. In contrast, our study characterizes the direct effect of the mutations on myocardial function and generates key information on the early pathogenesis of DCM. 2) It is difficult to control the expression levels of mutant proteins in transgenic studies. Our method ensures nearly 100% replacement with mutant proteins, as demonstrated by: SDS-PAGE [25], [26], [28], [45]; electron microscopy [24], [26], [28]; light microscopy [24], [26], [45]; mechanical tension [25], [26], [28], [45]; rate constant measurements [25], [28], [45], which reproduced 107±4% of isometric tension, 98±6% of rate constant 2 Π b and 92±4% of the rate constant 2 Π c (reviewed by [24]). Also, perfect reproduction of Ca2+ sensitivity and cooperativity has been reported [26].
In the present pCa-tension study, E40K and E54K showed a Ca2+ sensitivity similar to that of WT, in the presence of 8 mM Pi in high-IS solution. However, in the absence of added Pi in low-IS solution, the Ca2+ sensitivity decreased. The latter results are similar to those obtained previously [22], [23], [40]. It has been reported, based on studies using rabbit psoas fibers, that elevating IS from 128 mM to 201 mM decreases pCa50 by about 0.35 units [70], and that an increase in [Pi] from 0 to 7.5 mM decreases pCa50 by 0.23 units [71]. Other researchers reported a similar effect of [Pi] on pCa50 in cardiac fibers [47], [72]. The physiological concentration of ATP is reported to be ∼4.5 mM, and Pi to be ∼6 mM [53], [73]. The IS under physiological conditions may be as large as 215 mM [54]. It is apparent that our high IS solution is a better match to the physiological conditions.
E40K and E54K also showed similar T HC to that of the WT in the low IS solution, while these mutants significantly decreased T HC in the high IS. This is probably due to the dramatically increased number of strongly attached cross-bridges when lowering [Pi] and ionic strength [74], [75]. Sinusoidal analysis showed that E40K maintained similar THC/(cross-bridge) with that of the WT and caused a ∼17% decrease in the number of actively cycling cross-bridges (decreased Att). E54K showed a ∼13% decrease in THC/(cross-bridge) and a ∼6% decrease in Att. However, these changes are much smaller when compared with the effect of [Pi] and IS. The reconstituted fibers generated about 2.5×tension in low IS solution than in high IS solution (Fig. 3E vs. 3A). Therefore, the effect of mutations on cardiac contractility may not be as evident in the low IS solution as in the high IS solution.
Because previous studies have indicated that DCM-related mutations in sarcomeric proteins are associated with decreased pCa50 [11], it has been thought that decreased pCa50 may be the critical determinant of DCM pathogenesis. However, a recent transgenic study has indicated that familial DCM is not always associated with decreased pCa50 [76], and some earlier studies even reported increased pCa50 in the DCM myocardium [12], [13]. It also has been reported that Ca2+ sensitivity of the myocardium can be altered by other non-genetic factors such as remodeling [77] and exercise [78] without causing DCM or HCM. The results we report here also indicate that decreased pCa50 may not be the key molecular cause of DCM pathogenesis at the physiological Pi concentration and ionic strength (Fig. 3C). Therefore, we conclude that for the Tm E40K and E54K mutants, changes in pCa50 within muscle fibers may not be the key factor in disease pathogenesis.
A decrease in contractility may be the main cause for the systolic dysfunction in the context of DCM-related Tm mutants. Both E40K and E54K showed significantly decreased T HC and T act in the reconstituted myocardium (Fig. 3A), which could well explain the systolic dysfunction in the DCM heart. At the molecular level, our results indicate that the number of strongly attached cross-bridges decreased in both mutants (Fig. 7); in addition, force/cross-bridge was decreased in E54K. This is consistent with previous findings that DCM-related Tm mutations may decrease the proportion of strongly bound actomyosin complexes [79]. Recent studies in transgenic mice have shown that the E54K mutant caused a decrease in maximum tension in addition to a series of typical DCM phenotypes [40], suggesting that the decreased myocardial contractility may be the direct trigger of subsequent clinical manifestations, including dilation of the LV and deformation of the extracellular matrix. Therefore, cardiac remodeling events that lead to defects such as myocyte disarray and enlargement of the heart may be maladaptive compensation for the decrease in contractility.
The decrease in T LC caused by E40K and E54K may contribute to the filling pattern in DCM patients, which is defined using the ratio E/A [80], where E represents the degree of filling due to relaxation of the LV, and A the degree of filling due to contraction of the left atrium (LA). The decrease in T LC in DCM lowers the LV diastolic pressure, increases the early ventricular filling velocity, and results in an increase in the E/A ratio. Cardiac muscle relaxation is an energy consuming process [81]. The diminished process B (diminished magnitude B) of the Nyquist plot at pCa 7.0 (Fig. 4C and D) implies a diminished energy output during the early stages of diastole. We previously studied three HCM related Tm mutants (V95A, D175N, and E180G) [17] using the same techniques as in this report. Table 1 compares the effects of HCM- and DCM-related Tm mutants on isometric tension and cross-bridge kinetics, demonstrating that the effects are diverse and complex. These results suggest that the underlying molecular mechanism may be different for each mutant, and that they must be dealt with individually.
Table 1. Comparison of HCM- and DCM-related Tm mutations on isomeric tension and cross-bridge kinetics.
| Parameters (unit) | HCM mutants (V95A, D175N and E180G) | DCM mutants (E40K and E54K) |
| T LC (Ta) | All ↑ (2–3 fold) | Both ↓ (∼50%) |
| T HC (Ta) | E180G ↑ (∼17%) | Both ↓ (∼20%) |
| T act (Ta) | V95A and D175N ↓ (∼28%) | Both ↓ (∼20%) |
| Y LC (Ta) | All ↑ (2–2.5 fold) | Both ↓ (∼5%) |
| Y HC (Ta) | E180G ↑ (∼33%) | Both ↓ (∼33%) |
| Y act (Ta) | D175N ↓ (∼33%) | Both ↓ (∼20%) |
| pCa50 | V95A and E180G ↑ | Remain similar in high-IS solution Both ↓ in low-IS solution |
| Hill Factor (Cooperativity) | All ↓ (∼38%) | E54K ↑ (∼20%) |
| K 0 (mM−1) | V95A↓ (∼40%) and E180G ↑ (∼35%) | E40K ↓ (∼70%) |
| K 1 (mM−1) | V95A↓ (∼50%) | E40K ↑ (∼6 fold) E54K ↑ (∼3 fold) |
| K 2 | V95A↓ (∼33%) | E40K ↑ (∼84%) |
| K 4 | All remain similar to WT | E40K ↓ (∼67%) |
| K 5 (mM−1) | V95A and D175N ↓ (∼60%) | E40K ↓ (∼50%) |
| k 2 (s−1) | D175N ↑ (∼30%) | E40K ↑ (∼70%) E54K ↓ (∼30%) |
| k- 2 (s−1) | V95A ↑ (∼60%) | E54K ↓ (∼20%) |
| k 4 (s−1) | V95A ↑ (∼40%) | Both remain similar to WT |
| k -4 (s−1) | All remain similar to WT | E40K ↑ (∼2 fold), E54K ↑ (∼50%) |
Notes: ↑ increase, ↓ decrease; Only significant changes are indicated; All percent change is in comparison to WT. The data on HCM are from earlier publication [17].
E40K and E54K are both located at the “e” position of the heptad repeat and respectively interact with R35 and K49 of another chain of Tm molecule to stabilize the coiled-coil structure [19]. The opposite charge (from E to K) introduced by these mutants can destabilize the local coiled-coil structure. Previous differential scanning calorimetry (DSC) study has shown that both E40K and E54K caused a significant loss of the stability in Tm [23]. It has been suggested that the destabilization of Tm’s coiled-coil structure promotes the Tm-actin binding [82]. Therefore, a likely scenario is that E40K and E54K enhance the Tm-actin binding to over-inhibit the actin-myosin interaction.
One generalization we can make is that Tm mutation-related HCM causes an elevation in T LC, whereas Tm mutation related DCM causes a decrease in T LC. These results demonstrate regulatory dysfunction in two opposite directions. Elevated T LC observed in HCM may be directly responsible for the impaired relaxation and may lead to the disease phenotype [17]. Decreased T LC is a sign that mutant Tm over-inhibits the actomyosin interaction in the absence of Ca2+, as suggested by a decrease in the number of actively cycling cross-bridges (Fig. 3B, decreased Y LC). This over-inhibition effect is also evidenced at pCa 4.66 (Fig. 3B, decreased Y HC; Fig. 7, decreased Att) and causes a decrease in T HC (Fig. 3A).
The “three state model” proposed by McKillop and Geeves [83] may explain this over-inhibition effect. In the absence of Ca2+, enhanced binding of Tm and actin at the “blocked state” leads to an inadequate exposure of actin to myosin for interaction, leading to decreased T LC. In the presence of Ca2+, Tm azimuthally moves ∼25° to allow a moderate actin-myosin binding (closed state), then Tm moves further ∼5° to induce the strong actin-myosin binding (open state) [83]. Due to the enhanced Tm-actin binding, the two mutants are likely to shift the balance among the three states towards the “blocked state” and the “closed state”, thereby reducing the number of actively cycling cross-bridges at the “open state” to result in a decreased T HC.
At the same time, these two adjacent and similar mutants exhibited differences in force generation and cross-bridge kinetics. The most notable difference was that E54K decreased T HC/(cross-bridge) compared to that of WT, whereas E40K did not, indicating that E54K diminished the strong binding of actin and myosin. This phenomenon can be explained by the mechanism in which E40K and E54K not only affect the local structure of Tm, but also affect the overall structure in a different way, leading to two different effects in force generation and cross-bridge kinetics. Electron microscopy and molecular dynamics simulations have shown that Tm mutants D175N and E180G caused a change in the overall structure of Tm [84]. Therefore, we conclude that E40K and E54K may change the overall flexibility of Tm in their own specific ways, thus leading to a DCM phenotype through two different mechanisms.
Conclusion
We conclude that over-inhibition of the actomyosin interaction by Tm mutants E40K and E54K, which leads to decreased force generation under both high [Ca2+] (pCa 4.66) and low [Ca2+] conditions (pCa 7.0), is the primary cause of Tm mutation-related DCM pathogenesis.
Acknowledgments
We are indebted to Dr. Potter and Dr. Dweck of the University of Miami, Department of Pharmacology, for providing the regulatory proteins and insightful discussions. We would like to thank Ms. Hannah Caster for performing some of the experiments reported in this paper.
Funding Statement
This work was in part supported by a grant National Institute of Health HL70041. No additional external funding was received for this study. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Fuster V, Gersh BJ, Giuliani ER, Tajik AJ, Brandenburg RO, et al. (1981) The natural history of idiopathic dilated cardiomyopathy. Am J Cardiol 47: 525–531. [DOI] [PubMed] [Google Scholar]
- 2. Codd MB, Sugrue DD, Gersh BJ, Melton LJ (1989) Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy: a population-based study in Olmsted County, Minnesota, 1975–1984. Circulation 80: 564–572. [DOI] [PubMed] [Google Scholar]
- 3. Gillum RF (1986) Idiopathic cardiomyopathy in the United States, 1970–1982. Am Heart J 111: 752–755. [DOI] [PubMed] [Google Scholar]
- 4. Meyer M, Schillinger W, Pieske B, Holubarsch C, Heilmann C, et al. (1995) Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation 92: 778–784. [DOI] [PubMed] [Google Scholar]
- 5. Michels VV, Moll PP, Miller FA, Tajik AJ, Chu JS, et al. (1992) The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med 326: 77–82. [DOI] [PubMed] [Google Scholar]
- 6. Grunig E, Tasman JA, Kucherer H, Franz W, Kubler W, et al. (1998) Frequency and phenotypes of familial dilated cardiomyopathy. J Am Coll Cardiol 31: 186–194. [DOI] [PubMed] [Google Scholar]
- 7. Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, et al. (2009) Genetic evaluation of cardiomyopathy–a Heart Failure Society of America practice guideline. J Card Fail 15: 83–97. [DOI] [PubMed] [Google Scholar]
- 8. Lakdawala NK, Dellefave L, Redwood CS, Sparks E, Cirino AL, et al. (2010) Familial dilated cardiomyopathy caused by an alpha-tropomyosin mutation: the distinctive natural history of sarcomeric dilated cardiomyopathy. J Am Coll Cardiol 55: 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Olson TM, Kishimoto NY, Whitby FG, Michels VV (2001) Mutations that alter the surface charge of alpha-tropomyosin are associated with dilated cardiomyopathy. J Mol Cell Cardiol 33: 723–732. [DOI] [PubMed] [Google Scholar]
- 10. Seidman JG, Seidman C (2001) The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell 104: 557–567. [DOI] [PubMed] [Google Scholar]
- 11. Willott RH, Gomes AV, Chang AN, Parvatiyar MS, Pinto JR, et al. (2010) Mutations in Troponin that cause HCM, DCM AND RCM: what can we learn about thin filament function? J Mol Cell Cardiol 48: 882–892. [DOI] [PubMed] [Google Scholar]
- 12. Lee S, Lu R, Muller-Ehmsen J, Schwinger RH, Brixius K (2010) Increased Ca2+ sensitivity of myofibrillar tension in ischaemic vs dilated cardiomyopathy. Clin Exp Pharmacol Physiol 37: 1134–1138. [DOI] [PubMed] [Google Scholar]
- 13. Wolff MR, Buck SH, Stoker SW, Greaser ML, Mentzer RM (1996) Myofibrillar calcium sensitivity of isometric tension is increased in human dilated cardiomyopathies: role of altered beta-adrenergically mediated protein phosphorylation. J Clin Invest 98: 167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McConnell BK, Jones KA, Fatkin D, Arroyo LH, Lee RT, et al. (1999) Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice. J Clin Invest 104: 1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mandinov L, Eberli FR, Seiler C, Hess OM (2000) Diastolic heart failure. Cardiovasc Res 45: 813–825. [DOI] [PubMed] [Google Scholar]
- 16. Dweck D, Reynaldo DP, Pinto JR, Potter JD (2010) A dilated cardiomyopathy troponin C mutation lowers contractile force by reducing strong myosin-actin binding. J Biol Chem 285: 17371–17379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bai F, Weis A, Takeda AK, Chase PB, Kawai M (2011) Enhanced active cross-bridges during diastole: molecular pathogenesis of tropomyosin’s HCM mutations. Biophys J 100: 1014–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brown JH, Kim K-H, Jun G, Greenfield NJ, Dominguez R, et al. (2001) Deciphering the design of the tropomyosin molecule. Proc Natl Acad Sci 98: 8496–8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smille LB (1999) Tropomyosins. In: Kreis T, Vale R, editors. Guidebook to the Cytoskeletal and Motor Proteins. London: Oxford University Press. 159–164. [Google Scholar]
- 20. McLachlan AD, Stewart M (1976) The 14-fold periodicity in alpha-tropomyosin and the interaction with actin. J Mol Biol 103: 271–298. [DOI] [PubMed] [Google Scholar]
- 21. Holthauzen LM, Correa F, Farah CS (2004) Ca2+-induced rolling of tropomyosin in muscle thin filaments: the alpha- and beta-band hypothesis revisited. J Biol Chem 279: 15204–15213. [DOI] [PubMed] [Google Scholar]
- 22. Chang AN, Harada K, Ackerman MJ, Potter JD (2005) Functional Consequences of Hypertrophic and Dilated Cardiomyopathy-causing Mutations in α-Tropomyosin. J Biol Chem 280: 34343–34349. [DOI] [PubMed] [Google Scholar]
- 23. Mirza M, Robinson P, Kremneva E, Copeland On, Nikolaeva O, et al. (2007) The Effect of Mutations in α-Tropomyosin (E40K and E54K) That Cause Familial Dilated Cardiomyopathy on the Regulatory Mechanism of Cardiac Muscle Thin Filaments. J Biol Chem 282: 13487–13497. [DOI] [PubMed] [Google Scholar]
- 24. Kawai M, Ishiwata S (2006) Use of thin filament reconstituted muscle fibres to probe the mechanism of force generation. J Muscle Res Cell Motil 27: 455–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fujita H, D.Sasaki, Ishiwata S, Kawai M (2002) Elementary steps of the cross-bridge cycle in bovine myocardium with and without regulatory proteins. Biophys J 82: 915–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fujita H, Yasuda K, Niitsu S, Funatsu T, Ishiwata S (1996) Structural and functional reconstitution of thin filaments in the contractile apparatus of cardiac muscle. Biophys J 71: 2307–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lu X, Heeley DH, Smillie LB, Kawai M (2010) The role of tropomyosin isoforms and phosphorylation in force generation in thin-filament reconstituted bovine cardiac muscle fibres. J Muscle Res Cell Motil 31: 93–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lu X, Tobacman LS, Kawai M (2003) Effects of tropomyosin internal deletion Δ23Tm on isometric tension and the cross-bridge kinetics in bovine myocardium. J Physiol 553: 457–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Spudich JA, Watt S (1971) The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J Biol Chem 246: 4866–4871. [PubMed] [Google Scholar]
- 30. Holroyde MJ, Robertson SP, Johnson JD, Solaro RJ, Potter JD (1980) The calcium and magnesium binding sites on cardiac troponin and their role in the regulation of myofibrillar adenosine triphosphatase. J Biol Chem 255: 11688–11693. [PubMed] [Google Scholar]
- 31. Monteiro PB, Lataro RC, Ferro JA, Reinach Fde C (1994) Functional alpha-tropomyosin produced in Escherichia coli. A dipeptide extension can substitute the amino-terminal acetyl group. J Biol Chem 269: 10461–10466. [PubMed] [Google Scholar]
- 32. Kawai M, Lu X, Hitchcock-DeGregori SE, Stanton KJ (2009) MichaelW.Wandling (2009) Tropomyosin Period 3 Is Essential for Enhancement of Isometric Tension in Thin Filament-Reconstituted BovineMyocardium. J Biophys 2009: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kawai M, Brandt PW (1980) Sinusoidal analysis: a high resolution method for correlating biochemical reactions with physiological processes in activated skeletal muscles of rabbit, frog and crayfish. J Muscle Res Cell Motil 1: 279–303. [DOI] [PubMed] [Google Scholar]
- 34. Kawai M, Saeki Y, Zhao Y (1993) Crossbridge scheme and the kinetic constants of elementary steps deduced from chemically skinned papillary and trabecular muscles of the ferret. Circ Res 73: 35–50. [DOI] [PubMed] [Google Scholar]
- 35. Wannenburg T, Heijne GH, Geerdink JH, Dool HWVD, Janssen PM, et al. (2000) Cross-bridge kinetics in rat myocardium: effect of sarcomere length and calcium activation. Am J Physiol 279: H779–790. [DOI] [PubMed] [Google Scholar]
- 36. Martyn DA, Chase PB, Regnier M, Gordon AM (2002) A simple model with myofilament compliance predicts activation dependent crossbridge kinetics in skinned skeletal fibers. Biophys J 83: 3425–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang GW, Ding W, Kawai M (1999) Does Thin Filament Compliance Diminish the Cross-Bridge Kinetics? A Study in Rabbit Psoas Fibers. Biophys J 76: 978–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kawai M, Halvorson HR (1991) Two step mechanism of phosphate release and the mechanism of force generation in chemically skinned fibers of rabbit psoas. Biophys J 59: 329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mirza M, Marston S, Willott R, Ashley C, Mogensen J, et al. (2005) Dilated cardiomyopathy mutations in three thin filament regulatory proteins result in a common functional phenotype. J Biol Chem 280: 28498–28506. [DOI] [PubMed] [Google Scholar]
- 40. Rajan S, Ahmed RPH, Jagatheesan G, Petrashevskaya N, Boivin GP, et al. (2007) Dilated Cardiomyopathy Mutant Tropomyosin Mice Develop Cardiac Dysfunction With Significantly Decreased Fractional Shortening and Myofilament Calcium Sensitivity Circ Res. 101: 205–214. [DOI] [PubMed] [Google Scholar]
- 41. Saeki Y, Kawai M, Zhao Y (1991) Comparison of cross-bridge dynamics between intact and skinned myocardium from ferret right ventricles. Circ Res 68: 772–781. [DOI] [PubMed] [Google Scholar]
- 42. Zhao Y, Swamy PM, Humphries KA, Kawai M (1996) The effect of partial extraction of troponin C on the elementary steps of the cross-bridge cycle in rabbit psoas muscle fibers. Biophys J 71: 2759–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kentish JC, Terkeurs HEDJ, Ricciardi L, Bucx JJJ, Noble MIM (1986) Comparison between the Sarcomere Length-Force Relations of Intact and Skinned Trabeculae from Rat Right Ventricle - Influence of Calcium Concentrations on These Relations. Circulation Research 58: 755–768. [DOI] [PubMed] [Google Scholar]
- 44. Strang KT, Sweitzer NK, Greaser ML, Moss RL (1994) Beta-adrenergic receptor stimulation increases unloaded shortening velocity of skinned single ventricular myocytes from rats. Circ Res 74: 524–529. [DOI] [PubMed] [Google Scholar]
- 45. Fujita H, Lu X, Suzuki M, Ishiwata S, Kawai M (2004) The effect of tropomyosin on force and elementary steps of the cross-bridge cycle in bovine myocardium. J Physiology 556: 637–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu X, Tobacman L, Kawai M (2006) Temperature-Dependence of Isometric Tension and Cross-Bridge Kinetics of Cardiac Muscle Fibers Reconstituted with a Tropomyosin Internal Deletion Mutant. Biophysical Journal 91: 4230–4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kentish JC (1986) The effects of inorganic phosphate and creatine phosphate on force production in skinned muscles from rat ventricle. J Physiol 370: 585–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kawai M, Wray JS, Güth K (1990) Effect of ionic strength on crossbridge kinetics as studied by sinusoidal analysis, ATP hydrolysis rate and x-ray diffraction techniques in chemically skinned rabbit psoas fibres J Muscle Res Cell Motil. 11: 392–402. [DOI] [PubMed] [Google Scholar]
- 49. Kentish JC (1984) The inhibitory effects of monovalent ions on force development in detergent-skinned ventricular muscle from guinea-pig. J Physiol 352: 353–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Harrison SM, Bers DM (1989) Influence of temperature on the calcium sensitivity of the myofilaments of skinned ventricular muscle from the rabbit. J Gen Physiol 93: 411–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ranatunga KW (1999) Endothermic force generation in skinned cardiac muscle from rat. J Muscle Res Cell Motil 20: 489–496. [DOI] [PubMed] [Google Scholar]
- 52. Fujita H, Kawai M (2002) Temperature effect on isometric tension is mediated by regulatory proteins tropomyosin and troponin in bovine myocardium. J Physiol (Lond) 539: 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Opie LH, Mansford KR, Owen P (1971) Effects of increased heart work on glycolysis and adenine nucleotides in the perfused heart of normal and diabetic rats. Biochem J 124: 475–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Godt RE, Maughan DW (1988) On the composition of the cytosol of relaxed skeletal muscle of the frog. Am J Physiol 254: C591–604. [DOI] [PubMed] [Google Scholar]
- 55. Dec GW, Fuster V (1994) Idiopathic dilated cardiomyopathy. N Engl J Med 331: 1564–1575. [DOI] [PubMed] [Google Scholar]
- 56. Hershberger RE, Siegfried JD (2011) Update 2011: clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol 57: 1641–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sagawa K, Maughan L, Suga H, Sunagawa K (1988) Cardiac contraction and the pressure-volume relationship: Oxford University Press. [Google Scholar]
- 58.Fozzard HA, Haber E, Jennings RB, Katz AM (1992) The heart and cardiovascular system: Scientific foundations (2nd ed.). New York: Raven Press. [Google Scholar]
- 59. Solaro RJ (1999) Integration of myofilament response to Ca2+ with cardiac pump regulation and pump dynamics. Am J Physiol 277: S155–S163. [DOI] [PubMed] [Google Scholar]
- 60. Solaro RJ, Tombe PPd (2008) Sarcomeric proteins as key elements in integrated control of cardiac function. Cardiovasc Res 77: 616–618. [DOI] [PubMed] [Google Scholar]
- 61.Solaro RJ, Wolska BM, Arteaga G, Martin AF, Buttrick P, et al.. (2002) Modulation of thin filament activity in long and short term regulation of cardiac function. In: Solaro RJ MR, editor. Molecular Control Mechanisms in Striated Muscle Contraction. The Netherland: Kluwer Academic Publishers. 291–327. [Google Scholar]
- 62. Davis JP, Tikunova SB (2008) Ca2+ exchange with troponin C and cardiac muscle dynamics. Cardiovasc Res 77: 619–626. [DOI] [PubMed] [Google Scholar]
- 63. Hanft LM, Korte FS, McDonald KS (2008) Cardiac function and modulation of sarcomeric function by length. Cardiovasc Res 77: 627–636. [DOI] [PubMed] [Google Scholar]
- 64. Hamdani N, Kooij V, Dijk Sv, Merkus D, Paulus WJ, et al. (2008) Sarcomeric dysfunction in heart failure. Cardiovasc Res 77: 649–658. [DOI] [PubMed] [Google Scholar]
- 65. Sivasubramanian N, Coker ML, Kurrelmeyer KM, MacLellan WR, DeMayo FJ, et al. (2001) Left ventricular remodeling in transgenic mice with cardiac restricted overexpression of tumor necrosis factor. Circulation 104: 826–831. [DOI] [PubMed] [Google Scholar]
- 66. Marian AJ, Wu Y, Lim DS, McCluggage M, Youker K, et al. (1999) A transgenic rabbit model for human hypertrophic cardiomyopathy. J Clin Invest 104: 1683–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. James J, Zhang Y, Osinska H, Sanbe A, Klevitsky R, et al. (2000) Transgenic modeling of a cardiac troponin I mutation linked to familial hypertrophic cardiomyopathy. Circ Res 87: 805–811. [DOI] [PubMed] [Google Scholar]
- 68. Abraham TP, Jones M, Kazmierczak K, Liang HY, Pinheiro AC, et al. (2009) Diastolic dysfunction in familial hypertrophic cardiomyopathy transgenic model mice. Cardiovasc Res 82: 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, et al. (2001) A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest 107: 813–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kawai M, Cox RN, Brandt PW (1981) Effect of Ca ion concentration on cross-bridge kinetics in rabbit psoas fibers. Evidence for the presence of two Ca-activated states of thin filament. Biophys J 35: 375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Brandt PW, Cox RN, Kawai M, Robinson T (1982) Effect of cross-bridge kinetics on apparent Ca2+ sensitivity. J Gen Physiol 79: 997–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Solaro RJ, Holroyde MJ, Herzig JW, Peterson J (1980) Cardiac relaxation and myofibrillar interactions with phosphate and vanadate. Eur Heart J Suppl A: 21–27. [DOI] [PubMed] [Google Scholar]
- 73. Roth K, Hubesch B, Meyerhoff DJ, Naruse S, Gober JR, et al. (1989) Noninvasive Quantitation of Phosphorus Metabolites in Human-Tissue by Nmr-Spectroscopy. Journal of Magnetic Resonance 81: 299–311. [Google Scholar]
- 74. Hibberd MG, Dantzig JA, Trentham DR, Goldman YE (1985) Phosphate Release and Force Generation in Skeletal-Muscle Fibers. Science 228: 1317–1319. [DOI] [PubMed] [Google Scholar]
- 75. Julian FJ, Moss RL (1981) Effects of Calcium and Ionic-Strength on Shortening Velocity and Tension Development in Frog Skinned Muscle-Fibers. Journal of Physiology-London 311: 179–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Song W, Dyer E, Stuckey D, Leung MC, Memo M, et al. (2010) Investigation of a transgenic mouse model of familial dilated cardiomyopathy. J Mol Cell Cardiol 49: 380–389. [DOI] [PubMed] [Google Scholar]
- 77. Wang J, Flemal K, Qiu Z, Ablin L, Grossman W, et al. (1994) Ca2+ handling and myofibrillar Ca2+ sensitivity in ferret cardiac myocytes with pressure-overload hypertrophy. Am J Physiol 267: H918–924. [DOI] [PubMed] [Google Scholar]
- 78. Wisloff U, Loennechen JP, Currie S, Smith GL, Ellingsen O (2002) Aerobic exercise reduces cardiomyocyte hypertrophy and increases contractility, Ca2+ sensitivity and SERCA-2 in rat after myocardial infarction. Cardiovasc Res 54: 162–174. [DOI] [PubMed] [Google Scholar]
- 79. Borovikov YS, Avrova SV, Karpicheva OE, Robinson P, Redwood CS (2011) The effect of the dilated cardiomyopathy-causing Glu40Lys TPM1 mutation on actin-myosin interactions during the ATPase cycle. Biochem Biophys Res Commun 411: 496–500. [DOI] [PubMed] [Google Scholar]
- 80. Pinamonti B, Di Lenarda A, Sinagra G, Camerini F (1993) Restrictive left ventricular filling pattern in dilated cardiomyopathy assessed by Doppler echocardiography: clinical, echocardiographic and hemodynamic correlations and prognostic implications. Heart Muscle Disease Study Group. J Am Coll Cardiol 22: 808–815. [DOI] [PubMed] [Google Scholar]
- 81. Zile MR, Brutsaert DL (2002) New concepts in diastolic dysfunction and diastolic heart failure: Part II: causal mechanisms and treatment. Circulation 105: 1503–1508. [DOI] [PubMed] [Google Scholar]
- 82. Hitchcock-DeGregori SE, Singh A (2010) What makes tropomyosin an actin binding protein? A perspective. Journal of Structural Biology 170: 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. McKillop DFA, Geeves MA (1993) Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys J 65: 693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Li XE, Suphamungmee W, Janco M, Geeves MA, Marston SB, et al. (2012) The flexibility of two tropomyosin mutants, D175N and E180G, that cause hypertrophic cardiomyopathy. Biochem Biophys Res Commun 424: 493–496. [DOI] [PMC free article] [PubMed] [Google Scholar]