

Abstract
In prostanoid biosynthesis, the first two steps are catalyzed by cyclooxygenases (COX). In mice and humans, deregulated expression of COX-2, but not of COX-1, is characteristic of epithelial tumors, including squamous cell carcinomas of skin. To explore the function of COX-2 in epidermis, a keratin 5 promoter was used to direct COX-2 expression to the basal cells of interfollicular epidermis and the pilosebaceous appendage of transgenic mouse skin. COX-2 overexpression in the expected locations, resulting in increased prostaglandin levels in epidermis and plasma, correlated with a pronounced skin phenotype. Heterozygous transgenic mice exhibited a reduced hair follicle density. Moreover, postnatally hair follicle morphogenesis and thinning of interfollicular dorsal epidermis were delayed. Adult transgenics showed a body-site-dependent sparse coat of greasy hair, the latter caused by sebaceous gland hyperplasia and increased epicutaneous sebum levels. In tail skin, hyperplasia of scale epidermis reflecting an increased number of viable and cornified cell layers was observed. Hyperplasia was a result of a disturbed program of epidermal differentiation rather than an increased proliferation rate, as reflected by the strong suppression of keratin 10, involucrin, and loricrin expression in suprabasal cells. Further pathological signs were loss of cell polarity, mainly of basal keratinocytes, epidermal invaginations into the dermis, and formation of horn perls. Invaginating hyperplastic lobes were surrounded by CD31-positive vessels. These results demonstrate a causal relationship between transgenic COX-2 expression in basal keratinocytes and epidermal hyperplasia as well as dysplastic features at discrete body sites.
The cyclooxygenase (COX) isoenzymes COX-1 and -2 catalyze the first two steps in prostanoid biosynthesis (1). Using arachidonic acid as a substrate, the intermediate product prostaglandin (PG) H2 is converted by prostanoid synthases into the biologically active prostanoids PGE2, PGF2α, PGD2, PGI2, and thromboxane A2 in a cell-type-specific manner (2–4). In many tissues, COX-1 is expressed constitutively, whereas COX-2 is induced by a variety of stimuli such as growth factors, cytokines, and hormones. In fact, COX-2 is thought to act as a proinflammatory emergency enzyme upon tissue irritation and damage (5).
In normal epidermis, the balance between cell proliferation in the basal layer, cell differentiation in the suprabasal spinous and granular layers, and cell death at the transition of stratum granulosum and stratum corneum is strictly regulated. Besides this spatial separation, there is also a temporal separation of proliferating from differentiating cells, which implies that cells leaving the basal layer lose their proliferative capacity and simultaneously enter the stage of terminal differentiation. Under these circumstances, COX-1 is produced constitutively in keratinocytes throughout the living epidermis, whereas COX-2 is not expressed (6).
A transient COX-2 induction has been observed upon treatment of skin (6–8) and other tissues (9) with tumor-promoting agents. Permanent up-regulation of COX-2 already has been found in early stages of carcinogenesis and appears to be a consistent feature of neoplastic development in a wide variety of human and animal epithelia (10–13). This holds true also for human squamous cell carcinogenesis, because aberrant COX-2 expression was found consistently in both preneoplastic cells of actinic keratoses and tumor cells of squamous cell carcinomas of skin (14). Using the experimental protocol of multistage carcinogenesis in mouse skin (9), we found aberrant COX-2 expression to be associated with the promotion stage (12, 15), where initiated keratinocytes harboring a Ha-ras mutation clonally expand into preneoplastic papillomas, some of which subsequently develop into carcinomas. The close interrelation between COX-2 expression and inflammation (16), wounding (7), and skin carcinogenesis (14, 15, 17) prompted us to analyze the possible consequences of an aberrant overexpression of COX-2 in epidermal tissue in vivo.
For this purpose, we developed transgenic mouse lines, which, under the control of a bovine keratin 5 (K5) promoter, constitutively express COX-2 in the basal cells of the interfollicular epidermis and the pilosebaceous unit. The transgenic mice exhibit a distinct skin phenotype characterized by delayed hair follicle morphogenesis, reduced hair follicle density, sebaceous gland hyperplasia, and profound hyperplasia in scale epidermis of the tail with foci exhibiting signs of dysplasia.
Materials and Methods
Materials.
Affinity-purified rabbit polyclonal antibodies directed against mouse keratin 10, as well as loricrin, were from Babco (Richmond, CA); purified monoclonal rat anti-mouse CD31 antibody was from Becton Dickinson; rabbit anti-mouse Ki-67 and alkaline phosphatase-conjugated goat anti-rabbit IgG were from Dianova (Hamburg, Germany); goat polyclonal anti-COX-1 (SC1754) and anti-COX-2 (SC1745) antibodies, peroxidase-conjugated goat anti-rabbit IgG, goat anti-rat IgG, and donkey anti-goat IgG were from Santa Cruz Biotechnology; ELISA-BSA and goat serum were from Sigma; and PGE2, PGF2α, and 6-keto-PGF1α enzyme immunoassays were from Cayman Chemicals (Ann Arbor, MI).
Generation of Transgenic Mice.
The K5 promoter and its 3′ untranslated region were excised from the bovine KIII/KIV minilocus (18) and cloned into a pBluescript KS Vector (pK5), and a full-length COX-2 cDNA without 3′ untranslated region (19) was subcloned downstream of the K5 promoter (Fig. 1A). The orientation of the COX-2 sequence was verified by sequencing. The linearized and purified KpnI fragment of the recombinant plasmid containing the K5-COX-2 sequences (Fig. 1A) was used for microinjection into pronuclei of fertilized NMRI oocytes. Two-cell embryos were transferred into the oviducts of pseudopregnant foster NMRI mice. Integration of transgenic DNA was checked by Southern blot analysis and PCR of genomic tail DNA. For PCR analysis, primers (Fig. 1A, set a/a′) specific for the 3′ end of the K5 promoter (5′-CCT AGA TAA CAG AGC CGC TTT C-3′) and the 5′ end of the COX-2 cDNA sequence (5′-TTT CAC CAT AGA ATC CAG TCC G-3′), as well as primers (Fig. 1A, set b/b′) specific for the 3′ end of the COX-2 cDNA sequence (5′-GCAATAATGTGAAGGGGTGTCC-3′) and the 5′ end of the polyadenylation sequence (5′-GCTAGAAACAGTGGGACTCTGA-3′), were used to amplify 332- and 380-bp fragments, respectively. Founders were backcrossed to NMRI from RCC (Füllinsdorf, Switzerland). Without exception, sex- and body-site-matched tissues of wild-type (wt) littermates were used as controls.
Figure 1.

K5-COX-2 DNA construct, identification of K5-COX-2 founders, and analysis of COX-2 expression. (A) Scheme of the K5-COX-2 DNA transgene consisting of a bovine K5 promoter, a mouse COX-2 cDNA, and a bovine K5 3′ genomic sequence. (B) Detection of transgene integration by PCR of genomic tail DNA by using primer sets a/a′ and b/b′. Data from transgenic founders and an unaffected littermate 664 are shown. Neg, PCR without DNA. (C) RT-PCR of transgenic COX-2 mRNA from epidermis of back, abdomen, tail, and foot sole and from forestomach, esophagus, heart, kidney, lung, pancreas, spleen, brain, colon, and testis (lanes 1–14) of a transgenic mouse (trA) and a wt littermate by using primer set c/c′. NC, PCR without cDNA; PC, PCR with cDNA from BMGE+ cells transfected with the K5-COX-2 construct. As an internal control, β-actin was amplified. (D) COX-2 and COX-1 protein in wt and transgenic (trA) tissues. Epidermal homogenates of back, abdomen, tail, ear, tongue, and forestomach (lanes 1–6) were immunoprecipitated and immunoblotted by using COX-2-specific (Upper) and COX-1-specific (Lower) antibodies. IgG cross-reacted with the secondary antibody. PC, positive control (9). (E–H) Localization of COX-2 protein in tail skin of a 10-month-old wt littermate (E) and three K5-COX-2 transgenics from lines A and B (F–H) by immunohistochemistry. Note the uniform COX-2 expression in basal keratinocytes (arrows) of interfollicular epidermis (e in F and G), the focal expression in suprabasal keratinocytes of hyperplastic lobes (F Inset), the outer root sheath (H, *), and sebaceous gland epithelium (H, Δ), but not sebocytes (s in H). Only few ORS cells were COX-2-positive in hair follicles (hf) of wt mice (E). [×400 (E–G) and ×630 (H and I Inset).] (I–K) PGE2 (solid bars) and PGF2α (open bars) levels in tail (I) and dorsal (J) epidermis of 10-month-old transgenic (trA and trB) and wt mice and in plasma of 3-month-old mice (K). n = 5, P < 0.003 (J) and < 0.04 (K).
Reverse Transcription–PCR (RT-PCR) Analysis.
Total RNA was extracted from pulverized frozen tissues by using RNA Clean (AGS, Heidelberg, Germany), and RT-PCR was performed with the cDNA cycle kit (Invitrogen) as described (14). Using primers 5′-ATTCCTGCAGCCCAACATCTGT-3′ and 5′-AGGTTCTGCTTTGGGTTTGGG-3′ (Fig. 1A, set c/c′), a 460-bp fragment diagnostic for 3′ untranslated region of transgenic mRNA was amplified. β-Actin (fragment size, 430 bp) was amplified with primers 5′-AAACTGGAACGGTGAAGGC-3′ and 5′-GCTGCCTCAACACCTCAAC-3′.
Immunoblot Analysis.
Immunoprecipitation of COX-2 and COX-1 as well as immunoblot analysis were carried out by using isoenzyme-specific antisera as described (6, 15).
Histochemistry and Immunohistochemistry.
For hematoxylin/eosin (H&E) staining, skin was fixed in PBS-buffered 4% paraformaldehyde for 16 h before embedding into paraffin. Epidermal thickness was determined at regular intervals of H&E-stained sections by means of the axiovision 2.05 software (Zeiss). COX isozymes were immunodetected by using 5-μm cryosections (6, 15). Ki-67, keratin 10, loricrin, and CD31 stainings were performed according to the same protocol, except that 1% ELISA-BSA in PBS was used as blocking solution and sections were fixed in acetone for 10 min. Primary antibodies were diluted 1:50 and peroxidase-conjugated secondary antibodies were diluted 1:100 in blocking solution. In vertically sectioned skin, Ki-67-positive cells were counted in 25 successive scale regions. For immunofluorescence analysis of K5 and involucrin, frozen sections were fixed with acetone at −20°C for 10 min (K5) or left unfixed (involucrin). Polyclonal guinea pig anti-mouse K5 antiserum was diluted 1:250 in PBS, containing 1% ELISA-BSA, and polyclonal rabbit anti-mouse involucrin was diluted 1:800 in PBS containing 10% goat serum. Incubation was performed for 1 h at room temperature. Secondary antibodies were Alexa Fluor 488-labeled goat anti-guinea pig IgG (Molecular Probes) and Cy3-conjugated goat anti-rabbit IgG (Dianova) for K5 and involucrin, respectively. For lipid staining by oil red, frozen skin embedded in Tissue Tek was cut into 5-μm sections and fixed in 1% Baker's formol.
PG Analysis.
PG levels in epidermis, skin, and platelet-poor plasma were quantitated by means of commercially available enzyme immunoassays as described (12).
Statistics.
Data represent mean values ± SD. Using Student's t test, P values < 0.05 were regarded as significant.
Results
Generation of K5-COX-2 Transgenic Mice.
To explore the role of COX-2 in skin, transgenic mice were generated, expressing COX-2 under the control of the bovine K5 promoter, which directs the expression of the transgene to basal keratinocytes of interfollicular epidermis and basal cells of the pilosebaceous unit. Microinjection of the purified K5-COX-2 construct (Fig. 1A) into fertilized NMRI oocytes yielded an offspring of 93 mice, 8 of which harbored the transgene according to PCR analysis with different primer sets (Fig. 1 A and B) and confirming Southern blot hybridization of tail DNA (data not shown). The transgenic mouse 666 died without offspring at the age of 3 months; founders 663, 667, 675, and 678 died at the age of 10 months; and founders 4, 19, and 29 are still alive and 19–23 months old at present. All founders (four males, three females) were bred successfully. Gender distribution was similar in wt and transgenic mice. Data from heterozygous mice are presented.
Expression and Activity of the Transgenic COX-2.
Transgene expression was verified by RT-PCR analysis by using primer set c/c′ (Fig. 1 A and C). The diagnostic COX-2 3′ untranslated region DNA amplicon of 460 bp in size was amplified from RNA of stratified epithelial tissues including epidermis from different body sites, forestomach, and esophagus (a representative result is shown in Fig. 1C) as well as tongue (not shown). Among various other organs analyzed, surprisingly, lung expressed COX-2 transgenic mRNA. The absence of the diagnostic amplicon in RT-PCR assays with RNA of wt tissues confirmed the specificity of the primers for the transgene-specific mRNA (Fig. 1C).
Immunoblot analysis revealed the epidermal expression of COX-2 protein as a 72- to 74-kDa signal in all transgenic lines that was strongest in tail, followed by back, abdomen, and ear. COX-2 protein was not detectable in tongue and forestomach (representatively shown in Fig. 1D), even though transgene-specific mRNA signals were seen. In wt epidermis with synchronized resting hair follicles or other stratified epithelia, COX-2 protein was not detectable (representatively shown in Fig. 1D). COX-1 expression was not altered in transgenic mice (Fig. 1D). For further analysis, the high-expresser lines 667 (trA) and 675 (trB) were selected.
By means of immunohistochemistry, COX-2 protein was found in keratinocytes of the interfollicular stratum basale, the outer root sheath (ORS) cells of hair follicles, and the epithelial lining of sebaceous glands as shown for tail skin of adult transgenics (Fig. 1 F–H). This distribution corresponded to K5-promoter activity in skin. In addition, COX-2 signals occasionally were visible in suprabasal keratinocytes within hyperplastic epidermal lobes (Fig. 1F Inset; see also below). In epidermis of wt mice, endogenous COX-2 was detectable only in a few ORS cells and occasionally in interfollicular suprabasal keratinocytes (Fig. 1E). The expression in ORS cells was strongest during the anagen, weaker during the catagen, and undetectable during the resting phase of the hair cycle. The same held true for basal epithelial cells of the sebaceous gland (data not shown).
The levels of PGE2 and PGF2α were increased in tail and dorsal epidermis of transgenic compared with wt mice (Fig. 1 I and J). In addition, the skin content of 6-keto-PGF1α, the stable metabolite of PGI2 [122 ± 19 (SE) vs. 64 ± 10 (SE) pg/mg protein] and plasma levels of PGE2 were elevated significantly in transgenics (Fig. 1K).
Gross Phenotype of Transgenic Mice.
Transgenic COX-2 expression and increased PG levels correlated with marked phenotypic changes. All transgenic lines exhibited an exceptional nail growth especially of the hind feet (Fig. 2 A and B). Showing a normal appearance at birth, transgenics began to differ from wt mice at the age of 8 days in that they were still hairless (Fig. 2C). By the age of 7 weeks, transgenic mice had developed a furry coat, but it remained less dense for their lifetime and looked greasy and coarse as compared with wt mice (Fig. 2 D–F). This hair phenotype showed body-site variations that were most pronounced on back skin. In addition, three of the seven transgenic lines developed a severe pancreatitis at the age of about 6–10 months, although no transgenic COX-2 expression was found in the pancreas. Histological examination of H&E-stained skin sections of transgenic mice indicated remarkable defects in epidermal differentiation and skin maturation, depending on age and body site.
Figure 2.
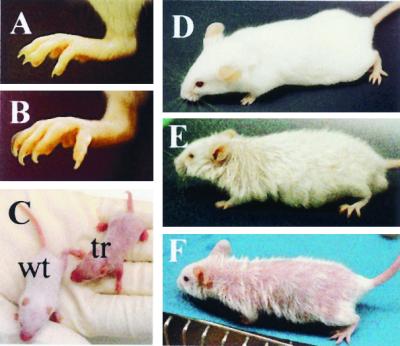
Gross phenotype of K5-COX-2 transgenic and wt mice. Hind foot of a 7-week-old wt (A) and a transgenic mouse (B) with long nails. (C) Hair coat in the transgenic neonatal (tr) 8 days after birth, showing an almost naked appearance compared with a wt control. (D–F) Twelve-week-old wt (D) and moderately (E) and severely (F) affected transgenic mice, the latter with much less hair especially on back skin. Hair, particularly of the neck, looked greasy.
Delayed Hair Follicle Development.
At day 9 postnatally, almost all pelage hair follicles of wt mice had entered the final stage of hair follicle morphogenesis (Fig. 3 J and K), i.e., hair follicles had acquired their maximal length and prominent hair shafts had broken through the epidermis (20). In 9-day-old transgenics, however, most hair shaft tips had just left the inner root sheath and entered the hair canal, the majority of hair follicles had not yet reached the s.c. panniculos carnosus, i.e., they were only half as long as the wt hair follicles, and, in line with this, the transgenic back skin was thinner than wt skin (Fig. 3 L and M). The transgenic follicles were aligned nearly perpendicularly to the skin surface, whereas wt follicles formed a 45° angle. Hair follicle density appeared to be reduced. Whereas at day 9 the interfollicular wt epidermis had already thinned toward the typical, three-layered adult phenotype with the more disorganized spinous and granular layers (Fig. 3K), transgenic epidermis still exhibited the neonatal morphology characterized by full stratification (Fig. 3M). Expression of keratins 5 and 10, involucrin, and loricrin was regular (data not shown). Whereas adult (>7 weeks) interfollicular epidermis was similar in wt and transgenic mice, differences still were seen with respect to sebaceous gland morphology (see below) and hair follicle density [39 ± 5 vs. 18 ± 3 hair follicles per visual field (P < 0.05)] in 7-week-old wt mice and in moderately affected transgenics, respectively (see moderate phenotype in Fig. 2E).
Figure 3.

Skin histology. H&E-stained longitudinal sections of tail (A–I) and dorsal (J–M) skin of K5-COX-2 transgenic (C–I, L, and M) and wt (A, B, J, and K) mice; tail skin from 7-week- (A, C, and G), 14-week- (D and H), 6-month- (E), and 10-month-old (F and I) transgenics and wt mice. Note, in tail, the hyperplasia in scale epidermis (C–F, H, and I), the papillary structure (D–F, H, and I), the intradermal epithelial invagination with horn perls (I), the sebaceous gland hyperplasia (E and F), the loss of cell polarity in the basal layer (G and H), and the irregular orientation of epidermal cells (G and H). Cryosections of dorsal skin from 9-day-old wt (J and K) and transgenic mice (L and M). Note, in transgenic skin, the reduced hair follicle density, the retarded formation, and emergence of hair shafts, and inappropriate orientation (90° angle) of hair follicles (L), and delayed thinning of interfollicular epidermis (M). [×160 (A–F, J, and L), ×400 (H and I), ×630 (G).]
Hyperplastic Scale Epidermis of Tail.
Compared with tail epidermis of adult wt mice (age >7 weeks, Figs. 3 A and B, 1E, and 4A), transgenic scale epidermis was hyperplastic reflecting first a hyperkeratosis and, later, additionally an increase in the number of viable cell layers, both leading to a doubling of epidermal thickness (Fig. 3 C–F). The normal columnar alignment of basal keratinocytes and the more flattened arrangement of suprabasal cells (Figs. 3A, 1E, and 4A) had been lost (Fig. 3 G and H). Frequently, invaginations of COX-2-positive hyperplastic epidermis into the dermis were observed (Figs. 3 D–I, 1, 4, and 5), which sometimes contained horn perls in the center (Fig. 3I). In the immediate vicinity of these hyperplastic lobes, blood vessels could be visualized in papillary dermis by decorating endothelial cells with anti-CD31 antibody (Fig. 4L). In the upper dermis, vessel density appeared to be increased compared with controls (Fig. 4K). Epidermal papillary hyperplasia was restricted to tail and not seen in dorsal or ventral skin of transgenics, whereas mild hyperplasia was observed in COX-2-immunopositive specimens of transgenic tongue, esophagus, and forestomach (data not shown). No symptoms of inflammation were seen in any skin site of transgenic mice.
Figure 4.

Expression of marker proteins of epidermal differentiation and proliferation-associated Ki-67 antigen and visualization of endothelial cells in skin of 10-month-old K5-COX-2 transgenics (B, D, F, H, J, and L) and wt tails (A, C, E, G, I, and K). Immunofluorescence (C, D, G, and H) or immunoperoxidase staining (A, B, E, F, and I–L) with anti-Ki-67 (A and B), anti-K5 (C and D), antikeratin 10 (E and F), anti-involucrin (G and H), anti-loricrin (I and J), and anti-CD31 (K and L) antisera. Note suprabasal Ki-67-positive keratinocytes in hyperplastic transgenic epidermis (B), the local extension of K5 (D), the local reduction of keratin 10 (F), the restriction of involucrin to the uppermost spinous layer (H), the regular, although less intensive loricrin staining (J), and the subepidermal vessels reaching into the upper papillary dermis surrounding the epidermal lobes that have protruded deeply into the dermis (L, arrows). [×400 (C–L) and ×630 (A and B).]
Impairment of Terminal Differentiation in Scale Epidermis of Tail.
By counting Ki-67-immunopositive cells, the proliferation rate in scale epidermis of tail was found to be similar in transgenic and wt mice, i.e., 43.3 ± 5 and 47.5 ± 8%, respectively (P > 0.05). Whereas in wt epidermis, proliferating cells were located almost exclusively in the basal layer (Fig. 4A), in transgenics, proliferating cells also were found in the suprabasal compartment (Fig. 4B), indicating a disturbed terminal differentiation. To test this hypothesis, the expression of K5, keratin 10, involucrin, and loricrin, i.e., marker proteins of proliferation or immediate early, early, and late epidermal differentiation (21, 22), was analyzed by immunohistochemistry. In wt epidermis, K5 signals were confined to the basal cell compartment (Fig. 4C), whereas the differentiation-associated keratin 10 was expressed throughout the suprabasal compartment (Fig. 4E), involucrin in the upper spinous layers (Fig. 4G), and loricrin in a single layer of the uppermost stratum spinosum (Fig. 4I). By contrast, in invaginating hyperplastic lobes of transgenics, K5 expression was found in up to eight suprabasal layers (Fig. 4D) and keratin 10 expression was focally lacking in the suprabasal compartment, whereas in other regions it was regular, although partly less intensive (Fig. 4F). Involucrin expression was most drastically affected, i.e., was found only in patches mainly within the uppermost suprabasal cell layers (Fig. 4H), and loricrin expression seemed to be reduced (Fig. 4J). These data provide evidence for an aberrant terminal differentiation of transgenic scale epidermis of tail. A disturbance of the characteristic parakeratotic differentiation pattern (23) of the scale regions was not observed, however (Figs. 4 B, F, and J and 1G).
Sebaceous Gland Hyperplasia.
With increasing age, the transgenics showed a sebaceous gland hyperplasia that was most pronounced in dorsal and tail skin (Figs. 3 E and F and 5 A–D). In the latter, sebaceous gland lobules, grouped around an enlarged gland duct, were increased in number and consisted of well differentiated sebocytes. Glands were enclosed in a single layer of K5-positive basal cells showing an increased rate of proliferation (61 ± 9% vs. 39 ± 6% Ki-67-positive cells in transgenic and wt mice, respectively; P < 0.001). Oil-red staining revealed lipid deposition in these glands and the ramified sebaceous duct (Fig. 5B). Likewise, the stratum corneum and the surface of transgenic epidermis were stained more intensively than epidermis of wt mice. In fact, epicutaneous sebum concentrations were increased strongly in transgenic neck and tail skin compared with wt mice (Fig. 5 E and F).
Figure 5.

Lipid content in skin of 10-month-old K5-COX-2 transgenic (B and D, trA and trB) and wt tails (A and C). (A–D) Oil-red staining: note sebaceous gland hyperplasia in transgenic skin (B and D). Ramified sebaceous ducts (B and D), stratum corneum (B), and hair canal (B and D) of transgenic epidermis were stained more intensely than in controls (A and C). [×160 (A and B), ×400 (C and D).] Sebum accumulation on tail surface (E) and shaved neck skin (F) of transgenic (trA and trB) as compared with age-matched wt mice was determined by a sebumeter (SM810; Courage and Khazaka, Cologne, Germany). n = 5; P < 0.05.
Discussion
In this study, the functional consequences of transgenic COX-2 expression, driven by a K5 promoter in basal cells of the interfollicular epidermis and the pilosebaceous unit of mouse skin, were analyzed. Increased levels of PGE2, PGF2α, and PGI2 found in transgenic mice most probably were due to the constitutively expressed COX-2, because a marked change in COX-1 protein levels was not detected. This indicates a functional coupling in transgenic epidermis between COX-2 and other enzymes involved in PG biosynthesis, such as phospholipase A2 (24, 25) and prostanoid synthases (2–4), which deserves further investigation. Although COX-2 has been described as a proinflammatory enzyme (26, 27), the transgenic animals did not exhibit symptoms of inflammation, indicating that COX-2 expression alone is not sufficient to initiate an inflammatory response in mouse skin.
Although no abnormal skin morphology has been reported for COX-2 knockout mice (28, 29), a characteristic phenotypic change was observed in the K5-COX-2 transgenics. In these animals, morphogenesis of pelage hair follicles was delayed and the number of hair follicles was reduced, the latter most probably a result of an impairment of hair follicle induction rather than hair follicle degradation, because no degenerated hair follicles were observed and, compared with wt mice, fewer hair follicles were present 9 days after birth. Hair follicle morphogenesis is known to be initiated by distinct epithelial–mesenchymal interactions at a definite time point after gestation (30). A possible role of COX-2 in these processes remains to be explored. Also, a later stage of hair follicle morphogenesis (20), i.e., the differentiation of matrix cells into the mature hair shaft, was delayed in the transgenics. Because hair matrix cells were COX-2-negative, a paracrine-inhibitory effect of PG liberated from COX-2-positive basal cells may be postulated. Delayed hair follicle morphogenesis correlated with a delayed postnatal thinning of interfollicular epidermis of back skin (31) while the program of terminal differentiation (i.e., the coordinated expression of keratins 5 and 10, involucrin, and loricrin) remained unaffected.
Aberrant COX-2 expression in basal gland epithelial cells correlated with sebaceous gland hyperplasia, which was characterized by the presence of numerous lobules consisting of well differentiated sebocytes grouped around an enlarged sebaceous gland duct. Increased sebum accumulation at the skin surface resulted in greasy-looking hair. Recently, peroxisome proliferator-activated receptor (PPAR) isoforms α and γ have been shown to play a role in the differentiation of sebocytes (32). Therefore, it is intriguing, that PGI2, known to bind to and activate PPAR-α (33), was found to be elevated in transgenic skin. The putative roles of PGI2 and 15-deoxyΔ12,14-PGJ2 [an activator of PPAR-γ (34)] in sebocyte differentiation presently are being investigated.
Epidermal differentiation was disturbed by the transgenic COX-2 expression, particularly in tail skin. This might be due to the unscheduled COX-2 expression, the high COX-2 and prostaglandin levels, and the expression of prostaglandin receptors (35), but also to the unique epidermal structure. Although the alternating pattern of orthokeratotic interscales and parakeratotic scales remained unaffected in transgenic mice, COX-2 overexpression was accompanied by hyperplasia in the scale epidermis, reflecting both an increase of cell number and hyperkeratosis. Moreover, proliferating keratinocytes were not confined to the basal layer but also were found in suprabasal layers. However, the total number of proliferating cells was comparable to wt epidermis. Thus, this type of hyperplasia clearly differs from hyperproliferation-associated hyperplasia as found in hairless mouse skin (36) or other hyperproliferative skin diseases and in neoplasias of man (37, 38). Furthermore, in hyperplastic areas of transgenics, an expanded expression pattern of K5 protein was observed upon immunohistochemical analysis. This may be a result of epitope unmasking in suprabasal cells, because K5 protein is known to persist in a masked form in suprabasal normal keratinocytes (39). The reduced and disturbed expression of keratin 10, an immediate early marker of epidermal differentiation, involucrin, a marker protein of early differentiation normally expressed in the upper spinous layers (21), and loricrin, a marker of late differentiation (22), indicates that hyperkeratosis of transgenic scale epidermis resulted from a decelerated desquamation of cornified cells rather than an accelerated cornification. Thus, COX-2 overexpression seems to favor the expansion of the pool of proliferative keratinocytes by preventing their entrance into the postmitotic state, which normally is coupled with switching on the expression of the above-mentioned differentiation-associated proteins. It remains to be elucidated whether cell–cell and cell–matrix interactions required for epithelial differentiation (40) become disturbed upon COX-2 overexpression, as was observed in intestinal cells (41). Similar alterations may occur in COX-2 transgenic hyperplastic-scale epidermis, leading to a loss of keratinocyte polarity. The phenotypical changes observed in transgenic tail epidermis are quite similar to early epidermal dysplasia during mouse skin tumorigenesis (42). The epithelial lobes deeply protruding into the dermis of transgenic mouse tails, consisting of keratin lamellae surrounded by rather regularly stratified layers including an intact basal cell layer, resemble seborrheic keratosis of man. COX-2-positive papillary hyperplastic lobes were surrounded by vessels, and vessel density near these lesions was increased. This observation indicates that constitutive COX-2 expression in basal epidermal cells may suffice to stimulate vessel growth into previously avascular regions. In line with this are recent reports showing COX-2 expression to be crucial for tumor angiogenesis (10, 43).
In conclusion, some of the histological changes observed in COX-2 transgenic mice, including the locally apparent signs of dysplasia within hyperplastic lobes of tail epidermis, may be typical for premalignant lesions in humans and mice. In fact, there is ample evidence that pharmacological (12, 15, 17) or genetic (44) inactivation of COX-2 may result in a strong inhibition of mouse skin carcinogenesis, pointing to a causal relationship between COX-2 expression and the development of squamous cell carcinomas in skin.
Acknowledgments
We thank M. Blessing (University of Mainz, Germany) for providing the K5-promoter construct, D. L. DeWitt (Michigan State University, East Lansing, MI) for the COX-2 full-length cDNA, F. Watt (Imperial Cancer Research Fund, London) for the anti-involucrin antibody, L. Langbein for the anti-K5 antibody, K. Goerttler for evaluating tissue specimens, and J. Schweizer for helpful discussions (L. Langbein, K. Goerttler, and J. Schweizer are all from the German Cancer Research Center, Heidelberg), and S. Pfrang, B. Steinbauer, A. Pohl-Arnold, D. Kucher, U. Beckhaus, M. Henrich, and U. Klotz for excellent technical assistance.
Abbreviations
- COX
cyclooxygenase
- PG
prostaglandin
- ORS
outer root sheath
- K5
keratin 5
- RT-PCR
reverse transcription–PCR
- wt
wild type
- H&E
hematoxylin/eosin
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Marnett L J, Rowlinson S W, Goodwin D C, Kalgutkar A S, Lanzo C A. J Biol Chem. 1999;274:22903–22906. doi: 10.1074/jbc.274.33.22903. [DOI] [PubMed] [Google Scholar]
- 2.Martin C, Ullrich V. In: Prostaglandins, Leukotrienes and Other Eicosanoids: From Biogenesis to Clinical Application. Marks F, Fürstenberger G, editors. New York: Wiley; 1999. pp. 89–108. [Google Scholar]
- 3.Murakami M, Naraba H, Tanioka T, Semmyo N, Nakatani Y, Kojima F, Fueki M, Ueno A, Oh-ishi S, Kudo I. J Biol Chem. 2000;275:32783–32792. doi: 10.1074/jbc.M003505200. [DOI] [PubMed] [Google Scholar]
- 4.Tanioka T, Nakatani Y, Semmyo N, Murakami M, Kudo I. J Biol Chem. 2000;275:32775–32782. doi: 10.1074/jbc.M003504200. [DOI] [PubMed] [Google Scholar]
- 5.Vane J R, Bakhle Y S, Botting R M. Annu Rev Pharmacol. 1998;38:97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- 6.Müller-Decker K, Scholz K, Neufang G, Marks F, Fürstenberger G. Exp Cell Res. 1998;242:84–91. doi: 10.1006/excr.1998.4068. [DOI] [PubMed] [Google Scholar]
- 7.Scholz K, Fürstenberger G, Müller-Decker K, Marks F. Biochem J. 1995;309:263–269. doi: 10.1042/bj3090263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buckman S Y, Gresham A, Hale P, Hruza P, Anast J, Masferrer J, Pentland A P. Carcinogenesis. 1998;19:723–729. doi: 10.1093/carcin/19.5.723. [DOI] [PubMed] [Google Scholar]
- 9.Marks F, Fürstenberger G. Eur J Cancer. 2000;36:314–329. doi: 10.1016/s0959-8049(99)00318-4. [DOI] [PubMed] [Google Scholar]
- 10.Masferrer J L, Leahy K M, Koki A T, Zweifel B S, Settle S L, Woerner B M, Edwards D A, Flickinger A G, Moore R J, Seibert K. Cancer Res. 2000;60:1306–1311. [PubMed] [Google Scholar]
- 11.Charyalertsak S, Sirikulchayanonta V, Mayer D, Kopp-Schneider A, Fürstenberger G, Marks F, Müller-Decker K. Gut. 2000;48:80–86. doi: 10.1136/gut.48.1.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Müller-Decker K, Scholz K, Marks F, Fürstenberger G. Mol Carcinogen. 1995;12:31–41. doi: 10.1002/mc.2940120106. [DOI] [PubMed] [Google Scholar]
- 13.Oshima M, Dinchuk J E, Kargman S L, Oshima H, Hancock B, Kwong E, Trazaskos J M, Evans J F, Taketo M M. Cell. 1996;87:803–809. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- 14.Müller-Decker K, Reinerth G, Krieg P, Zimmermann R, Heise H, Bayerl C, Marks F, Fürstenberger G. Int J Cancer. 1999;82:648–656. doi: 10.1002/(sici)1097-0215(19990827)82:5<648::aid-ijc6>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 15.Müller-Decker K, Kopp-Schneider A, Seibert K, Marks F, Fürstenberger G. Mol Carcinogen. 1998b;23:36–44. doi: 10.1002/(sici)1098-2744(199809)23:1<36::aid-mc5>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 16.Wang H Q, Smart R C. J Cell Sci. 1999;112:3497–3506. doi: 10.1242/jcs.112.20.3497. [DOI] [PubMed] [Google Scholar]
- 17.Fischer S M, Lo H, Gordon G B, Seibert K, Keloff G, Lubet R A, Conti C J. Mol Carcinogen. 1999;25:231–240. [PubMed] [Google Scholar]
- 18.Blessing M, Nanney L B, King L E, Jones C M, Hogan B L. Genes Dev. 1993;7:204–215. doi: 10.1101/gad.7.2.204. [DOI] [PubMed] [Google Scholar]
- 19.Meade E A, Smith W L, DeWitt D L. J Lipid Med. 1993;6:119–129. [PubMed] [Google Scholar]
- 20.Paus R, Müller-Röver S, Van der Veen C, Maurer M, Eichmüller S, Ling G, Hofmann U, Foitzik K, Mecklenburg L, Handjiski B. J Invest Dermatol. 1999;113:523–532. doi: 10.1046/j.1523-1747.1999.00740.x. [DOI] [PubMed] [Google Scholar]
- 21.Eckert R L, Yaffe M B, Crish J F, Murthy S, Rorke E A, Welter J F. J Invest Dermatol. 1993;100:613–617. doi: 10.1111/1523-1747.ep12472288. [DOI] [PubMed] [Google Scholar]
- 22.Mehrel T, Hohl D, Rothnagel J A, Longley M A, Bundman D, Cheng C, Lichti U, Bisher M E, Steven A C, Steinert P M, et al. Cell. 1990;61:1103–1112. doi: 10.1016/0092-8674(90)90073-n. [DOI] [PubMed] [Google Scholar]
- 23.Schweizer J. In: Molecular Biology of the Skin: The Keratinocyte. Blumenberg M, Darmon M, editors. San Diego: Academic; 1993. pp. 33–77. [Google Scholar]
- 24.Li-Stiles B, Lo H H, Fischer S M. J Lipid Res. 1998;39:569–582. [PubMed] [Google Scholar]
- 25.Schadow A, Scholz K, Lambeau G, Gelb M H, Fürstenberger G, Pfeilschifter J, Kaszkin M. J Invest Dermatol. 2001;116:31–39. doi: 10.1046/j.1523-1747.2001.00179.x. [DOI] [PubMed] [Google Scholar]
- 26.Seibert K, Zhang Y, Leahy K, Hauser S, Masferrer J, Perkins W, Lee L, Isakson P. Proc Natl Acad Sci USA. 1994;91:12013–12017. doi: 10.1073/pnas.91.25.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanchez T, Moreno J J. Biochem Pharmacol. 1999;58:877–879. doi: 10.1016/s0006-2952(99)00169-0. [DOI] [PubMed] [Google Scholar]
- 28.Dinchuk J E, Car B D, Focht R J, Johnston J J, Jaffee B D, Covington M B, Contel N R, Eng V M, Collins R J, Czerniak P M, et al. Nature (London) 1995;23:406–409. doi: 10.1038/378406a0. [DOI] [PubMed] [Google Scholar]
- 29.Morham S G, Langenbach R, Loftin C D, Tiano H F, Vouloumanos N, Jennette J C, Mahler J F, Kluckman K D, Ledford A, Lee C A, et al. Cell. 1995;83:473–482. doi: 10.1016/0092-8674(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 30.Hardy M H. Trends Genet. 1992;8:159–166. doi: 10.1016/0168-9525(92)90350-d. [DOI] [PubMed] [Google Scholar]
- 31.Weiss L W, Zelickson A S. Acta Dermatovener. 1975;55:431–442. [PubMed] [Google Scholar]
- 32.Rosenfield R L, Kentsis A, Deplewski D, Ciletti N. J Invest Dermatol. 1999;112:226–232. doi: 10.1046/j.1523-1747.1999.00487.x. [DOI] [PubMed] [Google Scholar]
- 33.Hertz R, Berman I, Keppler D, Tana J. Eur J Biochem. 1996;235:242–247. doi: 10.1111/j.1432-1033.1996.00242.x. [DOI] [PubMed] [Google Scholar]
- 34.Forman B M, Tontonoz P, Chen J, Brun R P, Spiegelman B M, Evans R M. Cell. 1995;83:803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 35.Goetzl E J, An S, Smith W L. FASEB J. 1995;9:1051–1058. doi: 10.1096/fasebj.9.11.7649404. [DOI] [PubMed] [Google Scholar]
- 36.Berton T R, Mitchell D L, Fischer S M, Locniskar M F. J Invest Dermatol. 1997;109:340–347. doi: 10.1111/1523-1747.ep12335984. [DOI] [PubMed] [Google Scholar]
- 37.Lu S, Tiekso J, Hietanen S, Syrjanen K, Havu V K, Syrjanen S. Acta Derm Venereol. 1999;79:268–273. doi: 10.1080/000155599750010634. [DOI] [PubMed] [Google Scholar]
- 38.Kawahira K. Arch Dermatol Res. 1999;291:413–418. doi: 10.1007/s004030050431. [DOI] [PubMed] [Google Scholar]
- 39.Lloyd C, Yu Q, Cheng J, Turksen K, Degenstein L, Hutton E, Fuchs E. J Cell Biol. 1995;129:1329–1344. doi: 10.1083/jcb.129.5.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fuchs E, Dowling J, Segre J, Lo S H, Yu Q-C. Curr Opin Gen Dev. 1997;7:672–682. doi: 10.1016/s0959-437x(97)80016-0. [DOI] [PubMed] [Google Scholar]
- 41.Tsujii M, DuBois R N. Cell. 1995;83:493–501. doi: 10.1016/0092-8674(95)90127-2. [DOI] [PubMed] [Google Scholar]
- 42.Aldaz C M, Conti C J, Klein-Szanto A J P, Slaga T J. Proc Natl Acad Sci USA. 1987;84:2029–2032. doi: 10.1073/pnas.84.7.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prescott S M. J Clin Invest. 2000;11:1511–1513. doi: 10.1172/JCI10241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Langenbach R, Loftin C, Lee C, Tiano H. Biochem Pharmacol. 1999;58:1237–1246. doi: 10.1016/s0006-2952(99)00158-6. [DOI] [PubMed] [Google Scholar]